Prev Up Top Next Contents
9.D Chimica del carsismo
La classificazione dei fenomeni carsici basata sul numero delle componenti
conivolte nel processo carsico distingue fenomeni di carsismo (a tre
componenti), ipocarsismo (con meno di tre componenti) e ipercarsismo
(piu` di tre).
Infatti il caso piu` comune delle grotte nel calcare coinvolge tre componenti:
carbonato di calcio, anidride carbonica ed acqua.
Esempi di processi ipocarsici (con meno di tre componenti) sono:
-
grotte in quarzite e in tufo (bradicarsismo) (due componenti)
-
grotte in gesso e in salgemma (tachicarsismo) (due componenti)
- grotte nel ghiaccio (un componente)
- grotte di scorrimento lavico (un componente)
- bolle di gas nella lava (zero componenti)
- grotte di origine tettonica e da erosione (zero componenti)
Quando invece sono coinvolti piu` di tre componenti si parla di
ipercarsismo [
864] [
865] [
866] .
Questi fenomeni sono comunemente ascrivibili ad ambienti idrotermali
ma possono avere rilevanza anche in situazioni "normali".
In genere i processi ipercarsici coinvolgono altri sali oltre alla
CaCO3, che intervengono nella chimica del processo
carsico. Questi sali possono avere uno ione in comune con la sostanza
carsificata, oppure intervengono solo indirettamente influenzando la
forza ionica della soluzione.
In generale l'aggiunta di un sale con uno ione in comune con il soluto
in soluzione diminuisce la solubilita di questo. Tuttavia cio` non e` sempre
vero: l'aggiunta di carbonato di magnesio aumenta la solubilita`
della calcite (effetto Picknett).
Questa e` la ragione dei grandi vuoti al contatto fra calcare e dolomia.
La solubilita` della CaCO
3 cresce al crescere della salinita`
(forza ionica). Ad elevata salinita` (-7, come nel mare) la solubilita`
di CaCO
3 supera 210 ppm, indipendentemente dalla concentrazione
della CO
2 nell'atmosfera.
A basse salinita` (0, come nell'acqua piovana) la solubilita` dipende
invece dalla CO
2 ed e` dell'ordine di 100 ppm.
Valori tipici dei sali disciolti:
- 200 - 500 mg/l nei calcari;
- 2500 - 3000 mg/l nei gessi;
- 360 gl/l per il sale.
La diminuzione della solubilita della sostanza gia` presente, ne provoca la
precipitazione.
Pertanto si possono avere sia fenomeni di accentuata corrosione
che fenomeni di deposizione, di concrezionamento e mineralizzazione.
L'ipercarsismo termale e` dovuto alla miscelazione di acque calde in
risalita con acque fredde di origine meteorica.
9.D.1 Carsismo nel calcare
Il carsismo dipende essenzialmente dalla dissoluzione della roccia
(sovente calcare, percio` principalmente carbonato di calcio, CaCO
3)
da parte delle acque.
L'acqua pura scioglie poco il carbonato di calcio (16 mg/l).
Pero` l'acqua in contatto con l'atmosfera contiene disciolta una piccola
quantita` di CO
2 che e` sufficiente ad aumentare la solubilita`
del carbonato ci calcio
(70 mg/l a 0°C, e 50 mg/l a 15°C).
Nel suolo, sotto una copertura vegetale o sotto la neve,
l'acqua si arricchisce ancor piu` di anidride carbonica, e puo` disciogliere
anche alcune centinaia di mg/l di carbonato di calcio.
La tabella sotto riporta lacuni valori tipici delle concentrazioni di
CaCO
3 disciolta in acqua
[
867] [
868] [
869] [
710] .
| CaCO3 disciolta in acqua
|
| Alta montagna |
100 mg/l
|
| Regioni temperate |
240 mg/l
|
| Regioni mediterranee |
200 mg/l
|
| Regioni intertropicali |
170 mg/l
|
Nelle regioni fredde il gelo inibisce la corrosione carsica superficiale
per parecchi mesi l'anno.
Pero` sotto la neve si formano sacche d'aria ricche di anidride
carbonica che rende l'acqua di fusione piu` aggressiva.
In clima temperato la corrosione e` frenata dall'aumento della
temperatura, ma e` favorita dalla presenza di vegetazione.
Nelle regioni calde e secche invece la corrosione superficiale e`
ostacolata dalla alta temperatura e dalla scarsita` di copertura
vegetale. In queste regioni pero` la corrosione profonda e` possibile
poiche` le acque di condensazione sono molto piu` fredde sotto terra
che in superficie.
Infine nelle regioni calde tropicali la corrosione e` intensa tanto in
superficie quanto in profondita`, a causa principalmente della
vegetazione.
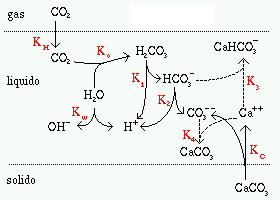
Fig. 359. Chimica del carsismo
9.D.1.1 Equilibrio CO2 - H2O
La quantita` di anidride carbonica disciolta in acqua e` proporzionale
alla pressione parziale di essa nell'atmosfera (legge di Henry)
che esprime l'attivita` della CO
2 nella fase liquida in funzione della
pressione parziale della stessa nella fase gassosa,
aCO2 = mCO2 gCO2 = KH PCO2
dove il coefficiente
KH e` la costante di Henry per la CO
2.
Questa costante varia da gas a gas, e in genere cresce con decrescere
della temperatura.
La molalita`
m rappresenta il numero di moli di CO
2
disciolte per massa di acqua,
m =
n/(
na Ma ),
dove
M denota la massa molare.
Il coefficiente di attivita` g
i tende all'unita` per soluzioni molto
diluite.
Spesso si usa la costante di Henry effettiva, H = K
H/g
H, percui
questa relazione si scrive
mCO2 =
H pCO2.
Altre quantita` utilizzate sono la molarita`, la frazione molare, e la
frazione di massa. La molarita` e` il numero di moli per volume di
acqua,
[i] = ni / V
= ni d / ( na Ma + ∑nj Mj )
dove
d e` la densita` del liquido. La molarita` viene anche
denotata
ci. Essa e` utile nel calcolo di reazioni chimiche
ma la molalita` e` piu` utile in calcoli di termodinamica.
La frazione molare varia fra 0 ed 1,
xi = ni / ( na + ∑nj )
Infine la frazione di massa e`
ni Mi / ( na Ma + ∑nj Mj )
La frazione di anidride carbonica disciolta puo` essere espressa in
litri per litro d'acqua o in milligrammi (il rapporto fra questi due
vale 1.964 gr/l =44/22.4), secondo la tabella sotto riportata
che contiene i coefficienti di solubilita` alla pressione di una
atmosfera.
Quando la pressione parziale della CO2 e` inferiore,
la quantita` disciolta in acqua e` ottenuta moltiplicando questi
coefficienti per la sua pressione parziale (per esempio in aria
questa vale circa 0.0003 atm).
La percentuale di anidride carbonica nelle cavita` e` superiore.
Il tasso di CO2 nel sottosuolo e nel reticolo di fratture
vale tra 2 e 6 % . Nella zona delle gallerie aerate scende a 0.1 - 2 % .
Nella zona profonda di dissoluzione ha valori generalmente compresi
fra 0.2 e 4 % .
Nel caso di dissoluzione di acidi deboli HY bisogna tener conto anche della
dissociazione in soluzione in ioni H
+ e Y
- per cui si ha
la costante d'equilibrio
Ka = aH+ aY- / aHY
= ( mH+ gH+ mY- gY- ) / mHY gHY
La quantita` totale di gas disciolto e` maggiore che se non ci fosse
dissociazione,
mHY + mY- = KH pHY
( gHY-1 + Ka (gH+ gY- mH+)-1 )
= H pXY
avendo usato la costante di Henry effettiva.
Per gli acidi forti HX la dissociazione e` praticamente completa,
la "costante di Henry" vale
K'
a = K
H K
a = a
H+ a
X- / p
HX.
In tal caso la costante di Henry effettiva diviene
H = K'a ( gH+ gY- mH+)-1.
La soluzione di anidride carbonica in acqua provoca la formazione di ioni
carbonato e bicarbonato in soluzione.
CO32- con H+ secondo le reazioni
schematizzate in figura, le cui costanti di equilibrio sono
riportate nella tebella sotto.
