|
SEMINARIO 5 |
ESTRUCTURA CELULAR, PURIFICACION: METODOS Y CRITERIOS
1) Se desea separar tres proteínas de la semilla de soja, para lo cual se sigue el siguiente protocolo: Después del procedimiento de extracción con buffer de la harina de semillas de soja, se obtiene 200 ml de un extracto crudo. Al mismo se le hace una precipitación isoeléctrica a pH=4,5 y se lo centrifuga, resuspendiéndose el pellet en 200 ml de buffer fosfato pH=6,0. Se toman muestras de 1 ml de cada fracción, y a la correspondiente al extracto crudo se le agregan 9 ml de agua destilada, mientras que a la proveniente del resuspendido, 1 ml de agua destilada. Un ml de cada una de estas diluciones se procesa adecuadamente para estimar su contenido de proteínas por el método de Biuret. También se realiza la curva de calibración para dicho método, arrojando los siguientes valores:
|
Contenido de proteínas (mg) |
|||||
|
BLANCO |
1.0 | 2.0 | 3.0 | 4.0 | |
|
DO (550) |
0.053 | 0.109 | 0.150 | 0.191 | 0.262 |
| 0.049 | 0.091 | 0.142 | 0.200 | 0.242 | |
a) Grafique la curva de calibración. Diga qué contenido de proteínas había en el extracto crudo y en el resuspendido de la precipitación isoeléctrica si los valores de DO550 observados fueron 0,147 y 0,223 respectivamente. Calcule el rendimiento.
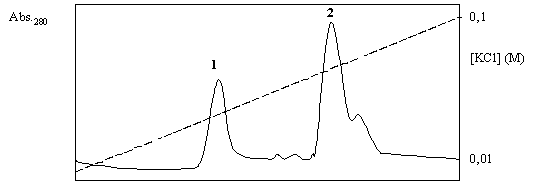
b) Se corre una cromatografía de intercambio iónico del resuspendido en Q-Sepharosa (un intercambiador de aniones) a pH 6,0 y se eluye con un gradiente de KCl de 0,01 a 0,1 M. Se obtiene el cromatograma que se muestra en la Figura 1.5.
Luego, se toman muestras de los picos 1 y 2 y se los corre en PAGE con (Figura 1.6a) y sin (Figura 1.6b) SDS a pH=8.
Analice y explique estos resultados. ¿Cómo obtendría por separado las proteínas que se observa?. Fundamente su respuesta y explique cómo se cercioraría del éxito de la separación.
Figura 1.5
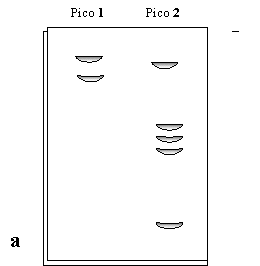
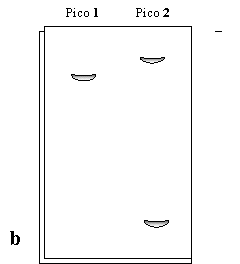
Figura 1.6
2) Se purificó una proteína a homogeneidad, y en una caracterización preliminar de su estructura se realizó una cromatografía de exclusión molecular en condiciones nativas (Figura 1.7b, pág. siguiente) o en presencia de ß-mercaptoetanol (Figura 1.7c). En la Figura 1.7a se muestra el cromatograma de los patrones de peso molecular, que fueron: albúmina bovina (66.000 Da), anhidrasa carbónica bovina (29.000 Da), citocromo c de caballo (12.400 Da) y aprotinina bovina (6.500 Da). El volumen muerto se determinó con azul dextrano (2.000.000 Da) y fue de 5 ml.
Paralelamente, se corrió un PAGE-SDS con (Figura 1.8a) y otro sin (Figura 1.8b) ß-mercaptoetanol. En las calles de la izquierda de cada gel se sembraron los patrones de peso molecular, que fueron: albúmina bovina (66.000 Da), albúmina de huevo (45.000 Da), gliceraldehído 3-P deshidrogenasa de conejo (36.000 Da), anhidrasa carbónica bovina (29.000 Da), tripsinógeno bovino (24.000 Da), inhibidor de tripsina de soja (20.100 Da) y a -lactalbúmina bovina (14.200 Da). En lass calles de la derecha de cada gel se sembró la proteína, tratada (Figura 1.5a) o no (Figura 1.5b) con ß-mercaptoetanol.
¿Cuál es la estructura cuaternaria de la proteína?
