|
SEMINARIO 6 |
PURIFICACION: METODOS Y CRITERIOS, ANALISIS PARAMETROS
1) Una enfermedad desconocida hasta hace poco venía causando la muerte de niños menores de diez años, particularmente en las cercanías de la cuenca del Río Reconquista. Tras una serie de estudios se llegó a la conclusión de que la patología era causada por una especie bacteriana mutante, surgida en dicho río debido a su fuerte contaminación con desechos industriales. Si unas pocas de estas bacterias eran ingeridas en el agua o a través de ciertos alimentos (verduras), su potente toxina conducía a la sintomatología característica, y a la muerte, en alrededor de una semana.Se descubrió que cierta enzima que puede extraerse de las hojas del filodendro hidroliza específicamente esta toxina, pudiendo administrarse por vía endovenosa sin efectos colaterales. Se sabe que la misma es un monómero de 95 kDa, tiene un pI de alrededor de 5,7 y se encuentra normalmente en el citoplasma, constituyendo alrededor del 0,5% de las proteínas totales. Para aislarla, se ensayó el protocolo tentativo que se muestra en la Tabla 1.5
a) Calcule la recuperación y el rendimiento y grafíquelos en función del No de etapa de purificación. ¿Cuántas veces se purificó dicha enzima?
b) De acuerdo a los resultados obtenidos por Ud. en (a), ¿cree que el protocolo de purificación es óptimo, o puede mejorarse?
c) Ya que obtener una pureza de 100% en el producto con un alto rendimiento es un objetivo demasiado ambicioso, debe programarse una solución de compromiso adecuada al uso que se va a hacer de la proteína. En el presente caso, ¿Cuál es el rendimiento máximo que teóricamente podría esperarse?. Y el rendimiento que se obtuvo ¿fué satisfactorio en relación con aquél?. Defina (en los términos mencionados) cuál sería su objetivo para esta purificación. ¿Cree que el protocolo mostrado lo cumpliría?. ¿Cómo lo evaluaría?.
d) Suponga que Ud. posee un análogo no hidrolizable de la toxina (capaz de unirse específicamente al sitio activo). ¿Cómo lo utilizaría para mejorar el esquema de purificación?
Tabla 1.5
| ACT.ENZ. | PROTEINAS | VOL | |
| (mUE/ml) | (mg/ml) | (ml) | |
| Extracción en PBS pH=7.5 | 345.2 | 50.0 | 1000 |
| Pptación. con SO4(NH4) 60% saturación | 581.7 | 12.0 | 500 |
| Sephadex G-200* | 17252.5 | 50.0 | 10 |
| Diálisis (corte: 50 kDa) | 6.922.0 | 25 | 20 |
| Q-Sepharosa** | 34570.0 | 50 | 2 |
* Exclusión molecular, rango de retención: 5-600 kDa
** Intercambiador de aniones
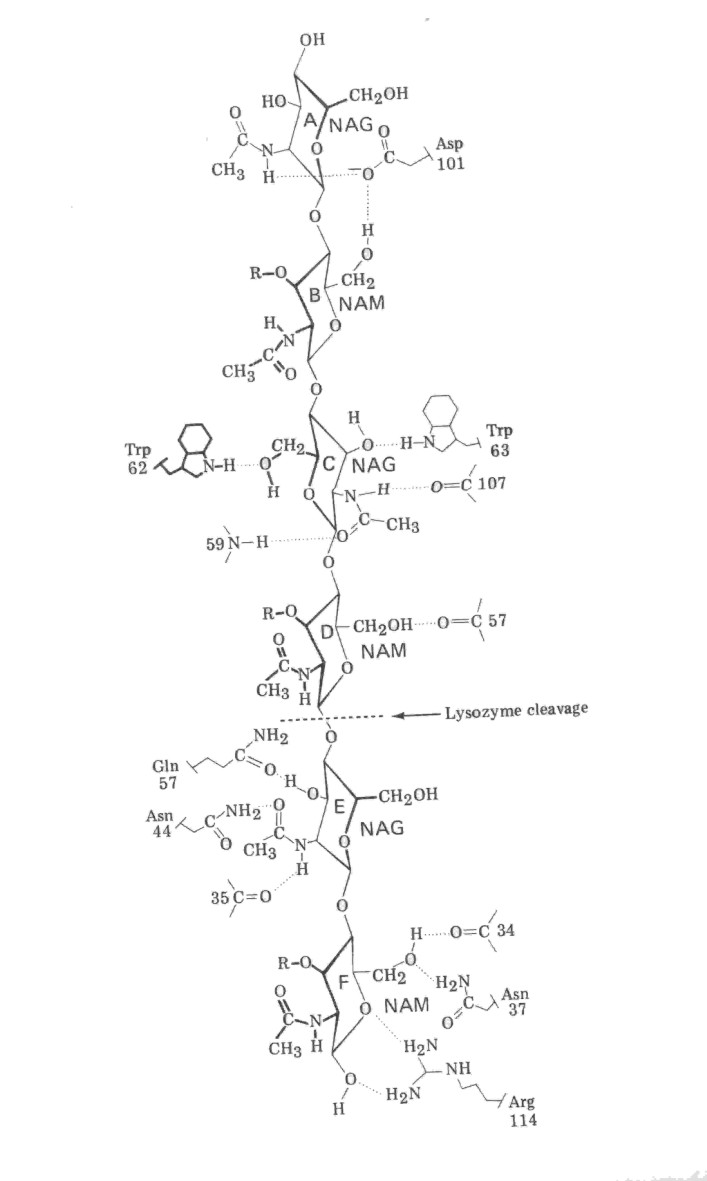
2) La lisozima es una enzima presente en grandes cantidades en la clara de huevo, cuya función consiste en hidrolizar uniones glicosídicas de polisacáridos (lo que le permite degradar paredes celulares bacterianas).