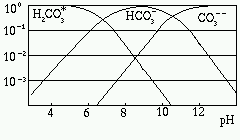
Fig. 360. Frazioni ioniche
La maggior parte della CO
2 in soluzione rimane come molecola lineare
O-C-O idrata. Una parte forma il complesso CO
32- + 2 H
+.
Questi due sono sovente raggruppati introducendo la
concentrazione effettiva della CO
2 disciolta,
come somma di quella libera in soluzione e quella legata,
[ H2CO3* ]
= [ CO2 ] + [ H2CO3 ]
= ( 1 + Ko ) [ H2CO3 ]
Il valore di 1/
Ko e` piccolo, circa 0.003, pertanto
solo lo 0.3% della CO
2 in soluzione e` dovuta all'acido carbonico.
Le costanti di equilibrio delle reazioni sono espresse in termini delle
attivita`,
ax, dei reagenti.
Queste sono legate alle concentrazioni, [x], tramite i coefficienti di
attivita`
gx =
ax / [x], i quali dipendono dalla
forza ionica della soluzione,
I = (1/2) ∑Zx2 [x]
secondo la legge di Debye-Huckel (ed estensioni empiriche a concentrazioni
oltre 0.01 mol Kg
-1 che tentano di tener in conto interazioni
fra gli ioni in soluzione),
log(
gx) = - A I
1/2 Z
x2
/ ( 1 +
B I
1/2 r
x ), dove
rx e`
il raggio ionico effettivo del reagente ( che vale 9 per [H
+],
4.5 per [CO
32-], 4 per [HCO
3-], e 3 per [OH
-] ).
I coefficienti
A e
B valgono
A=0.4883 + 8.074 10
-4 T[°C] e
B=0.3241 + 1.6 10
-4 T[°C].
L'equilibrio di queste reazioni e` governato dal pH della soluzione
oltre che dalla pressione della CO2 nell'atmosfera.
La figura a lato riporta le frazioni molari di
H2CO3*, HCO3- e
CO32- rispetto alla concentrazione totale di
atomi di carbonio. Da notare che per pH inferiori a 8.3 (come e`
generalmente il caso per le acque carsiche che sono solitamente leggermente
basiche avendo pH compreso fra 7 e 8-8.5), la frazione di
[ CO32- ] e` trascurabile.
La cinetica di questo equilibrio e` dominata dalla reazione
H2O + CO2 ->H2CO3,
che e` abbastanza lenta. Tipicamente ci vogliono 50 s per arrivare
al 50% dell'equilibrio e 100 s per il 99
9.D.1.2 Equilibrio CO2 - H2O - CaCO3
In soluzione acquosa il carbonato di calcio e` ionizzato
CaCO
3 ->Ca
2+ + CO
32-
e la CO
32- si combina con H
+ per
formare HCO
3-. Si tratta di tre reazioni scrivibili
globalmente
CaCO3 + H2O + CO2
->CaCO3 + H+ + HCO3-
->Ca2+ + 2 HCO3-
Il prodotto (solubilita`) delle concentrazioni dei due ioni all'equilibrio e`
Kc = [Ca
2+]
eq [CO
32-]
eq
e dipende solo dalla temperatura.
I valori di
Kc sono molto piccoli e decrescono leggermente
con la temperatura.
La saturazione rispetto al calcio e` il rapporto rispetto a
Kc delle effettive concentrazioni
[Ca2+] [CO32-] / Kc
Gli ioni Ca2+ in soluzione reagiscono con
HCO3- e con CO32-.
Queste due reazioni hanno costanti d'equilibrio molto piccole e possono
essere trascurate nelle condizioni dei carsi naturali.
La dissoluzione del carbonato di calcio aumenta [CO32-]
e diminuisce [H+] e [H2CO3*], cioe` essa consuma
anidride carbonica
(una molecola di CO2 per ogni molecola di CaCO3).
L'evoluzione di queste concentrazioni dipende dal rapporto fra i volumi
di atmosfera e liquido, X = Vg / Vaq.
Quando l'acqua scorre nelle fessure senza atmosfera o nella
zona allaganta (sistema chiuso) X=0. Quando scorre nelle gallerie
il sistema e` aperto e X e` molto grande.
L'equilibrio chimico e` governato dalle leggi di massa (tabella sotto),
dalla condizione di neutralita` elettrica,
2 [Ca2+] + [H+] = 2 [CO32-]
+ [HCO3-] + [OH-]
(che essenzialmente si riduce a 2 [Ca
2+] =
[HCO
3-], e quindi la forza ionica e` circa
I = 3 [Ca
2+] ), dalla condizione di conservazione
degli atomi di carbonio,
[HCO3-] + [H2CO3*]
+ [CO32-] + X [CO2(g)]
= [Ca2+] + [HCO3-]i
+ [H2CO3*]i
+ [CO32-]i
+ X [CO2(g)]i
Da notare che [CO
32-]
i e` trascurabile,
e [H
+]
i = [HCO
3-]
i
quindi per la legge di massa,
[HCO3-]i =
( [H2CO3*]i K1 / cH,i cHCO3,i
)1/2.
Queste equazioni messe assieme permettono di ottenere una relazione
fra [Ca
2+] e [H
+], dopodiche` le concentrazioni
delle altre specie ioniche possono essere ricavate.
Queste relazioni descrivono l'andamento chimico della soluzione
al variare della concentrazione di ioni calcio disciolti.
La cinetica delle reazioni chimiche e` dominata dalla reazione piu`
lenta, percio` e` il passaggio in soluzione del carbonato di calcio
che determina la velocita` d'insieme di questo sistema di equazioni
chimiche.
Il tasso di dissoluzione "teorico" alla superficie e` dato dalla somma
delle attivita` moltiplicate per le costanti cinetiche delle reazioni
dirette meno quella inversa (equazione di Plummer, Wigley, Parkhurst)
FPWP = k1 (H+)
+ k2 (H2CO3*)
+ k3
- k4 (Ca2+) (HCO3-)
Questa equazione pero` da' valori troppo grandi, di un ordine di
grandezza, perche` non tiene conto della diffusione degli ioni
nell'acqua e del ruolo della CO
2.
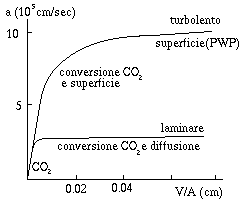
Fig. 361. Coefficente di dissoluzione
Il tasso di dissoluzione alla superficie e` proporzionale ad una funzione
della differenza fra la concentrazione degli ioni alla superficie,
[x]s, e quella di equilibrio:
Fs = as f( [x]eq - [x]s ).
Questo deve essere pari alla diffusione nel liquido,
Fd = (D/h) ( [x]s - [x]l ),
dove D e` il coefficiente di diffusione (tipicamente 10-5
cm2/s), h e` lo spessore
dello strato limite, e [x]l e la concentrazione nel liquido.
La cinetica e` dominata dalla diffusione quando rs e`
grande, e dalla dissoluzione alla superficie quando e` piccolo.
Nel processo pero` interviene anche la conversione di CO2
in H+ e HCO3-, il cui tasso e`
fCO2 A/V, dove A e` l'area della superficie
e V e` il volume del liquido. Questo deve egualiare Fs.
Quando il volume V e` molto piccolo questa e` la reazione determinante
la cinetica.
In condizioni chiuse con flusso laminare (fessure e microfratture) il tipo
di cinetica dipende dall'ampiezza H della fessura.
Per H<2 10-5m la cinetica e` dominata dalla
conversione di CO2 e la dissoluzione cresce linearmente
con H.
Per valori di H superiori cresce piu` lentamente ed la cinetica
e` governata ssia dalla conversione di CO2 che dalla diffusione.
Per H >1 mm il tasso di dissoluzione resta costante e quando H
supera 5 cm predomina solo la diffusione.
Se il flusso e` turbolento (strato limite inferiore a 10-4 cm)
la diffusione e` determinata dai vortici: in pretica il coefficiente di
diffusione e` 104 volte piu` grande. La dissoluzione
e` controllata dalla conversione di CO2.
Per H >1 mm il tasso di dissoluzione diventa costante e la cinetica
e` controllata dalla reazione alla superficie.
