Projet de fin d'études
Master Professionnel OPExEtude, classification par spectroscopie proche infra rouge (NIR) des produits cosmétiques
Juan ROSAS
Cédric LACOUR
Matériels et méthodes
I. Spectroscopie UV visible et proche infrarouge
a.- Zone spectrale en infrarouge.
Principe
On appelle « infrarouge » le rayonnement correspondant aux longueurs d’onde directement supérieures à celles du spectre de la lumière visible. Conventionnellement les limites du proche infrarouge se situent entre 800 et 2500 nm.
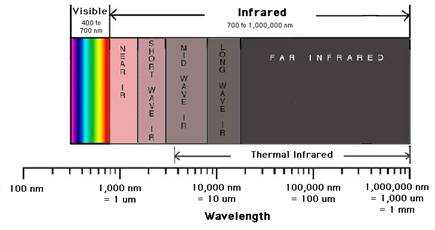
Figure 1: domaine de longueur d’onde
Les domaines de longueur d’onde de rayonnement attribués au proche infrarouge sont de 4000-15 600cm-1, alors que l’UV couvre de 200 – 400 nm.
L’absorption des rayonnements par les échantillons dépend de la composition de la matière organique. En effet les liaisons chimiques sont capables d’absorber les rayonnements correspondant à certaines fréquences particulières. C’est cette propriété des liaisons chimiques de la matière organique que va utiliser la spectroscopie dans le proche infrarouge pour établir un lien entre l’absorption de la lumière et la composition de l’échantillon.
Pour faire cette analyse, l’échantillon va être éclairé à différentes fréquences (ou longueurs d’onde). L’absorption de la lumière à chacune de ces longueurs d’onde constitue le « spectre » de l’échantillon. Ce spectre peut être constitué de plusieurs centaines de longueurs d’onde pour chacune desquelles on a mesuré l’absorption de la lumière.
Le spectre est caractéristique d’un échantillon car il regroupe des informations (quantité et caractéristiques) de chacun de ses constituants organiques (protéines, matières grasses, fibres, etc.).
Cette richesse d’information constitue l’avantage et la difficulté de l’analyse SPIR : beaucoup d’informations sont présentes dans un spectre, mais elles sont complètement emmêlées ! Pour surmonter cette difficulté il faut faire appel à des méthodes statistiques complexes, qui vont permettre de relier les spectres et les analyses chimiques : c’est la phase de « calibration ». Chaque équation de calibration est spécifique d’un paramètre chimique pour une matière première donnée : ce travail doit donc être répété pour chaque nouvelle matière première.
b.- La spectrométrie à transformé de Fourier
Dans le but de réduire le temps d’analyse par échantillon, nous utilisons la spectroscopie proche infrarouge à transformée de Fourier. La source infrarouge est modulée par un interféromètre. Elle va traverser ou s réfléchir su l’échantillon. Puis elle arrive le détecteur proche. Le signal, récupéré par le détecteur, est retranscrit sous forme d’un interférogramme qui va être numérisé et converti par transformation de Fourier en spectre.
c.-Technique de réflexion diffuse
L’étude en moyen infrarouge est réalisée par transmission au travers de pastilles de KBr. La spectroscopie proche infrarouge permet de mesure directement les échantillons en poudre en utilisant la technique de réflexion diffuse.
Dans la technique de réflexion diffuse, le rayon infrarouge est réfléchi par la surface de l’échantillon. Dans le cas de poudres, l’énergie ayant pénétré la matière, est réfléchie dans toutes les directions et on effectue la récolte de la lumière diffusée à l’aide d’un miroir hémisphérique (accessoire Nicolet Near Updrift)
II.- Méthode statistique
Analyse Qualitative
Les méthodes chimiométriques de classification dont le but est de faire ressortir des groupements de points sont réparties en deux catégories [11] : les méthodes non supervisées et les méthodes supervisées.
Pour les premières, on dispose d'un certain nombre de variables et il faut regrouper les échantillons similaires pour former des agrégats, puis les identifier. Les méthodes les plus couronnent utilisées pour la classification non supervisée sont l'analyse en composante principale ACP et les méthodes de classification hiérarchique.
Les méthodes de classification peuvent être regroupées suivant différentes caractéristiques. Ainsi on distingue les méthodes linéaires et les méthodes non linéaires.