El esquema de la Figura 1.10 representa al sitio activo de esta enzima con un análogo de su sustrato (el NAG-NAM)3 unido a él. Las líneas de puntos representan puentes de hidrógeno y las líneas llenas los enlaces covalentes (las más gruesas simbolizan posiciones por encima del plano del papel). No se ha representado la cadena polipeptídica completa, sino sólo las posiciones relativas en el espacio correspondientes a los aminoácidos que juegan un papel en el proceso catalítico.
La Figura 1.11 es un esquema del mecanismo de la reacción, donde las flechas indican transferencia de electrones.
a) Calcule la energía de ligado (pegado, binding) del (NAG-NAM)3 a la lizsozima, suponiendo que la energía de enlace promedio para un puente de hidrógeno es de 5 kcal/mol, y no tomando en cuenta las interacciones de van der Waals.
b) Esquematizando el proceso de catálisis como constando de dos etapas: ligado e hidrólisis, una mutación cuya consecuencia sea la sustitución de un aminoácido por otro puede afectar una o ambas de dichas etapas. Explique, en este marco qué consecuencias tendrá:
b1) La sustitución de Glu35 por Gln
b2) La sustitución de Asn44 por Phe
b3) La sustitución de Asp52 por Arg
Figura 1.10
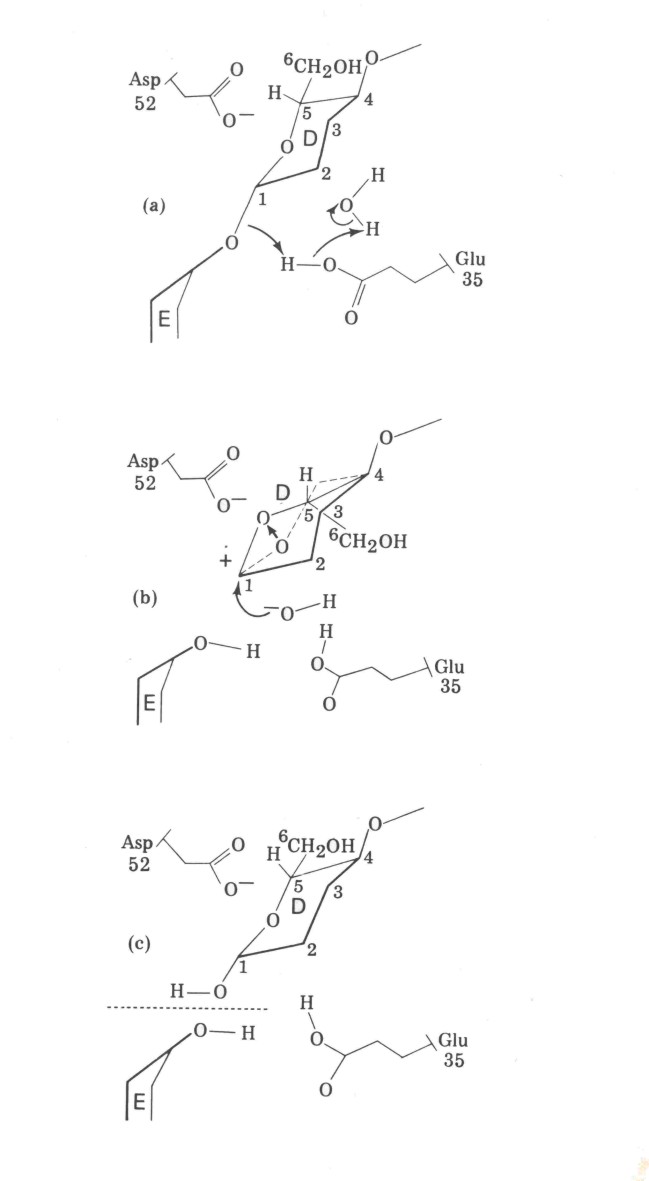
Figura 1.11
3) Se desea purificar la a -galactosidasa de arveja, para lo cual se llevó a cabo el siguiente protocolo: 500 g de harina de semilla se extrajeron con 750 ml de buffer y se centrifugaron a 5.000 xg 10 min. El sobrenadante se llevó a pH=3,0 con ácido cítrico, manteniéndose en esta condición a 4
0C durante la noche con agitación. Luego se eliminó el precipitado por centrifugación, y el sobrenadante se llevó a pH=4,8. Se eliminaron los ácidos nucleicos mediante un tratamiento enzimático. Posteriormente se realizó una precipitación con acetona 30% (v/v final), eliminándose el precipitado por centrifugación. El sobrenadante se llevó a 60% (v/v) de acetona, se centrifugó y el precipitado se resuspendió en 20 ml de buffer pH=4,8. Finalmente, el resuspendido se dializó exhaustivamente contra buffer pH=4,8 siendo el volumen final de 5 ml.La Tabla 1.6 muestra los resultados experimentales obtenidos para las distintas etapas de la purificación.
