The Disease
Chapter 3.: Consequences
![]() Poison, destruction, but it is serious that!!
Poison, destruction, but it is serious that!!
Yes, it is serious, but all depends on the age of the patients:
After the end of the fast development of the brain, the mode must be continued. The toxic effects of high rates of phenylalanine and its derived products are of two types:
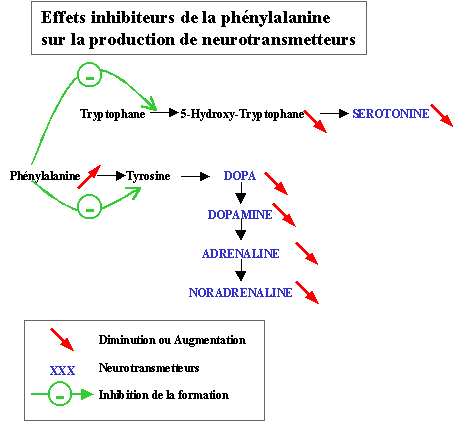
This reduction explains the changes of behavior and performance observed a
few hours after a meal rich in proteins absorptive by a patient phenylcetonuric:
The fall of the phenylalanine rates will allow a rapid standardization of the performances .
The results of cerebral imageries practised by nuclear magnetic
resonance showed anomalies of the white substance when a dietetic control in the long run
of the disease was not satisfactory.
These results are discussed , but while waiting for complementary studies,
one should encourage the continuation of the processing lasting all the life .
Finally the clean experiment of the adolescent and young patients adult who noticed that their performances were better when they were subjected to a strict control of the blood phenylalanine rates also contributed to the development of new recommendations.
The maternal hyperphenylalaninemy exposes:
It appeared that a strict control of the maternal phenylalanine rates before the design was essential, not only in the phenylcetonuric mothers, but also in the hyperphenilalaninemic mothers, i.e. who have a rate moderately raised of phenylalanine which did not have, until the pregnancy, not required particular dietetic processing.
![]() Much of people are reached by this disease?
Much of people are reached by this disease?
The risk to put at the world a phenylcetonuric child for carrying parents is 1
out of 4 .
The frequency in Belgium of this disease is 1 for 15' 000 births ,
with the result that it nait each year in Belgium, on average, approximately 3 or 4
phenylcetonuric children.
![]() How is it known if one is reached by it?
How is it known if one is reached by it?
To the 4 2nd day of life each new-born baby undergoes a blood
test, which makes it possible to seek a certain number of hereditary diseases, before they
appear by clinical signs, and which could be prevented by a processing appropri�.Ce test
of tracking bears the name of its inventor, Dr. Guthrie, an American pediatrist.
Since 1965, this test of Guthrie made it possible to measure
the phenylalanine rates to the birth at more than 2 million and half of new-born babies.
were detected allowing an early processing by a diet.
| Tracking with the
birth of the hereditary metabolic Diseases in Switzerland of 1965 to 1998 |
|||
|---|---|---|---|
| Diseases | Detected cases | Incidence | |
| 1998 | 1965 to 1998 | ||
| ph�nylc�tonurie | 7 | 173 | 1/15' 000 |
| Other hyperph�nylalanin�mies | 7 | 139 | 1/19' 000 |
| Gal-1-P Uridyltransf�rase: total deficit | 1 | 47 | 1/52' 000 |
| Gal-1-P Uridyltransf�rase: Partial deficit | 8 | 458 | 1/5' 300 |
| Galactokinase | 0 | 1 | |
| UDP-Gal-4-�pim�rase | 0 | 17 | |
| Congenital Hypothyr�ose (since 1978) | 13 | 478 | 1/3' 600 |
| Biotinidase: Total deficit (since 1978) | 3 | 11 | 1/92' 000 |
| Biotinidase: Partial deficit | 1 | 21 | 1/48' 000 |
| Congenital Hyperplasy suprarenals | 11 | 77 | 1/7' 648 |
The tracking of a deficiency in BH4 must be practised at the new-born babies with
phenylalanine rates raised with the birth since the assumption of responsibility of these
patients is different from that of the traditional ph�nylc�tonurie.
![]() Don't Ph�nyc�tonurie, hyperphenylalaninemy, moreover, everyone have the
same disease?
Don't Ph�nyc�tonurie, hyperphenylalaninemy, moreover, everyone have the
same disease?
Not! The concept of tolerance to phenylalanine for a given patient is significant
:
-106-