In generale il tasso di dissoluzione risulta proporzionale alla differenza
fra la concentrazione e quella di equilibrio,
F = a ( [x]eq - [x] )
dove il coefficiente
a dipende da temperatura, pressione parziale
di CO
2, rapporto
V/A, spessore dello strato limite, e
tipo di flusso.
La figura a lato mostra il coefficiente
a al variare di
V/A
per flusso laminare e turbolento (
T = 10°C,
PCO2 = 0.05 atm).
La concentrazione di ioni calcio varia nel tempo
d[x]/dt = (A/V) F([x]).
Poiche'
F([x]) e' lineare [x] cresce esponenzialmente verso
il valore di equilibrio (la costante di tempo e' dell'ordine di 100 s).
Queste formule sono in buon accordo coi dati sperimentali per concentrazioni
abbbastanza lontane da [x]
eq. Per valori prossimi alla
concentrazione di equilibrio l'andamento di
F([x]) non e' piu' lineare
(come secondo l'equazione PWP) ma ha un andamento del tipo
F([x]) = k ( 1 - [x]/[x]eq )n [x] <[x]o
F([x]) = k' ( 1 - [x]/[x]eq )n' x[] >[x]o
dove [x]
o rappresenta una soglia di transizione.
I valori degli esponenti
n e
n' e dei coefficienti
k e
k'
dipendono dalla roccia. In genere
n' >
n.
La discrepanza dalla legge lineare (PWP, che comunque vale per la calcite
sintetica ultrapura) e' dovuta alla presenza di impurita' che vengono adsorbite
sulla superficie ed inibiscono la dissoluzione degli ioni calcio. Si puo' scrivere
F([x]) = FPWP([x]) ( 1 - T([x]) )
dove
T([x]) e' l'isoterma di Fowker Frumkin (l'entalpia di adsorbimento
cresce lineramente col grado di adsorbimento).
In conclusione [
870]
per flussi turbolenti e grandi (
V/A >1 mm)
la cinetica e' dominata dalle reazioni alla superficie e vale la legge PWP.
Per flussi laminari in fessure inferiori al mm
e per i veli sulle pareti la cinetica e'
lineare e dominata della reazione H
2O + CO
2 = H
2CO
3.
Per flussi laminari piu' grandi interviene anche la diffusione.
Vicino all'equilibrio le reazioni superficiali diventano predominanti
(a causa della inibizione).
| Reazione
|
K
|
(T in °K)
|
0°C
|
5°C
|
10°C
|
15°C
|
20°C
|
25°C
|
unita`
|
| H2O + CO2 ->H2CO3
|
Ko = (CO2) / (H2CO3) = KH / K'
|
Ko = 330 circa
Ko = 1.7 10-4 / K1
|
5.54 |
5.54 |
5.64 |
5.60 |
5.53 |
5.56 |
101
|
| |
K' = [H2CO3] / PCO2
|
|
7.58 |
6.30 |
5.37 |
4.57 |
3.89 |
3.38 |
10-2
|
| H2CO3 ->H+ + HCO3-
|
(H+) (HCO3-) = K1 (H2CO3*)
|
log(K1) = - 356.3094 - 0.06091964 T
+ 21834.37 / T - 126.8339 log(T) - 1684915 T-2 [M/l]
|
2.24 |
2.45 |
2.69 |
2.95 |
3.23 |
3.54 |
10-7 M/l
|
| HCO3- ->H+ + CO32-
|
(H+) (CO32-) = K2 (HCO3-)
|
log(K2 = - 107.8871 - 0.03252849 T
+ 5151.79 / T + 38.92561 log(T) - 56371.9 T-2 [M/l]
|
.... |
2.88 |
3.23 |
3.71 |
4.17 |
4.68 |
10-11 M/l
|
| CaCO3 ->Ca2+ + CO32-
|
Kc = (Ca2+) (CO32-)
|
log(Kc = - 171.9065 - 0.077993 T
+ 2839.319 / T + 71.595 log(T) [(M/l)2]
|
3.63 |
3.71 |
3.47 |
3.23 |
3.02 |
2.81
|
10-9 (M/l)2
|
| CO2(g) ->CO2(aq)
|
KH = (CO2) / PCO2
|
log(KH) = 108.3865 + 0.01985076 T
- 6919.53 / T - 40.45154 log(T) + 669365 T-2 [M/l atm]
|
1.71 |
1.42 |
1.19 |
1.02 |
0.88 |
0.76
|
ml(CO2) / ml(H2O) (riportati a 0°C)
|
| |
M = KH 2.144
|
|
3.67 |
3.04 |
2.55 |
2.19 |
1.89 |
1.63 |
gr / l(H2O)
|
| H2O ->H+ + OH-
|
Kw = (H+) (OH-)
|
log(Kw = 22.801 - 0.010365 T
- 4787.3 / T - 7.1321 log(T) [(M/l)2]
|
1.15 |
1.82 |
2.88 |
4.57 |
7.07 |
10.96
|
10-15 (M/l)2
|
| |
K1 Kc / K2 H
|
|
.... |
3.58 |
2.75 |
2.09 |
1.64 |
1.29 |
10-8
|
| Ca2+ + HCO3- ->CaHCO3+
|
K3 = (CaHCO3+) / (Ca2+) (HCO3-)
|
log(K3 = 1209.12 + 0.31294 T
- 34765.05 / T - 478.782 log(T) [M/l]
|
|
|
|
|
|
|
|
| Ca2+ + CO32- ->CaCO3
|
K4 = (CaCO3) / (Ca2+) (CO32-)
|
log(K4 = -1228.732 - 0.299444 T
+ 35512.75 / T + 458.818 log(T) [M/l]
|
|
|
|
|
|
|
|
Nella tabella sopra riportata i logaritmi sono in base 10.
Il coefficiente di 1/T e` legato all'entalpia di reazione,
HR, secondo l'equazione di van t'Hoff
(R e` la costante dei gas, pari a 0.082 l atm/M °K),
d log(K) / dT = (H / R) T-2.
Il coefficiente 2.144 e` ( 44 / 22.4 ) * (273+25)/273.
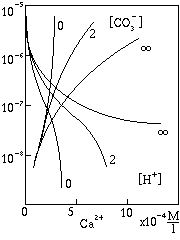
Fig. 362. Concentrazioni H e CO3
La variazione delle concentrazioni [H+] e
[CO32-] al variare della concentrazione di
Ca2+ in soluzione sono mostrate in figura, per differenti
valori di X, cioe` condizioni del sistema, da aperto (X
molto grande) a chiuso (X = 0).
La saturatione di calcio in soluzione varia in modo simile alla concentrazione
[CO32-].
[H2CO3*] decresce linearmente
con [Ca2+] con un coefficiente proporzionale a 1/X.
Nei sistemi carsici in cui l'acqua scorre in superficie
prima di infiltrarsi nella roccia, la dissoluzione
del calcio avviene in due modi: prima in condizioni aperte (lungo una
linea quasi orizzontale nel diagramma [H2CO3*]
- [Ca2+] ) poi in condizioni chiuse (lungo una linea molto
inclinata). Il risultato e` equivalente ad una dissoluzione
in condizioni chiuse con una maggiore pressione parziale iniziale di
CO2. Questo spiega le alte concentrazioni di calcio
(2 mM/l) in acque carsiche alimentate da un sistema diffuso.
Dalle equazioni del bilancio di massa e quella del bilancio della carica
(semplificata 2 [Ca
2+] = [HCO
3-] )
si ottiene la formula di Caro
per la concentrazione di equilibrio degli ioni H
+
e calcio in soluzione in funzione della pressione
parziale della CO
2:
[H+]3 = ( K12
K2 KH2 cCa )
/ ( Kc cH3 cHCO3 )
PCO22
[Ca2+]3 =
( K1 Kc KH ) /
( K2 cCa cHCO32 )
P(CO2)
Nei sistemi aperti PCO2 e` fissata e queste relazioni
permettono di ottenere la concentrazione d'equilibrio di Ca2+.
Nei sistemi chiusi e` fissata la PCO2 iniziale e quella finale
si esprime tramite essa e la concentrazione di calcio,
PCO2 = PCO2i - [Ca2+] /
( f KH [1 + 1/Ko] ). Si ottiene cosi` una equazione
cubica per la concentrazione degli ioni calcio.
Queste curve hanno una concavita` rivolta principalmente
verso il basso (cio` rende
conto della corrosione per miscelazione, effetto Bogli [v. sotto]).