Tabla 1.6
|
ETAPA |
VOLUMEN |
ACT.ENZ. |
PROTEINA |
|
(ml) |
(mUE/ml) |
(mg/ml) |
|
|
Extracción |
750 | 450 | 37.5 |
| Variación de pH | 800 | 274 | 4.0 |
| Degr. de Ac. nucleicos | 810 | 270 | 3.5 |
|
Pptación. con acetona |
20 | 6900 | 42.0 |
|
Diálisis |
5 | 16560 | 19.5 |
a) Con estos datos determine el número de veces que se purificó la enzima en cada etapa de purificación (con respecto a la anterior), y la recuperación y rendimiento con respecto a la etapa inicial.
b) Una alícuota del dializado se sembró en una columna de exclusión molecular obteniéndose el cromatograma de la Figura 1.12. Se tomaron muestras del pico I y el pico II y se corrió un gel de poliacrilamida con SDS, obteniéndose el resultado de la Figura 1.13.
Otra alícuota del dializado se cromatografió en la misma columna de exclusión molecular pero en presencia de Urea 8M, el cromatograma resultante se muestra en la Figura 1.14. (la actividad se midió después de dializar).
En un experimento independiente se estudió la estabilidad térmica de las fracciones correspondiente a los picos I y II de la Figura 1.12. Para ello se incubó cada pico durante 15 minutos a una temperatura comprendida entre 30 y 80oC (véase Figura 1.15). Inmediatamente después se ensayó la actividad enzimática a 30oC. En la Figura 1.15 se grafica la actividad mantenida en cada tratamiento respecto del inicial (antes de la preincubación).
En función de todos estos resultados desarrolle un modelo molecular para las proteínas del pico I y del pico II de la Figura 1.12.
4) En la purificación de una enzima se obtuvieron los resultados de la Tabla 1.7
Tabla 1.7
| FRACCION | ACT. ESPECIFICA (U/mg) | RENDIMIENTO (%) |
| 1) Extracto crudo | 0.41 | 100 |
| 2) Calentamiento (50ºC, 5 min) | 0.59 | 96 |
| 3) Pptac. salina | 3.67 | 55 |
| 4) DEAE-Sephadex eluído con gradiente de pH | 9.80 | 44 |
|
5) DEAE-Sephadex eluído con gradiente de ClK |
52.00 | 36 |
| 6) Filtración por BioGel P-100 | 185.00 | 34 |
| 7) Cromatografía en hidroxiapatita | 500.00 | 30 |
a) Calcule cuántas veces se purificó la enzima en cada paso.
b) ¿Cuál es el paso más eficiente?
c) ¿Qué paso descartaría? ¿Por qué?
d) ¿Por qué se usa la actividad específica para seguir la purificación?
e) Considere las veces que se purificó la enzima y el rendimiento en proteína. ¿Cree que debe darle preferencia a un parámetro sobre el otro?.
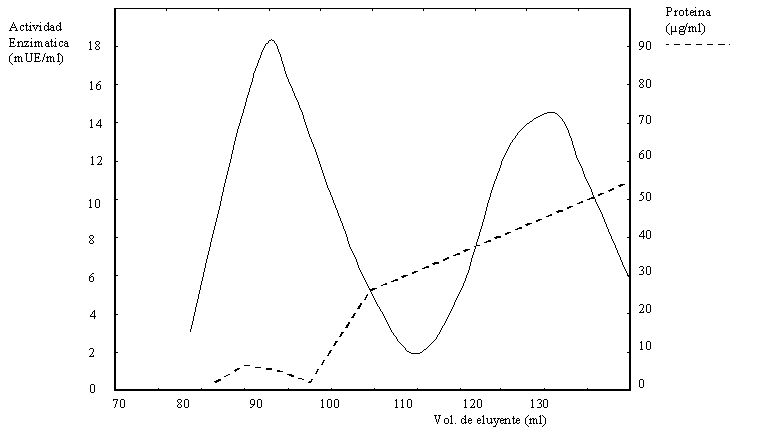
Figura 1.12
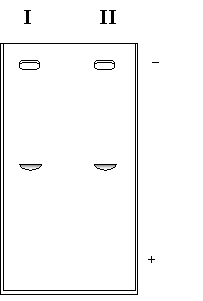
Figura 1.13
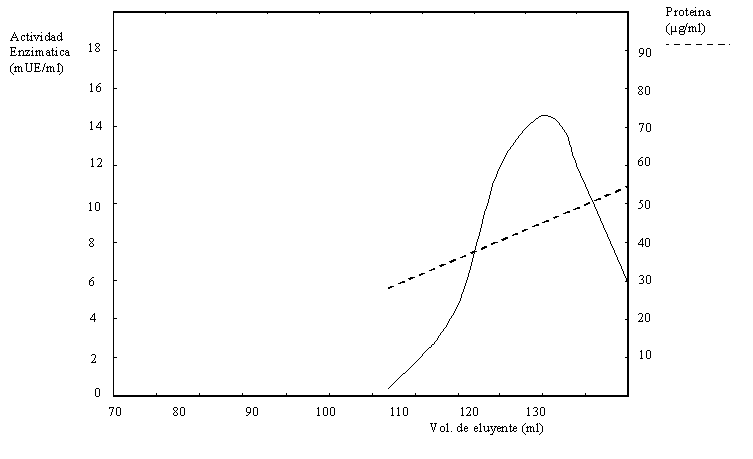
Figura 1.14
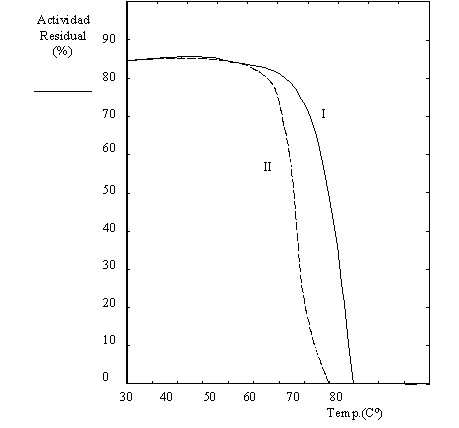
Figura 1.15