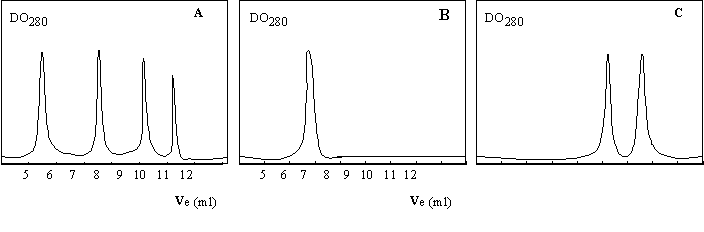
Figura 1.7
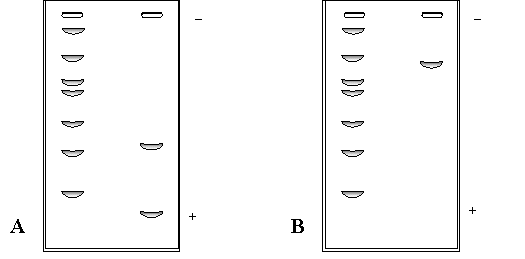
Figura 1.8
3) Se realizó la purificación y parcial caracterización de la enzima uroporfirinógeno I sintasa (UPGI-S) de eritrocitos humanos. Esta enzima cataliza la polimerización de cuatro moléculas de porfobilinógeno para formar uroporfirinógeno I.
El esquema de purificación fue el siguiente:
1.- Obtención de eritrocitos, lisis y centrifugación del material a 27.000 xg durante 90 min.
2.- El sobrenadante se eluyó por columna de DEAE-celulosa (buffer fosfato 0,03 M pH=7,3). La columna se lavó hasta eluír la hemoglobina. La celulosa se separó, agitó en buffer fosfato 0,13 M pH=7,4 y se centrifugó a 23.000 xg durante 10 min. con el objeto de recuperar la UPGI-S en el sobrenadante.
3.- El sobrenadante se precipitó con sulfato de amonio al 70% de saturación final. Se centrifugó y el precipitado se disolvió en fosfato 0,05 M pH 7,4/b -mercaptoetanol 0,01 M/glicerol 3%.
4.- El precipitado, disuelto en buffer, se eluyó por columna de Sephadex G-100.
5.- Las fracciones con actividad enzimática se combinaron y se trataron 15 min. a 60oC. Luego de enfriar en agua-hielo se centrifugó a 10.000 xg durante 10 min.
6.- El sobrenadante se eluyó por columna de DEAE-Sephadex A-50. El buffer de carga fue fosfato 0,05 M pH=7,4/b -mercaptoetanol 0,01 M. La columna se elluyó con un gradiente lineal de NaCl entre 0 y 0,2 M, en el mismo buffer.
Se dosó la actividad enzimática y la concentración de proteínas (ésta por el método de Lowry) a lo largo de la purificación.
Resultados obtenidos Tabla 1.2
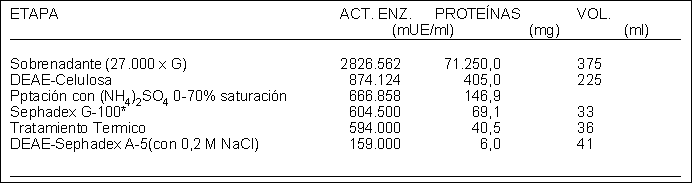
a) La elección de los métodos de separación empleados requirió de un previo conocimiento de algunas características fisicoquímicas de la UPGI-S. ¿Cuáles y por qué?
b) La elección de la secuencia en la que se emplean estos métodos requiere de algunos criterios. ¿Ud está de acuerdo con los criterios utilizados?. Fundamente sus acuerdos, críticas o dudas.
c) La modificación (o no) de un diseño experimental de purificación requiere del análisis crítico de los resultados obtenidos. Complete la tabla, calculando rendimientos y grados de purificación, y en función de sus cálculos decida si cree conveniente mantener (o no) este diseño para futuros experimentos. ¿Cree necesario añadir algún procedimiento analítico?
4) A partir de extractos de la levadura Trichosporon cutaneum se han purificado dos enzimas. Ambas catalizan la hidrólisis de la lactosa.
Una de las actividades enzimáticas corresponde a una b -galactosidasa y cataliza la hidrólisis de lactosa y otros b -galactósidos. La otra actividad correspponde a una b -glicosidasa e hidroliza tanto b -galactósidos como b -glucósidos.
Método de dosaje de las actividades enzimáticas: detección colorimétrica del producto 4-nitrofenol en medio alcalino. Sustratos de la reacción: 4-nitrofenil-b -D-galactopiranósido (PNP-gal) y 4-nitroofenil-b -D-glucopiranósido (PNP-glu).
Método de determinación de la concentración de proteínas: Lowry.
Purificación:
1.- El homogenado se centrifugó a 35.000 xg durante 60 min. El buffer empleado (buffer A) fue: fosfatos de sodio 8 mM; EDTA 1mM; DTT 1mM; pH=7,0.
2.- El sobrenadante se corrió en DEAE-Sepharose CL-6B equilibrada con buffer A+NaCl 0,1 M. Se eluyó con un gradiente lineal entre 0,1 y 0,25 M NaCl. En las fracciones se dosó la actividad enzimática y se determinó la absorbancia a 280 nm (Figura 1.9).
3.- El material de cada pico se concentró por ultrafiltración. Se corrió en Fast-Flow DEAE-Sepharose, en buffer A+NaCl 0,1 M. Se eluyó por gradiente lineal entre 0,1 y 0,4 M de NaCl.
4.- El material de cada pico se concentró por ultrafiltración. Se corrió en Mono Q (HR 5/5) equilibrado con buffer A+0,1 M NaCl. Se eluyó con gradiente lineal entre 0,1 y 0,4 M NaCl.