L'influenza di ioni estranei sull'equilibrio della calcite ha differenti
aspetti.
- Se il sale ha ioni che non rientrano nell'equilibrio della calcite
(per esempio Nacl), esso influenza solo la forza ionica della soluzione,
e quindi la solubilita` del calcio. Questo effetto e` solitamente piccolo;
- Se il sale aggiunge uno ione comune, Ca2+ oppure
CO32- si ha diminuzione della solubilita` della
calcite (anche se nel caso dello ione Ca2+ la quantita` totale di
calcio in soluzione aumenta);
- In presenza di ioni CaSO4 la concentrazione di
Ca2+ e` ridotta e la solubilita` della calcite cresce
(di circa 3% per 10-3 M/l di solfato).
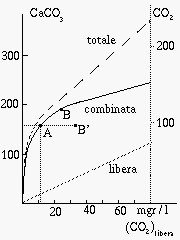
Fig. 363. CaCO3 e CO2 in soluzione
Nella CO2 in soluzione
(determinata dalla pressione di CO2 nell'atmosfera sovrastante),
si distinguono la porzione libera (in soluzione sotto forma di
acido carbonico)
e quella legata, combinata con i sali in soluzione perche` andata
a disciogliere il carbonato di calcio: la quantita` di carbonati
disciolti e` proporzionale alla quantita` di CO2 combinata,
in proporzione 2.273, pari al rapporto dei pesi molecolari,
100 e 44 rispettivamente: a 22 mgr/l di CO2 disciolta corrispondono
50 mgr/l di CaCO3 in soluzione.
All'equilibrio la soluzione non e` aggressiva.
Si ha una ben determinata quantita` di CO2 libera e
una di CO2
associata ai carbonati, e quindi un pH della soluzione ben preciso.
Dunque all'equilibrio c'e` una relazione che lega temperatura, pH,
e concentrazione di CaCO3- (v. tabella sotto e figura
a lato).
Se si discioglie una ulteriore quantita` di CO2
la soluzione diviene aggressiva: questo corrisponde ad un punto sotto la
curva di equilibrio, punto B' in figura.
La differenza B' - A rappresenta la CO2 aggressiva.
Il pH della soluzione e` inferiore a quello di equilibrio.
La soluzione si riporta allora all'equilibrio, aumentando sia la
CO2 libera (e quindi il pH) che quella combinata, portandosi
cioe` al punto B.
Per pH superiori al pH di
equilibrio la soluzione e` satura di carbonati e si ha deposizione.
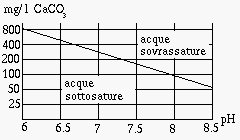
Fig. 364. Abaco di Roques
La misurazione del pH dell'acqua e della temperatura e` un indicatore della
aggressivita` dell'acqua.
Dalla misura della conducibilta` elettrica si risale alla concentrazione di
sali disciolti, che e` principalmente la CaCO
3 disciolta
(tabella sotto riportata, oppure abaco di Roques [
871] , in figura
e` riportata la curva per T=15°C).
Unitamente alla misura della pressione parziale della CO
2
nell'atmosfera (cioe` alla sua percentuale) questo permette di valutare
se gli equilibri delle reazioni chimiche sono spostati a sinistra oppure
a destra e quindi se l'acqua e` aggressiva (corrode la roccia) oppure
se tende a depositare i sali in essa disciolti.
Se la [CO
2] in soluzione (ottenuta dalla
P(CO
2) con la legge di Henry) e` inferiore al valore riportato
si ha deposizione di carbonato di calcio. Se il valore e` superiore
si ha corrosione della roccia.
Nell'impiego dell'abaco di Roques bisogna tener presente i suoi
limiti:
- riporta le concentrazioni, mentre gli equilibri chimici
dipendono dalle attivita`, percio` l'abaco va bene per acque con
debole forza ionica (contenuto di sali);
- si riferisce al solo sistema H2O - CaCO3 - CO2;
- non tiene conto degli altri ioni (in particolare Mg).
Il sistema di equazioni chimiche puo` essere impostato numericamente
e risolto col calcolatore (Fritz, 1981, "Etude thermodynamique et
modelisation des reactions hydrothermales e diagenetiques",
Mem. 65 Sc. Geol. Univ. Strasbourg).
Il pH e` definito come l'antilogaritmo della concentrazione di ioni
H
+: pH = - log( [H
+] ).
La soluzione e` acida quando il pH e` inferiore a 7; e` basica quando
e` superiore a 7. L'acqua del mare ha pH = 8.3 (cioe` e` basica); i danni
alla vita animale iniziano quando il pH scende sotto a 6.5 [
872] .
L'alcalinita` e` la capacita`
di reagire con ioni H+. Essa e` un indicatore del contenuto di
carbonati e bicarbonati (e alcali). Conoscendo il pH e la alcalinita`
si puo` ottenere la concentrazione di CO2 disciolta.
L'indice di saturazione della calcite
e` il rapporto fra le attivita` degli ioni
calcio e carbonato e la costante di equilibrio,
S = (Ca2+) (CO32-) / Keq
E` inferiore ad uno se la soluzione e` sottosatura, e l'acqua ha
capacita` di dissolvere la roccia. Se e` maggiore di uno
e` sovrassatura, e l'acqua tende a depositare calcite.
Alternativamente si usa il logaritmo di questa espressione
per cui si raffronta con 0. Il calcolo di questo indice dipende dalla
misura del pH e un errore in questa comporta un pari errore in log(S)
[
873] .
| CO2 libero (mgr/l)
|
CaCO3 in soluzione (mgr/l)
|
CO2 semicombinato (mgr/l)
|
pH
|
CaCO3 in soluzione (mgr/l)
|
CO2 semicombinato (mgr/l)
|
pH
|
| |
0°C
|
10°C
|
| 5 |
150 |
66.0 |
7.82 |
135 |
59.4 |
7.79
|
| 10 |
197 |
88.7 |
7.71 |
179 |
78.8 |
7.68
|
| 20 |
246 |
108 |
7.55 |
227 |
99.9 |
7.52
|
| 30 |
279 |
123 |
7.43 |
260 |
114 |
7.41
|
| 40 |
307 |
135 |
7.35 |
286 |
126 |
7.32
|
| 50 |
330 |
145 |
7.29 |
308 |
136 |
7.26
|
| 60 |
351 |
154 |
7.23 |
326 |
143 |
7.20
|
| 70 |
371 |
163 |
7.19 |
344 |
151 |
7.16
|
| 80 |
390 |
172 |
7.16 |
359 |
158 |
7.12
|
| 90 |
407 |
179 |
7.12 |
374 |
165 |
7.09
|
| 100 |
423 |
186 |
7.10 |
387 |
170 |
7.06
|
| 110 |
438 |
193 |
7.07 |
400 |
176 |
7.03
|
| 120 |
|
|
|
412 |
181 |
7.00
|
| 130 |
|
|
|
423 |
186 |
6.98
|
| 140 |
|
|
|
434 |
191 |
6.96
|
| 150 |
|
|
|
445 |
196 |
6.94
|
Ricapitolando:
la solubilita` della CO2 diminuisce con la temperatura,
mentre quella dalla CaCO3 aumenta.
L'aggressivita` dell'acqua cioe`
la capacita` di sciogliere il calcare (calcite e dolomite) dipende
dalla concentrazione di CO2.
L'acqua di origine meteorica che
passa nel terreno puo` arricchirsi di CO2 e diventare aggressiva.
Questo succede se l'acqua attraversa terreni ricchi di acidi organici
e in condizioni relativamente fredde, percio` gli ambienti piu` favorevoli
allo sviluppo del carsismo sono quelli coperte di
vegetazione e con clima temperato.
Pero` l'acqua di infiltrazione satura di CO2 diventa ben presto
satura di carbonato, quindi il suo effetto corrosivo e` pressoche` limitato
alla zona superficiale.
La concentrazione della CaCO3 disciolta nell'acqua in un
sistema carsico cresce andando dalle zone di assorbimento fino alle
risorgenze. Essa varia a seconda del regime idrico che ha regolato il
trasferimento dell'acqua e delle condizioni di scambio con la
roccia: fessure e vaschette con flussi lenti hanno acque molto sature,
condotti con flussi veloci portano acque con minor saturazione.