5) Las proteínas provenientes de la harina de soja están constituídas por fracciones identificadas por ultracentrifugación como 2S, 7S, 11S y 15S. La fracción 11S está formada por una proteína globular cuyo peso molecular, obtenido a través de cromatografía de exclusión molecular es de 350kD. Se observó en SDS-PAGE la aparición de una única banda de aproximadamente 58kD. No obstante, los resultados de una SDS-PAGE con b -mercaptoetanol mostraron el perfil quee se muestra en la Figura 1.16.
Para analizar dicha fracción, los extractos de harina de soja se centrifugaron y la fracción soluble se precipitó con sulfato de amonio en un rango de saturación de 40%-60%. El precipitado se solubilizó en un buffer adecuado y se realizó una cromatografía de intercambio iónico en Q-Sheparosa. Finalmente se llevó a cabo una crioprecipitación. En cada una de las fracciones purificadas se determinó la cantidad de proteína por el método de Biuret utilizando albúmina bovina como patrón. La actividad fue seguida utilizando una reacción específica para detectar la proteína 11S y expresada como Unidades 11S (U11S). Se observan los resultados en la Tabla 1.8.
Tabla 1.8. Purificación de la proteína 11S
|
Paso de purificación |
proteína (mg) |
actividad (U 11S) |
|
Sonicación |
8000 |
4800 |
|
Precipitación |
750 |
2730 |
|
Q-Sepharosa |
225 |
2200 |
|
Crioprecipitación |
35 |
1820 |
a) ¿Cuál es la estructura de la proteína 11S, qué tipos de enlaces participan en ella?
b)- Discuta los resultados de la Tabla 1.8 e indique si realizaría alguna modificación. Compare con la SDS-PAGE (Figura 1.16, calle Salv), considerando que el objetivo es utilizar esta proteína para análisis estructural.
Cuando se analizaron con SDS PAGE sin ß-mercaptoetanol los productos de la purificación de extractos de distintas variedades (Figura 1.17A) y las fracciones solubles e insolubles de la precipitación con sulfato de amonio (Figura 1.17B), se encontraron dos variedades (G1 y Ser2) que eran semejantes a la variedad Salv, en las cuales se supuso estaba modificada la composición aminoacídica.
c) Basándose en los resultados, identifique las mutaciones que llevan las variedades G1 y Ser2, y cómo cree usted que afectan éstas la estructura de la proteina 11S.
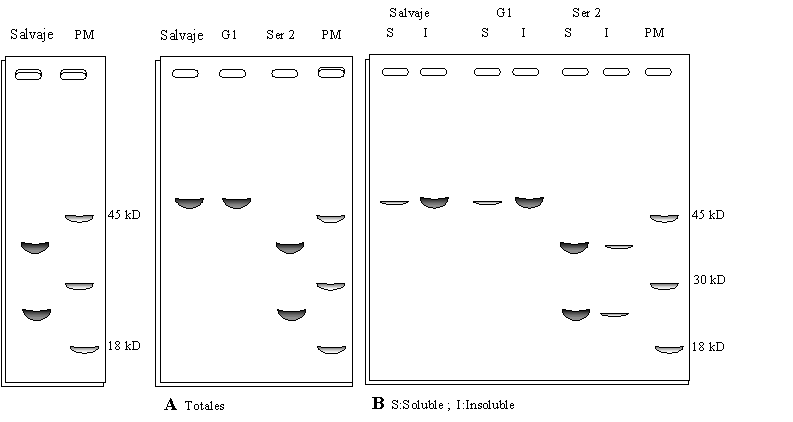
Figura 1.16 Figura 1.17
6) Se estudia una toxina bacteriana de naturaleza proteica y con actividad enzimática, la adenilato ciclasa (AC). Esta proteína proveniene de un microorganismo patógeno para el hombre, y su actividad patogénica consiste en alterar funciones metabólicas en células eucarióticas del hospedador a través de un incremento exacerbado del AMPc intracelular.
