And after does it become?
After, in the liver of the patients, phenylalanine is transformed by oxidation into tyrosin , thanks to a specific enzyme: The phenylalanine-hydroxylase which itself needs a cofactor, the t�trahydrobiopt�rine (BH4) .
It is like if in our liver, one had a small tiny workman: the enzyme, whose specific work is to cut phenylalanine of pieces. He has need for one assitant: The cofactor
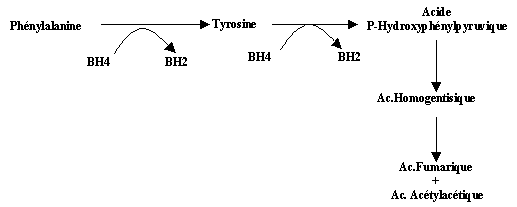
Then by transamination: one obtains acid p-hydroxyphenylpyruvic
By a continuation of transformation one leads to the formation of acetic acid hydroquinone (or ac homogentisic ).
At the end of the metabolism, one obtains fumaric acid and acetic acid ac�tyl .
All these products are used by the organization, or are eliminated by the kidneys if one does not need any.
Eh well, of the anomalies can occur in the normal metabolism of the phenylalanine whose
consequences are: appearance of serious illnesses such as: The
ph�nylc�tonurie .
Transfers producing the disease were described in gene coding the enzyme: phenylalanine-hydroxylase .
The absence of phenylalanine-hydroxylase in the liver of the patients,
involves the blocking of the transformation of tyrosin phenylalanine.
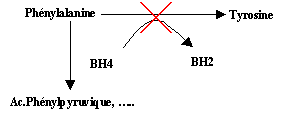
So the ph�nylanalanine is not degraded and accumulates in the blood and fabrics of the
patient, whereas a part is transformed into acid phenylpyruvic and
different catabolites:
This accumulation in the blood and the brain of phenylalanine and these derivatives is toxic for the nervous cells and involve a progressive destruction
of the brain.