La corrosione dei massicci nelle zone profonde ha luogo poiche`
le acque delle piene arrivano in profondita` in condizioni sottosature
a causa del veloce trasferimento all'interno del sistema.
La miscelazione di acque sature puo` produrre acqua sottosatura,
e quindi rinnovarne l'aggresivita` (questo e` chiamato effetto Bogli
[
874] ,
anche se in effetti e` stato proposto da Laptev trent'anni prima)
[
875] [
876] .
Questo effetto, certamente importante per acque marine, e` comunque debole
per le acque dolci.
La corrosione per miscela si realizza in assenza di atmosfera, altrimenti
la CO
2 si riequilibra con quella dell'aria.
Quindi questo meccanismo si applica a zone allagate e/o sature (reticolo
saturo). Le morfologie associate alla corrosione per miscelazione sono
slarghi dopo una confluenza e cupole alimentate da canali di piccole
dimensioni.
Quando l'acqua diviene sovrassatura si ha precipitazione di carbonato di
calcio. All'interno della grotta questo da` luogo agli svariati fenomeni
di concrezionamento.
Quando la precipitazione avviene alle risorgenze si generano
tufi e travertini.
9.D.2 Corrosione per condensazione
La condensazione di vapor acqueo sulle pareti produce acqua priva di
sali disciolti, e quindi aggressiva, cioe` in grado di corrodere la roccia.
Gli effetti sono evidenti quando c'e` una elevata differenza di temperatura,
ma anche quando l'acqua di condensazione e` particolarmente aggressiva per
per la presenza di acidi forti, oppure quando la quantita` di acqua
e` elevata come per l'aria in ingresso d'estate [
877] .
La corrosione di condensazione ha uogo anche in sistemi ipocarsici
(lava e ghiaccio) ove e` importante anche per l'influenza sulla genesi di
formazioni. Le tipiche morfologie attribuibili a questo fenomeno sono
duomi, cupole emisferiche, scallops.
Consideriamo [
878] una parete di roccia
coperta da un velo d'acqua, in equilibrio
termico con essa (temperatura T
r), e il cui spessore e` costante
se il flusso lungo la parete viene rimpiazzato dalla condensazione.
Si ha condensazione quando la pressione vapore nel fluido P
r
e` inferiore a quella nell'aria P
a.
Vicino alla parete l'aria ha uno strato limite di spessore
s in cui le molecole d'acqua diffondono (D coefficiente
di diffusione molecolare dell'acqua in aria 2.5 10-5 m2/s).
Il flusso di vapore attraverso lo strato limite e` descritto dalla
legge di Fick Fw = D/s (Ca - Cr),
dove C sono le concentrazioni di vapore in aria e sulla roccia.
In termini delle pressioni di vapore,
Fw = (D/s)(M/RTa)(Pa - Pr).
Il flusso di energia per conduzione dovuto alla differenza
di temperatura fra aria e roccia, DT=T
a - T
r, e`
F
c = K
a DT/s', dove
s' e` lo spessore
dello strato limite per la conduzione di calore, e K
a e` la
conduttivita` termica dell'aria (0.026 W/m K).
Il flusso di energia associato alla condensazione di vapore
e` dato dal prodotto di
Fw
per il calore latente di condensazione (2450 J/g),
e risulta circa 0.044 DT/s.
Il flusso totale di calore e` la somma di questi due contributi
Ftot = K DT/s = 0.07 [W/m K] DT/s.
In generale s'=s (Pr/Sc)
1/3, dove Pr e Sc sono i numeri di
Prandtl per la convezione, e di Schmidt per la diffusione. Per l'aria
entrambi sono 1 e s'=s.
La quantita` di acqua condensante
risulta Q = 1.8 10-5[g/m K] DT/s.
La soluzione ha un limite stazionario che viene raggiunto quando la roccia
raggiunge la temperatura T - T
r,0 = A/(1+A) (T
a - T
r,0),
dove A=(K/K
r) Z/s. Qui
Kr e` la conducibilita` termica
della roccia (1.3 W/m K per il calcare, 0.5 W/m K per il gesso), e
Z e` lo spessore della roccia (che dipende dalla scala temporale
del fenomeno).
Questo valore stazionario viene raggiunto
con un andamento esponenziale, con costante di tempo
t = k-1 (Z/b)2
dove
k e` la diffusivita` termica della roccia
(5.6 10
-7 m
2/s per il calcare, 3.6 10
-7 m
2/s
per il gesso), e
b e` la prima soluzione di b=A tg(b).
Poiche`
T risulta inferiore a T
a, anche in condizioni
stazionarie si ha sempre condensazione,
Fw,staz. = 1.8 10-5 (Ta - Tr,0)
/ (s + Z K/Kr ) [g m-2 s-1]
Per
Z abbastanza "grande" rispetto a
s, il flusso di
condensazione diventa indipendente da
s.
Questa analisi presuppone una parete piana;
per altre geometrie si ottengono risultati qualitativamente analoghi,
a meno di un fattore di forma che tien conto della geometria.
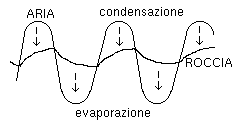
Fig. 365. Condensazione
Se la temperatura dell'aria e` alternata rispetto a quella della grotta
(ciclo diurno, ciclo stagionale), quando l'aria e` calda e umida si ha
condensazione, quando e` piu` fredda si ha evaporazione solo se l'acqua
non scorre via sulla roccia. Senza evaporazione il flusso di calore
roccia/aria e` solo quello dovuto alla conduzione termica (coefficiente
0.027 anziche 0.07) percio` il raffreddamento e` piu` lento del riscaldamento.
Ne risulta che la temperatura della roccia varia intorno ad un valore
piu` alto del valor medio della temperatura dell'aria (Figura).
Nelle variazioni stagionali, in estate entra aria calda e si ha condensazione,
in inverno entra aria fredda e la condensazione si ferma. La quantita` di
condensazione e` circa (per condensazioni con periodo superiore a
mezza giornata)
Q = ½(Ta - Tr)( 20 + tc1/2) [g/m2]
dove tc e` il periodo della condensazione (in secondi).
Per t=100 giorni Q=1500 g/m2 K, cioe` una dissoluzione di
3.5 10-5 mm/y K (senza tener conto della pressione parziale di
CO2:
bisognerebbe moltiplicare per (PCO2/0.00035)1/3 ).
Questo e` comunque un valore molto piccolo.
Nelle variazioni giornaliere, la condensazione da' risultati piu`
rilevanti (dell'ordine di 10-6 mm/y) pero` e` attiva solo
nelle prime parti della grotta.
La corrosione per condensazione puo` riguardare anche gli speleotemi
Il calore di condensaione viene trasferito al substrato roccioso.
L'equilibrio termico viene raggiunto dopo un tempo t=D/(4 b2 k);
D e` il diametro della concrezione, e b e` la prima radice
di tg(b)/b = 2 s Kr/(D K) (se D=30 cm, t=4 h).
Dopo 5t la condensazione e` essenzialmente finita (equilibrio termico).
Se D e` abbastanza piccolo la concrezione segue i cicli diurni
dell'aria.
La quantita` di condensazione si ricava dal bilancio energetico
(la condensazione corrisponde al 70 % circa del calore),
Qc Mc = 0.7 Cp M DT, quindi
F = M/(πD L) = 0.17 D DT [Kg/m2}, che corrsponde ad una
dissoluzione di 1.4 10-3 D DT [mm/y].
Concrezioni di diametro piu` grosso non raggiungono l'equilibrio termico, e
la loro temperatura oscilla intorno ad un valore intermedio fra in minimo e
il massimo dell'aria.
9.D.3 Carsismo ipogenico
Il carsismo ipogenico [
688] [
879]
risulta da aggressivita` delle acque
prodottasi in profondita` (CO
2 endogena, degassazione di magmi,
H
2S)
4) ed e` legato a flussi in risalita senza influenza diretta
da fattori esterni.
E` caratterizzato dall'assenza delle microforme gravitative/vadose (scallops,
fori, etc.) e dai tipici depositi (argille, ciotoli).
Le morfologie, dovute a gas ascendenti in ambiente freatico, sono
- canali di bolle ("bubble trails"), canali e tubi di corrosione, scavati
dalle bolle di gas in risalita;
- genarale copertura di calcite che si deposita dall'acqua sovrassatura,
eccetto che nei tubi di corrosione.