La enzima cataliza la siguiente reacción:
ATP -----------------> AMPc + PPi
Mediante una serie de ensayos se determinó que en el microorganismo dicha proteína se encuentra parte en el citosol y parte asociada a la membrana. Se pudo determinar que además del sitio de unión al ATP, la AC posee otro u otros para asociarse a una proteína eucariótica, la calmodulina.
El objetivo es determinar la estructura de la adenilato ciclasa, para lo cual previamente se desarrolló un protocolo de purificación.
A continuación se describen brevemente los pasos seguidos para la purificación de la proteína. Se adjunta además datos que se obtuvieron de la aplicación de cada uno de ellos:
1- Células bacterianas crecidas en medio líquido (2 l de medio) fueron centrifugadas a 6000 x g a 4
oC durante 20 min. El sobrenadante fue descartado y el sedimento celular fue resuspendido en 15 ml de tampón de extracción pH 8,02- luego de una incubación de una hora a 4
0C con agitación se realizó una centrifugación a 8000 xg y en este caso el sedimento fue descartado.3- se procedió a una precipitación con 2 volúmenes de acetona a -20
0C durante dos horas.4- luego de separar el sobrenadante del precipitado por centrifugación, el precipitado se disolvió con tampón pH 7,2, vol. final 2,0 ml.
5- dicho volumen se cromatografió en una columna de afinidad. De las fracciones colectadas se pudo detectar actividad en cuatro tubos cada uno de los cuales contenía 200 m l.
6- el volumen de los cuatro tubos se cromatografió mediante una columna de exclusión molecular (25.000-80.000). Sólo se pudo recuperar la actividad luego de una elución exhaustiva (Ve>>> Vo incluído)
Se concentró entonces la proteína y a través de distintas metodologías se determinó su peso molecular: 19.000 Da
Para evaluar este protocolo de purificación se midió en cada una de las etapas la actividad de la adenilato ciclasa a 30
0C (1U: 1 pmol de AMPc formado por min.) y proteínas por el método de Lowry. Para la determinación de proteínas se confeccionó una curva de calibración utilizando un patrón de albúmina de 300 m g/ml, en un volumen final de 1,3 ml obteniéndose los resultados que se muestran a continuación:| Tubo | 1 | 1´ | 2 | 2´ | 3 | 3´ | 4 | 4´ |
| Alb(ml) | 0 | 0 | 0.1 | 0.1 | 0.15 | 0.15 | 0.20 | 0.20 |
| DO-670nm | 0.013 | 0.015 | 0.060 | 0.075 | 0.100 | 0.904 | 0.126 | 0.130 |
Las determinaciones se realizaron a partir de 0,2 ml de muestra en un vol final de 1,3 ml
La Tabla 1.9 presenta los datos obtenidos del proceso de purificación
Tabla 1.9
| FRACCION | ACTIVIDAD AC. pmol AMP c/min.ml | DIL. P/DETERMINAR PROTEINAS | DO - 670 nm |
| Extracto | 128 | 1:20 | 0.110 |
| Ppdo. de acetona | 256 | 1:80 | 0.150 |
| Cromat. afinidad | 194 | 1:10 | 0.080 |
| Mezcla de fracciones:800ml Cromat. exclusión | 20 | 1:2 | 0.015 |
a) ¿Cómo realizaría el primer paso? Indique brevemente las condiciones, el tampón y las precauciones que debería tomar
b) Fundamente el paso 3
c) ¿Qué tipo de relleno emplearía para armar la columna de afinidad?
d) ¿Cuántas veces se purificó la adenilato ciclasa en cada uno de los pasos?
e) ¿Cómo mejoraría este protocolo? ¿Qué etapa agregaría o descartaría?
f) ¿Qué técnicas pueden usarse para la concentración de proteínas después del paso 6?
g) ¿Qué criterio utilizaría para establecer el grado de pureza de su preparación?
7) Se logró desarrollar una droga anticancerígena a partir del factor ß de necrosis tumoral humano (TNF-ß). Un estudio in vitro sirvió para demostrar que el TNF-ß tenía actividad citolítica y citotóxica provocando la muerte de células malignas, que son más susceptibles que las células normales.
El TNF-ß nativo (soluble y activo) fue purificado, obteniéndose dos fracciones proteicas, una pequeña fracción con la proteína activa y soluble y la otra con la proteína insoluble agregada en cuerpos de inclusión (CI) inactiva. A ambas fracciones se le realizaron electroforesis nativas y desnaturalizantes cuyos perfiles se observan en la Figura 1.18; en el gel A calle 2 no fue posible observar la fracción insoluble.