1. Méthodes de classification
Les principales techniques de classement supervise sont [7] :
1' analyse linéaire discriminante (LDA ou fonction discriminante de Fisher)
la comparaison directe (ou méthode du centroïde) ;
la stratégie des plus proches voisins (KNN- K Nearest Neighbours)
la méthode SIMCA ( Soft Independent Modeling of Class Analogy) ;
les réseaux de neurones (ANN);
Le tableau 1 résume les principales caractéristiques de ces méthodes. Les 4 premières approches sont couramment utilisées en routine pour l’exploitation des spectres proche infrarouge. Les autres méthodes, plus récentes, sont rarement implantées dans les logiciels de pilotage des spectromètres. Les études menées sur l’approche standard montrent toutefois qu'il n'existe pas de solution optimale. Le choix de la méthode est non seulement dépendant des conditions de la discrimination mais aussi de la structure même du jeu de données.
2. Traitement du signal
Il existe de nombreuses sources de variations spectrales, allant de l'échantillonnage à l' appareillage. Les plus fréquentes dans le domaine de la spectroscopie proche infrarouge sont:
Les interactions inter composés en particulier avec les liaisons hydrogènes intermoléculaire.
La diffusion de la lumière pour les échantillons solides ou les particules en suspension dans un liquide.
La faible reproductibilité du processus de mesure, en particulier dû à la variation du trajet optique.
Les distorsions spectrales dû à l'appareillage qui s'expriment par une dérive de la ligne de base, un déplacement des longueurs d'ondes, des effets dus a la non linéarité du détecteur, à la lumière parasite ou encore du bruit issu du détecteur.
Le but du traitement du signal brut est de réduire, éliminer ou standardiser I' impact des sources de variations spectrales, mentionnées ci-dessus, sans affecter l'information spectroscopique nécessaire a la réalisation du modèle de calibration. On s'affranchit ainsi de l'information parasite.
Dans notre étude, six prétraitements ont été appliques sur les différents groupes au moyen de l'assistant de calibration. Il s'agit de la correction diffuse MSC, de la normalisation par segment, de la normalisation par unité de grandeur, de la première dérivée et du lissage de Savitsky Golay.
a. Dérivée
Elle permet de réduire la dérive de la ligne de base, de séparer plus clairement les bandes d'absorption et d'exalter l'information spectrale. Il existe différentes méthodes pour calculer la dérivée :
La dérivée par intervalle calcule la dérivée sur un intervalle de points fixés par l' utilisateur. La dérivée basée sur la technique de la convolution de Savitsky et Golay [11]. Deux étapes sont nécessaires pour calculer la dérivée en un point: tout d'abord, un polynôme de degré k est ajuste sur au moins k + 1 points du spectre autour du point i. Ensuite la dérivée du polynôme en ce point est calculée.
Normalisation des spectres
Plusieurs types de normalisation existent.
i. Correction Multiplicative de diffusion (MSC)
La correction de diffusion MSC améliore la linéarité de la relation entre l'absorbance et la concentration [12]. Cette méthode est intéressante lorsque des régressions linéaires sont utilisées en particulier pour des analyses en réflexion diffuse sur des échantillons de poudres Pour effectuer la correction MSC, un spectre de référence est nécessaire, le cas échéant il peut s'agir du spectre moyen du set de calibration. Un modèle linéaire est mis en place entre le spectre et le spectre idéal. Le traitement MSC débute par la modélisation des q spectres cl p longueurs d'ondes sous la forme de droites tel que Xi = a+b Xi + ei avec a et b coefficient du modèle et ei erreur à la longueur d'onde i.
Le 'reste' ei correspond aux effets qui ne peuvent pas être modélisés par la constante ou le coefficient directeur de la droite. Ils correspondent en fait aux variations spectrales contenant l'information utile. La méthode MSC commence donc par estimer les constantes et les coefficients directeurs de ces droites par régression linéaire, pour chaque spectre.
Les spectres corriges sont ensuite obtenus par l'équation suivante :
Xcor,i=( Xi -a*i)/ b*i où a*i et b*i sont les estimations de ai et bi
ii. Standar Normalisation Variate (SNV)
Ce traitement s'applique à chaque spectre pris séparément, sans référence à l'ensemble des échantillons d'étalonnage.