L'acqua di origine profonda si decomprime durante la salita, quindi la
CO
2 disciolta si libera a genera le bolle di gas.
Le bolle si possono fermare in anfratti del soffitto, formando una
atmosfera ricca di CO
2, in cui la condensazione corrode il
sofitto ("boxwork") lasciando anche sporgenze pendenti ("piede d'elefante").
L'acqua degassata risulta soprassatura e quindi deposita calcite, generando
diverse morfologie,
- "folia" a soffitto: sono cornicioni invertiti, in cui si ha
deposizione lungo l'orlo e corrosione nella cavita`;
- coralloidi al suolo;
- "penitenti": torri coniche di deposizione subacquea;
- copertura calcitica a crescita rapida, percio` con molti vuoti
("popcorn", saccaroidi).
9.D.4 Carsismo nel gesso
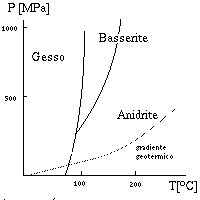
Fig. 366. Sistema gesso-anidrite
Il gesso e` la forma idratata del solfato di calcio
ed e` piu` stabile a temperatura e pressione normali.
L'anidrite predomina ad alte pressioni e/o temperature.
Percio` oltre una profondita` di circa 450 m si trova principalmente
anidrite.
La conversione di anidrite in gesso avviene durante l'emersione.
In essa il volume molare aumenta (di 1.626) ma il volume globale
diminuisce (teoricamente del 8.6in pratica di circa il 3Il processo di idratazione dipende dalla struttura della roccia,
dal chimismo dell'acqua e da pressione e temperatura.
Si presume che avvenga sia per dissoluzione/precipitazione che per diffusione
di acqua nel reticolo cristallino.
Le caratteristiche chimico-geologiche delle rocce gessose che ne influenzano
il carsismo sono
[
666] [
880] [
881] [
882] [
843]
- elevata solubilita` (2.53 g/l per il gesso, circa 2.0 g/l per l'anidrite;
entrambe leggermente crescenti con la temperatura), un ordine di grandezza
superiore a quella del calcare;
- facile erosibilita` meccanica;
- porosita` quasi nulla;
- bassa densita` di fratturazione;
- presenza di interstrati pelitici impermeabili.
Lo sviluppo del carsismo e` guidato dagli elementi strutturali (fratture,
interstrati, faglie) e dalle condizioni idrodinamiche. Le grotte nel gesso
vengono suddivise in quattro categorie:
- cavita` isometriche isolate,
- cavita` labirintiche (in 2 o 3 dimensioni),
- cavita` di attraversamento,
- cavita` a pozzo.
Le prima due sono legate ad acquiferi confinati (anche solo parzialmente),
con apporto laterale o basale (carsismo profondo). Le altre due sono legate
ad un carsismo di affioramenti il cui sviluppo e` praticamente vadoso
(o epifreatico).
La reazione di dissoluzione del gesso e`
CaSO4.2H2O <->Ca2+
+ SO42- + 2 H2O
Il
pK di questa reazione vale
4.667 - 5.197 10
-9 T + 1.133 10
-4 T
2
(dove
T e` in gradi centigradi).
L'indice di saturazione e` il rapporto fra le attivita` degli ioni
calcio e solfato e la costante di equilibrio,
S = (Ca2+) (SO42-) / Keq
E` inferiore ad uno se la soluzione e` sottosatura, e maggiore di uno
se e` sovrassatura (oppure si usa il logaritmo di questa espressione
per cui si raffronta con 0).
La solubilta` varia con la pressione sulla roccia: 1/S dS/dP vale
circa 400 Pa-1 per l'anidrite e 40 Pa-1 per il gesso.
Il gesso presenta differente solubilita` a seconda delle dimensioni dei
cristalli, con un massimo per 0.2-0.5 mm (per l'anidrite il massimo e`
a circa 2.8 mm).
La presenza di ioni estranei aumenta la solubilita`, per l'aumento della
forza ionica della soluzione. La presenza di uno ione comune diminuisce
la solubilita` di entrambi (come nel caso di CaCo3).
La cinetica del carsismo del gesso e` dominata dalla diffusione,
dc/dt = k A/V (cs - c)n
per il gesso e` del primo ordine (
n e` circa uno), per
l'anidrite del secondo ordine (
n circa due).
Il coefficiente
k dipende dalle condizione dello strato limite
(tipo di flusso, velocita` del flusso), proprieta` del minerale,
rugosita` superficiale, presenza di altri ioni, temperatura.
9.D.4.1 Sistema calcite gesso
Il gesso CaSO
4.2H
2O e il carbonato di calcio
CaCO
3 formano un sistema chimico con lo ione calcio
Ca
++ in comune [
883] .
Questo sistema comprende anche la dissoluzione della CO
2
presente nella fase gassosa e l'idrolisi dell'acido carbonico.
CaSO4 ->Ca2+ + SO42-
CaCO3 ->Ca2+ + CO32-
CO2 + H2O ->H+ + HCO3-
HCO3+ ->H+ + CO32-
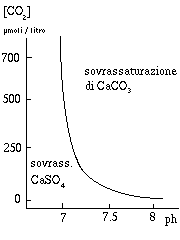
Fig. 367. Sistema calcite-gesso
Questo sistema chimico ci permette di capire fenomeni di dissoluzione e
concrezionamento delle rocce gessose. Per esempio,
la diffusione di CO2 (spostamento della terza reazione verso
destra) comporta un aumento di ione carbonato e quindi uno spostamento
della seconda reazione verso sinistra, cioe` una deposizione di
carbonato di calcio. Quindi la diminuzione di ione calcio in soluzione
favorisce la dissociazione del solfato, cioe` la dissoluzione del gesso.
In altre parole la diffusione di anidride carbonica rende la soluzione
aggressiva nei confronti del gesso (effetto Piknett).
L'analisi del sistema chimico fornisce curve di equilibrio in funzione
del pH della soluzione e della concentrazione della CO
2
disciolta [
884] . La curva varia a seconda della temperatura, in figura
e` riportata quella per 10°C.
La precipitazione di un qualsiasi sale rende la soluzione
sottosatura rispetto all'altro.
La precipitazione del gesso innalza il pH, quella della calcite lo
abbassa.
E` possibile riscontrare concrezioni calcitiche in grotte di gesso, come
pure concrezioni gessose in rocce calcaree.
La presenza di carbonati nelle rocce gessose e` dovuta a coperture,
intercalazioni, riepimenti, e anche all'azione della CO2 stessa.
La presenza di solfati in rocce calcaree e` dovuto a fenomeni indiretti:
ossidazione di solfuri da parte di batteri, solubilizzazione di gesso
o anidrite.
9.D.5 Carsismo nel sale
Il sale (halite) ha una solubilita` molto alta, 360 gr/l
[
885] .
La costante di equilibrio della dissoluzione di NaCl e`
K=(Na
+)(Cl
-) = 101.51 (a 0°C,
ma varia poco con la temperatura) [
886] .
La cinetica e` percio` controllata dalla diffusione.
Nelle pozze di acqua ferma si arriva all'equilibrio in alcune ore.
Nei veli sulle pareti in flusso laminare ci voogliono alcuni minuti.
I torrenti turbolenti restano sottosaturi per parecchie centinaia di metri.
Il maggior peso dell'acqua salata, rispetto a quella pura, provoca fenomeni di
stratificazione nell'acquifero.
9.D.6 Carsismo nel quarzo
Essenzialmente il carsismo nelle quarziti [
887]
[
888] [
889] [
890]
e` dominato dalla tettonica (fessurazioni, legate a fenomeni distensivi),
con forme a canion e reticoli di gallerie ad angolo retto.
La roccia e` trasformata dalla dissoluzione chimica in sabbia poco cementata,
facilmente asportabile dall'acqua. Sono presenti microforme carsiche epigee:
vaschette, canalicoli, docce e scanellature.
La solubilita` del quarzo e` troppo bassa (6 ppm a 25°C) e praticamente
indipendente dall'acidita`, per pH inferiori a 8. Altre forme della
silice SiO2 hanno solubilita` maggiori (calcedonio 17 ppm,
cristobalite 27 ppm, silice amorfa 115 ppm).