El objetivo ahora es lograr la purificación del TNF-ß a partir de los cuerpos de inclusión, para lo cual se utilizó los siguientes buffers:
|
A (pH=8) |
Tris |
B |
Buffer A |
C (pH=10.5) |
Na2B4O7 |
| ClNa | SDS | OHNa | |||
| EDTA | Lisozima | EDTA | |||
| OMSF | Urea |
El protocolo a seguir fue el siguiente (resumido en la Tabla 1.10), las bacterias K12HB101 clonadas con el gen rhTNF-ß (que codifica para la proteína de interés) en el plásmido pCG402 fueron crecidas en un fermentador automático a 30°C, se agregó un inductor IAA y se suplementó el medio con leucina y prolina. Se tomaron 1.000 ml del medio en la fase exponencial de crecimiento y se diluyó 3,5 veces para obtener una DO (600nm) de 0,8. Las bacterias fueron lavadas en buffer A y resuspendidas en 10 ml del mismo buffer. Posteriormente fueron sonicadas 3 veces (1 minuto cada vez) y centrifugadas a 17.400 xg por 30 minutos a 4°C. El pellet se resuspendió en 10 ml de buffer B y se incubó 20 minutos a temperatura ambiente. Luego se centrifugó a 17.400 xg por 15 minutos. El pellet lavado fue solubilizado en el buffer C (por cada gramo de pellet se usan 10 ml de buffer) ajustando el pH a 7,5 con HCl. 10 ml del resuspendido anterior se sembraron en una columna de intercambio catiónico (CM-Sepharosa), obteniéndose 2 picos. El pico II de la corrida anterior se sembró en una columna de intercambio aniónico (DEAE-Sepharosa) y se recolectó la fracción no unida. Como paso final se utilizó una técnica que permite la renaturalización de las proteínas, a lo que se llamó producto final. En cada uno de los pasos se determinó la cantidad de proteína utilizando el método de Lowry y la cantidad de TNF-ß por medio de un método específico.
Tabla 1.10
|
Pasos de purificación |
V(ml) |
Prot(mg) |
TNFß(mg) |
|
Extracto Crudo (1g) |
10 |
145 |
15,4 |
|
Sobrenadante lisis celular |
10 |
94 |
5,7 |
|
Pellet lisis celular |
10 |
51 |
9,7 |
|
Lavado Buffer B solubilizado C |
10 |
29,7 |
7,8 |
|
Después de intercambios iónicos |
29 |
5,8 |
5,3 |
|
Producto final |
30 |
3,1 |
2,9 |
Como comprobación del trabajo se realizó una cromatografía de exclusión molecular del producto final, para lo cual se calibró la columna con los siguientes patrones de peso molecular: Albúmina bovina (67 kDa), Ovoalbúmina (43 kDa), Quimotripsinógeno (25 kDa) y Citocromo C (12,4 kDa). Obteniéndose el perfil de la Figura 1.19 A y B.
a) Según los resultados de la Tabla 1.10 y la Figura 1.19 ¿se purificó satisfactoriamente el TNF-ß?.
b) Explique en forma breve y concisa cómo realizaría la cromatografía de exclusión molecular y qué información le entrega.
c) Utilizando la información general del problema y la Figura 1.18, ¿podría indicar cómo están formados los cuerpos de inclusión?.
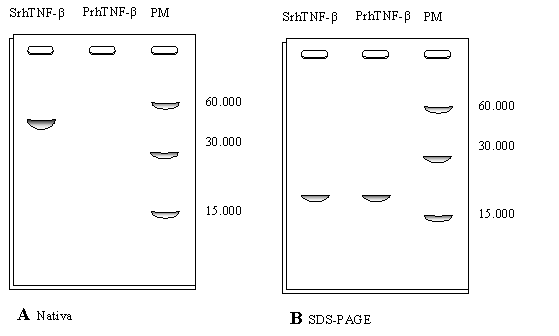
Figura 1.18
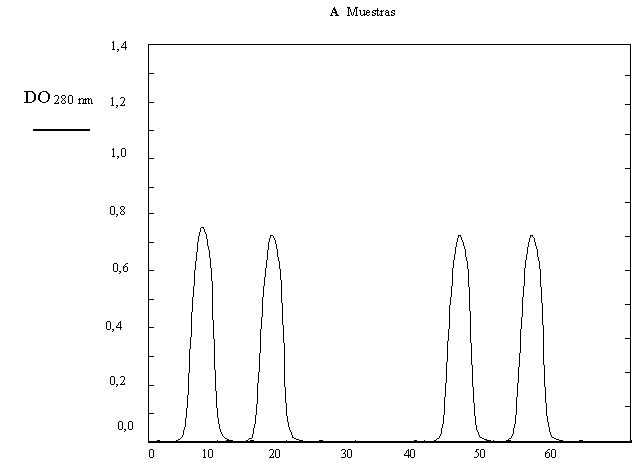
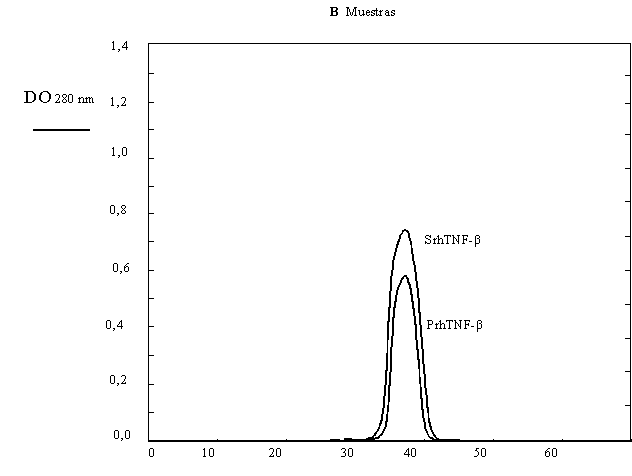
Figura 1.19
8) La bacteria Fusobacterium mortiferum coloniza el tracto oral y gastrointestinal de humanos y animales provocando absesos y necrosis, sensibilizando además dichos tejidos a infecciones por otros patógenos. Para controlar dicha bacteria se han iniciado estudios de sus requerimientos metabólicos. Se observó que durante la infección estas bacterias metabolizan la maltosa. La actividad mediante la cual pueden captar este disacárido es la fosforilación de la maltosa para dar maltosa 6-fostato y su posterior hidrólisis mediante una maltosa 6-fosfato hidrolasa (MalH). Esta enzima puede hidrolizar una variedad de glucósidos 6-fosforil-a -D ligados, entre ellos el 4-metilumbelliferil-a -D-glucopiranósido 6-fosfato (4MUa Glc6P), que al hidrolizarse libera 4-metilumbeliferona (4MU), la cual es fluorescente al ultravioleta.