5.- El material de cada pico se sembró en Phenyl Superose (HR 5/5) equilibrada con buffer A+(NH4)2SO4 1,4 M. Se eluyó con un gradiente lineal de 1,4 a 0 M (NH4)2SO4.
6.- El material de cada pico se corrió en S-300 HR en buffer A.
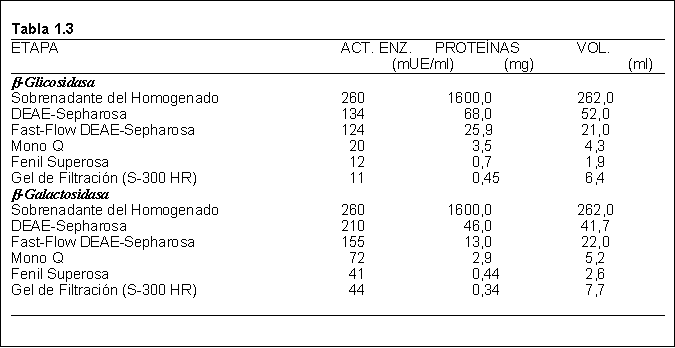
Los resultados obtenidos en las diferentes etapas de purificación se muestran en la Tabla 1.4. Ambas purificaciones enzimáticas fueron al menos 90% puras, según SDS-PAGE.
a) Describa brevemente el fundamento de los métodos cromatográficos de separación empleados. Indique qué parámetros fisicoquímicos de las macromoléculas determinan su velocidad de elución.
b) La purificación realizada aún puede ser optimizada. Analice los resultados obtenidos e indique sus acuerdos y/o desacuerdos con el esquema seguido. Si tiene modificaciones que proponer hágalo. Fundamente siempre y haga los cálculos que crea necesario.
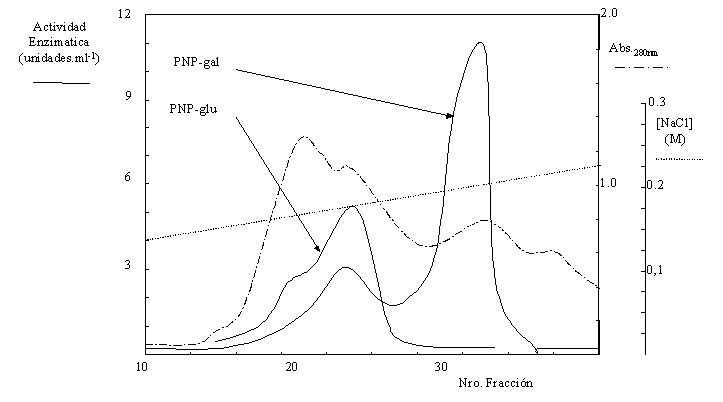
Figura 1.9
5) La fosforilasa es un tetrámero con subunidades de 96.000 Da unidas por interacciones hidrfóbicas. Describa su comportamiento con gráficos en una cromatografía de exclusión molecular en presencia de:
a) Urea 7 M
b) SDS 0,5%
c) EDTA 1 mM
En los mismos gráficos ubique las posiciones de la lisozima (14.000 Da), albúmina de suero bovino (68.000 Da) y b -galactosidasa (tetrámero con subunidadees de 116.000 Da).
¿Cuál/es de los siguientes dextranos podría usar como relleno de la columna para separar las distintas proteínas en los experimentos a, b y c?
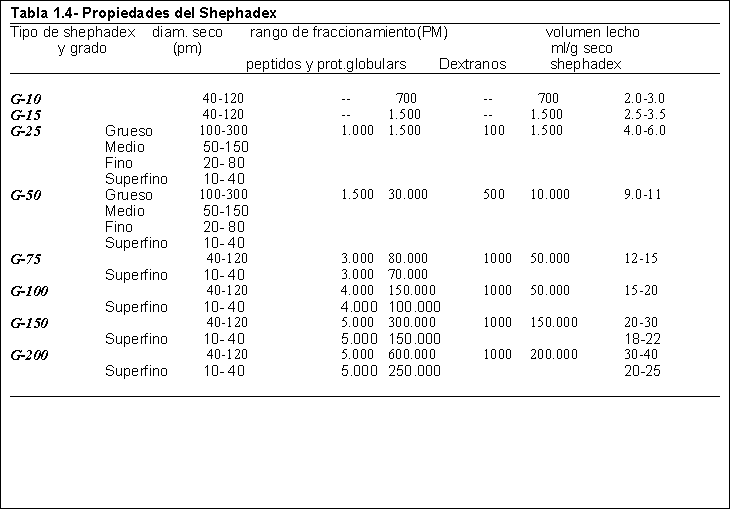
¿Cómo usaría este experimento para determinar el peso molecular de la fosforilasa a y el número de subunidades que forman la proteína nativa, si estos no fueran datos conocidos?.
¿Qué otros métodos podría usar para estimar el peso molecular de las subunidades y de la proteína nativa?.
![]()
Seminarios
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
![]()
Home Docentes Cronograma Programa Materia Programa TP Seminarios Links Publicaciones Bibliografia