La dissoluzione del quarzo e` un processo di idratazione,
SiO2(s) + 2 H2O ->H4SiO4(ac)
SiO2(s) + 2 H2O + OH- ->H3SiO4-
la prima reazione avviene in soluzione neutra, la seconda in soluzione
alcalina.
La costante di equilibrio della prima reazione
vale
K = (H
4SiO
4) = 1.1 10
-4 (a 25°C).
L'acido silicico si deidrata parzialmente formando polimeri e si
dissocia liberando ioni H
+.
La concentrazione di equilibrio e` circa
10 mgr/l. La cinetica e` controllata dalla rottura dei legami chimici
e dalla idratazione dei silicati ed e` molto lenta, a temperatura
ambiente (10
-17 M cm
2/s; puo` aumentare con la
presenza di cationi e composti organici).
La dissoluzione nella quarzite e` limitata alle superfici di separazione
fra i cristalli, e procede fino a che la rocce diventa sabbia.
L'acqua meteorica penetra tra giunti e superfici interstrato, dissolvendo
la quarzite ai lati. I maggiori condotti comunque si formano per
erosione meccanica, distaccamento dei grani uno ad uno, da parte
dell'acqua. Incominciano alle emergenza e procedono a ritroso nel
massiccio. Percio` le gallerie sono vadose.
Dalla prcipitazione si hanno concrezioni di opale.
Questa ha luogo solo dove la oluzione diventa sovrassatura,
quindi dove canalicoli raggiungono cavita` sotterranee aerate, e il substrato
resta stabile.
9.D.7 Carsismo nel ghiaccio
Il ghiaccio e` una formazione sedimentaria.
Il carsismo del ghiaccio
piu` che un fenomeno chimico e` un fenomeno fisico,
tuttavia lo incodo in questa sezione sui meccanismi di carsismo.
[
664]
[
891] , [
892] , [
893] , [
894] , [
895] ,
[
635] , [
896] ...
Le cavita` glaciali sono di diversi tipi:
I ghiacciai alpini sono "caldi" (temperati): la temperatura e` al
punto di fusione dell'acqua. Nei ghiacciai "freddi" (polari)
le uniche cavita` sono quelle di frattura, mentre in quelli caldi si
hanno anche cavita` generate dall'acqua:
- cavita` subglaciali, al contatto fra ghiaccio e roccia;
- condotti endoglaciali, cioe` interni alla massa di ghiaccio;
- crepacci, cavita` di distensione meccanica, che si formano
principalmente presso la zona terminale del ghiacciaio.
Estesi lateralmente e profondi al piu` alcune decine di metri.
- mulinelli glaciali: pozzi di assorbimento.
9.D.7.1 Fenomenologia
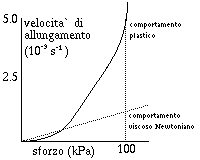
Fig. 368. Ghiaccio: deformazione
Il ghiaccio naturale presente sulla terra e` nello stato esagonale
(detto "Ih"). La linea di equilibrio fra acqua a ghiaccio Ih ha pendenza
negativa, cioe` il liquido (acqua) e` piu` densa del solido (ghiaccio).
La struttura atomica del ghiaccio e` esagonale, con ogni atomo di ossigeno
legato da quattro legami idrogeno agli atomi vicini posti ai vertici di
un tetraedro. Molte delle proprieta` fisiche del ghiaccio sono dovute
alle particolarita` del legami idrogeno.
Anomalie della struttura sono i difetti dei legami, dislocazioni cristalline,
e difetti superficiali (presenza di grani).
Il ghiaccio e` trasparente alla luce visibile. E` doppio rifrangente,
uniassiale, e otticamente positivo ma con birifrangenza molto piccola.
Ha una banda di assorbimento infrarossa larga.
Il comportamento meccanico del ghiaccio e` quello di un mezzo rigido
per valori dello sforzo di taglio bassi (quindi fratturabile), poi diventa
viscoso ed infine plastico.
Quando lo sforzo supera un determinato valore (circa 100 kPa) il
ghiaccio si deforma come se fosse creta.
Sotto la propria spinta idrostatica il ghiaccio e` gia` plastico alla
profondita` di una decina di metri.
La velocita` di scorrimento e` legata all'intensita` dello sforzo
s,
legge di Glen [
897]
du/dt = A sn exp( -Q/kT )
dove
T e` la temperatura (in gradi assoluti),
k la costante
di Boltzmann,
Q l'energia di attivazione. Per sforzi fra 100 e 250 kPa
A=5 10
-15 s
-1 kPa
-3,
Q = 139 kJ/mol e
n=3.
Per sforzi inferiori a 100 kPa
n=2.
Approssimativamente possiamo utilizzare
du/dt =
A sn
dove
s e` espresso in kPa.
L'esponente
n dipende dalla temperatura, e in genere varia
fra 1.9 e 4.5. Anche
A in questa approssimazione dipende
dalla temperatura.
Quando lo sforzo supera 400 kPa la velocita di scorrimento aumenta
sempre piu` velocemente e le precedenti formule non sono piu` valide.
Un sistema endoglaciale presenta della zone come un sistema
carsico nella roccia: zona di assorbimento superficiale
(in cui l'acqua entra nel ghiacciaio),
zona di trasferimento verticale
e zona di "falda" (con condotti orizzontali e grossi condotti
di riemergenza).
L'acqua di fusione scorre superficialmente sul ghiacciaio lungo
rivoli e rigagnoli. Quando penetra nel ghiacciaio scende generalmente
lungo pozzi verticali (mulinelli), separati da brevi meandri,
e impostati lungo discontinuita` meccaniche (fratture).
Le fratture della zona superiore (di trasferimento verticale)
si propagano all'interno della massa del ghiacciaio fino alla profondita`
limite a cui il ghiaccio diviene plastico e quindi si deforma in modo
da chiudere eventuali fratture. In linea di principio la pressione
dell'acqua potrebbe tener aperta una frattura anche oltre questa
profondita` limite, tuttavia i mulinelli possono raggiungere il substrato
roccioso solo se il ghiacciaio non e` spesso (meno di 50 m).
I mulinelli si formano in zone dove si ha fusione e scorrimento superficiale
ed in corrispondenza di discontinuita` meccaniche.
Possono arrivare a oltre 10 m di diametro, ma in genere sono di pochi metri
e scendopo per decine di metri.
Quindi in zone non crepacciate. Apparentemente i mulinelli hanno un ciclo
legato al ciclo di accumulo e ablazione del ghiacciaio.
Si generano sempre nello stesso posto, e poi migrano a valle trasportati
dallo scorrimento del
ghiacciaio fino a che non ricevono piu` acqua e collassano.
Il periodo di crescita e` in genere di uno o due anni, dopodiche`
inizia la senescenza e nal giro di pochi anni si chiudono
(sono stati osservati anche mulinelli di 20 anni).
L'anno successivo
si forma un nuovo proto-mulinello nella posizione originaria
che poi si sviluppa in un mulinello.
Il processo di formazione non e` chiaro; probabilmente determinato dallo
stato degli sforzi di distenzione all'interno del ghiaccio e dalla
conformazione superficiale (apporto idrico).
La morfologia dei mulinelli e` di due tipi
- pozzo a sezione ellittica con ruscellamento diffuso lungo le pareti, che
risultano liscie o con scanalature verticali;
- pozzo a cascata, con andamento a gradini, con terrazzi, marmitte;
hanno forme complesse con meandri e forre.
Il limite di profondita` dei pozzi glaciali sembra
essere attorno al centinaio di metri (50-80 m, ma in alcuni casi si sono
raggiunti fino a -200 m),
profondita` a cui il ghiaccio ha ormai comportamento plastico
e la pressione del ghiaccio
sovrastante e` tale da rendere i passaggi troppo stretti
(se non e` controbilanciata dalla pressione dell'acqua, nel qual caso
la galleria e` allagata) [
894] .
Il fondo dei pozzi e` solitamente pieno d'acqua (laghi endoglaciali oppure
il livello della zona satura).
Il livello della falda freatica (70-150 m)
e` soggetto a variazioni (anche veloci)
in funzione della alimentazione e della deformazione plastica del reticolo
di gallerie sommerse.
Nella stagione calda
il reticolo delle gallerie allagate si trova ad una profondita` di 100-150
m. D'inverno, quando cessa l'apporto idrico le gallerie collassano sotto la
spinta del ghiaccio a profondita` superiori a 50-60 m.