Para estudiar las características de esta enzima, un litro de cultivo de F. mortiferum fue lisado por sonicado y centrifugado y el sobrenadante separado, el que constituyó el extracto crudo. A dicho extracto se lo llevó a pH=4,9, se lo dejó equilibrando a 4oC una noche, se lo centrifugó y al precipitado resultante se lo resuspendió en 5 ml de buffer fosfatos pH=7,0. A este resuspendido se lo sembró en una columna de exclusión molecular Sephadex G-100 y se recogió el material correspondiente a la zona de entre 90-110 kDa, que eluyó en un volumen de 20 ml. A dicho eluído se lo precipitó con 3 volúmenes de acetona a -20oC durante 1 hora, se lo centrifugó y resuspendió en 1 ml de buffer fosfatos pH=7,0. Para medir la cantidad de proteínas totales y la actividad de MalH se tomó 1,0 ml del extracto crudo, 0,1 ml del resuspendido de la primera precipitación, 0,5 ml del eluído de la exclusión molecular y 0,03 ml del resuspendido de la segunda precipitación. A cada muestra se la llevó a un volumen final de 10 ml en buffer fosfatos pH=7,0. Luego, de las muestras así preparadas correspondientes al extracto crudo y al resuspendido de la primera precipitación, se las diluyó 1:10 en un volumen final de 5 ml cada una, y de las otras se tomó 5 ml de las respectivas preparaciones sin diluír y a cada muestra se la dividió en alícuatas de 1 ml. Con tres alícuotas de cada muestra se dosó proteínas totales (por triplicado) mediante el método de Bradford. En la Tabla 1.11 se dan los valores de DO obtenidos conjuntamente con los correspondientes a una curva de calibración realizada a partir de una solución stock de 10 mg/ml de albúmina. A cada una de las tres alícuotas restantes de 1 ml de cada muestra se agregó 1 ml de una solución que contenía la concentración apropiada de 4MUa Glc6P como para obtenerse concentraciones finales de 2,0; 7,0 y 15,0 mM en cada tubo. Cada solución de 4MUa Glc6P agregada a los tubos se preparó diluyendo adecuadamente un stock de 100,0 mM de 4MUa Glc6P. La reacción se inicó al momento de agregarse el 4MUa Glc6P y se siguió mediante la fluorescencia obtenida en 2; 5; 10 y 20 min de incubación a 37oC. Dichos valores fueron luego convertidos a m moles de 4MU por tubo de reacción. en la Tabla 1.12 se dan los valores obtenidos.
Se realizó además un análisis cualitativo de la purificación, cuyo resultado se muestra en la Figura 1.20.
Con la proteína obtenida en la muestra correspondiente al resuspendido de la última precipitación, se analizó el efecto de ciertos iones sobre su actividad (Tabla 1.13).
Se desarrolló un segundo ensayo de actividad en el cual 20 m l de extractos crudos de E. coli conteniendo 20 m g de proteína total se sembraron en geles de poliacrilamida nativos y luego de la corrida, los geles se incubaron con 4MUa Glc6P y se observaron en un transiluminador con luz ultravioleta, obteniéndose el resultado de la Figura 1.21A. Con este ensayo se determinó el efecto de la composición del medio de cultivo sobre la síntesis de MalH y de glucógeno (Figura 1.21B).
a) Explique el fundamento de cada una de las técnicas de purificación empleadas, como así también las características fisicoquímicas discriminadas.
b) ¿Qué diluciones se realizaron a partir del stock de albúmina para obtenerse los puntos de la curva de calibración para el método de Bradford?
c) En base a los datos de la Tabla 1.11, calcule la concentración de proteínas en cada muestra.
d) ¿Qué diluciones se realizaron para obtenerse las concentraciones apropiadas de 4MUa Glc6P a partir del stock con las que se realizaron los ensayos de actividad, y cuáles eran éstas?
e) Calcule el rendimiento, la recuperación y cuántas veces purificó la enzima.
f) Analice el resultado de la Figura 1.20 y sugiera un modelo molecular para esta enzima.
g) Explique cómo se realizó el experimento de la Tabla 1.13 y qué información obtiene de él.
h) ¿Qué conclusión obtiene de las Figuras 1.21A y B?
Tabla 1.11. Valores de DO para la curva de calibración y para las distintas muestras de la purificación de MalH
|
Muestra |
DO595 tubo1 |
DO595 tubo2 |
|
Calibr. Albúmina, 0 m g |
0.031 |
0.028 |
|
Calibr. Albúmina, 5 m g |
0.168 |
0.173 |
|
Calibr. Albúmina, 10 m g |
0.280 |
0.320 |
|
Calibr. Albúmina, 15 m g |
0.430 |
0.429 |
|
Calibr. Albúmina, 20 m g |
0.636 |
0.627 |
|
Muestra |
DO595 tubo1 |
DO595 tubo2 |
|
Extracto crudo |
0.528 |
0.531 |
|
1er. Precipitado |
0.381 |
0.380 |
|
Exclusión molecular |
0.433 |
0.429 |
|
2do. Precipitado |
0.479 |
0.482 |
Tabla 1.12. m moles de 4MU obtenidos a distintos tiempos con tres concentraciones de 4MUa Glc6P en las mezclas de reacción correspondientes a muestras del proceso de purificación de MalH (cada dato es promedio de tres determinaciones).
|
Sustrato (mM) |
2,0 |
|||
|
Tiempo (min) |
2 |
5 |
10 |
20 |
|
Extr. Crudo |
0.6 |
1.1 |
3.2 |
6.4 |
|
1er. Ppdo. |
11.7 |
29.3 |
58.5 |
117.1 |
|
Excl. Mol. |
89.4 |
223.5 |
447.1 |
894.3 |
|
2do. Ppdo. |
98.5 |
246.3 |
492.7 |
985.3 |
|
Sustrato (mM) |
7.0 |
|||
|
Tiempo (min) |
2 |
5 |
10 |
20 |
|
Extr. Crudo |
1.6 |
4.0 |
8.1 |
12.1 |
|
1er. Ppdo. |
29.1 |
72.8 |
145.6 |
218.5 |
|
Excl. Mol. |
222.5 |
556.3 |
999.2 |
1780.3 |
|
2do. Ppdo. |
245.2 |
613.0 |
1226.0 |
2084.2 |
|
Sustrato (mM) |
15.0 |
|||
|
Tiempo (min) |
2 |
5 |
10 |
20 |
|
Extr. Crudo |
2.3 |
5.9 |
11.8 |
16.5 |
|
1er. Ppdo. |
42.7 |
106.7 |
213.4 |
277.5 |
|
Excl. Mol. |
326.1 |
815.2 |
1141.3 |
1630.4 |
|
2do. Ppdo. |
359.3 |
898.2 |
1796.4 |
2694.6 |
Tabla 1.13. Estudio del efecto de iones y de la diálisis en la actividad con MaIH.
|
Determinación |
Act Sp (m mol de 4MUa Glc6P hidrolizado/mg de proteína/min) |
Actividad relativa |
|
Efecto de diálisis sobre la actividad enzimática |
||
|
Control antes de la diálisis |
0.117 |
1 |
|
Diálisis, sin agregado |
0.020 |
0.17 |
|
Diálisis + 1 mM DTT |
0.015 |
0.13 |
|
Diálisis + 1 mM Mn+2 |
0.603 |
5.15 |
|
Restauración de la act. enzimática por iones divalentes del extracto dializado |
||
|
Sin agregado de metales |
0.016 |
1 |
|
+1mM Ni+2 |
0.092 |
5.8 |
|
+1mM Mn+2 |
0.145 |
9.1 |
|
+1mM Co+2 |
0.181 |
11.1 |
|
+1mM Fe+2 |
0.241 |
15.1 |
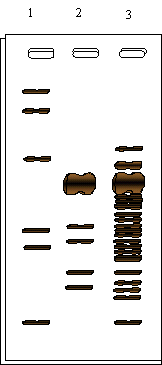
Fig 1.20. Análisis cualitativo de la purificación mediante SDS-PAGE y tincion con plata.
Calle1:Marcador PM Calle2:Producto final purificación Calle3:Extracto crudo
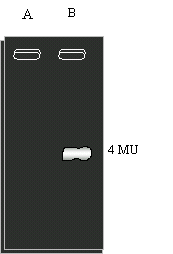
Fig 1.21 A. Actividad Mal II en geles de poliacrilamida .
CalleA: E.coli sin plasmido CalleB: E.coli con plasmido pCB4.11

Fig 1.21 B. Actividad Mal II en geles de poliacrilamida de extractos obtenidos en diferentes condiciones de cultivo.
Calle1: Alto contenido N y glucosa como única fuente de C. Calle2: Alto contenido N y maltosa como única fuente de C.
Calle3: Bajo contenido N y glucosa como única fuente de C. Calle4: Bajo contenido N y maltosa como única fuente de C.
![]()
Seminarios
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
![]()
Home Docentes Cronograma Programa Materia Programa TP Seminarios Links Publicaciones Bibliografia