La zona di scorrimento endoglaciale e` situata al limite fragile-plastico
del ghiaccio. L'acqua scorre in condotti sub-orizzontali, organizzati in
disposizioni dendritica che confluiscono via via in condotti di
dimensioni crescenti. La sezione dei condotti e` circolare similmente
alle gallerie freatiche carsiche.
Lo spessore della zona satura dipende dall'equilibrio fra apporto idrico
e portate uscenti. Il livello della zona satura oscilla nel corso
dell'anno, anche di parecchie decine di metri.
I ghiacciai temperati hanno una zona di scorrimento subglaciale
tra il ghiacciaio e il substrato roccioso.
Normalmente l'acquifero subglaciale e` indipendente da quello
endoglaciale, eccetto ove le fratture della zona superiore si possono
propagare fino al substrato, solitamente in prossimita`della fronte
del ghiacciaio dove lo spessore e` ridotto e la fratturazione e` piu`
intensa.
La acque subglaciali vengono a giorno in corrispondenza della fronte
stessa, formando torrenti che scorrono in condotti subglaciali
(cavita` di contatto, dette porte del ghiacciaio).
Questi sono sovente piu` d'uno.
Nelle cavita` di contatto e` importante speleogeneticamente
anche l'azione erosiva del
sedimento solido trasportato dall'acqua.
Tali cavita` hanno una evoluzione piu` irregolare dei mulinelli,
a causa anche delle instabilita` della volta (con collassi e distacco
di grandi lame di ghiaccio, "sfoliazioni").
Una particolare morfologia sono i grandi scallops sulle parete
generati dalle correnti d'aria, quando ci sono piu` ingressi
comunicanti.
9.D.7.2 Condotti glaciali
L'attrito dell'acqua che scorre produce calore che fonde il ghiaccio.
Percio` lo scorrimento tende ad allargare i condotti mentre la pressione
nel ghiacciaio tende a chiuderli.
I condotti maggiori si formano dunque a piccole profondita`.
Inoltre i condotti sono un fenomeno stagionale: tendono a chiudersi
d'inverno, quando viene meno l'apporto idrico, e migrano
con lo spostamentp della massa del ghiacciaio. Questa tende ad immergersi
scorrendo, quindi aumenta la pendenza dei condotti.
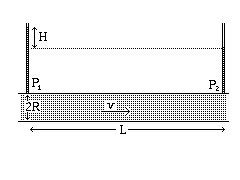
Fig. 369. Condotto glaciale
Consideriamo un condotto orizzontale, di raggio R e lunghezza L,
in cui fluisce acqua con velocita` v.
La differenza di pressione fra l'inizio e la fine e` proporzionale
all'altezza di carico, P
1 - P
2 = d g H, dove g e`
l'accelerazione di gravita` e
d la densita` dell'acqua.
In regime turbolento (
f e` il coefficiente di attrito, che
per i condotti glaciali vale circa 0.08),
H = f L v2 / ( 4 R g )
L'energia disponibile e` quella prodotta dagli attriti, percio`
il flusso di energia e`
dE/dt = dM/dt g H,
dove dM/dt = πR
2 v e` il flusso di massa.
D'altra parte l'acqua passando ad una pressione inferiore si scalda,
quindi assorbe calore, dT = q dP, dove q = 7.8 10
-11 °/ Pa.
Parte dell'energia se ne va quindi in flusso di calore assorbito dall'acqua,
dQa/
dt =
dM/dt c
a q dP
Pertanto il flusso netto di calore e`
dQ/dt = d g H dM/dt [ 1 - d ca q ]
Esso va a sciogliere il ghiaccio, quindi il condotto si allarga
con una velocita`
dR/dt = [ 1 - d ca q ] (d f)/(8 lf dg) v3
dove
lf e` il calore di fusione del ghiaccio
(80 Cal/Kg), e
dg e` la densita` del ghiaccio
(917 Kg/m
3).
Per esempio, con
v=1 m/s,
R=0.3 m, dR/dt = 1.6 10
-9 m/s.
Una immediata conseguenza (Shreve) e` che non si formano bypass.
In condotti in parallelo le velocita` sono
proporzionali a R1/2 dato che l'altezza di carico e`
la stessa (assumendo lunghezze comparabili).
Quindi condotti piu` grossi tendono ad accrescersi maggiormente
di condotti piccoli.
Ne risulta che il drenaggio ha una struttura ad albero.
Una seconda conseguenza e` il limite della pendenza che puo` avere
una condotta in salita. In tal caso l'energia disponibile e` la stessa,
ma la differenza di pressione contiene anche un termine dovuto alla
pendenza, P
1 - P
2 = d g ( H + L sin(a) ).
Quindi l'acqua si scalda maggiormente e resta meno energia disponibile
per sciogliere il ghiaccio.
Si raggiunge il limite quando tutta l'energia disponibile va a
scaldare l'acqua. Ne risulta una pendenza limite
sin(a) = 2.07 f v2 / (4 R g)
I condotti possono salire solo se hanno una alta impedenza, cioe` alte
perdite per attrito, e quindi molto energia dissipata e disponibile
per la fusione del ghiaccio.
L'allargamento del condotto porta ad una diminuzione dell'impedenza.
Si ha quindi un equilibrio fra impedenza e pendenza.
Quando il condotto e` in discesa non c'e` alcuna pendenza limite.
9.D.7.3 Stabilita` idrostatica di un mulinello
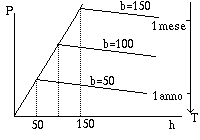
Fig. 370. Stabilita` dei mulinelli
Consideriamo un mulinello glaciale pieno d'acqua a partire da una
profondita`
b.
La pressione tra il ghiaccio e l'acqua e`
P = dg g h - d g (h-b) = ( b - 0.083 h ) d g
Ad elevate pressioni il ghiaccio e` plastico.
Quindi quando P e` positiva il ghiaccio tende a chiudere il condotto.
Il tempo di collasso e` (Nye, Hooke)
T = (4.5 105 / P)3 [year]
dove la pressione e` in Pa.
La velocita` di collasso,
dR/dt = R/T = 3.5 10-13 R (b - 0.083 h)3
decresce al crescere di
h fino ad annullarsi quando le pressioni
idrostatiche di acqua e ghiaccio si controbilanciano.
Una conseguenza e` che piu` b e` elevato, minore e` il tempo di
collasso. Per b=150 m il tempo e` dell'ordine di un mese.
Quindi strutture libere dall'acqua molto profonde hanno una esistenza
molto breve.
Questo e` in accordo con i valori riscontrati di profondita` limite
dei mulinelli glaciali.
9.D.7.4 Trasferimenti di calore
L'acqua scendendo lungo un condotto verticale aumenta la pressione
percio` si raffredda e cede calore che fonde il ghiaccio.
Quindi il condotto si allarga.
Viceversa l'acqua in risalita diminuisce la pressione, si scalda
ed assorbe calore con riformazione del ghiaccio.
9.D.7.5 Migrazione verticale dei condotti
La differenza di pressione fra pavimento e soffitto di un condotto
comporta lievi differenze di temperatura: il pavimento e` a pressione
maggiore, percio` a minor temperatura.
Quindi il calore disponibile tende ad andar piu` facilmente al
pavimento che al soffitto, e il ghiaccio fonde di piu` sul pavimento che
sul soffitto del condotto. Questo dunque migra verso il basso.
La differenza percentuale fra i flussi di calore a pavimento ed a
soffitto e` 2 R q g d / 273 = 2 10-3.
Quindi la velocita` di migrazione verticale e` dell'ordine di
10-12 m/s.
9.D.7.6 Deformazioni plastiche
Il ghiaccio all'interno del ghiacciaio ha un comportamento plastico,
percio` riprendiamo la teoria delle deformazioni plastiche
(v.
App. 10.h
)
Per semplicita` ci limitiamo a 2 dimensioni.
La condizione che definisce la plasticita` di un mezzo deformabile
e` una relazione fra le componenti del tensore degli sforzi,
per esempio (legge di Mises)
qij qij = 2 h2
dove
h e` una costante (di plasticita`), e
q
ij = s
ij - p a
ij e` il deviatore degli sforzi, cioe`
il tensore degli sforzi a meno della componente di pressione idrostatica.
La velocita` di deformazione e` proporzionale al deviatore degli sforzi
duij = dL qij + 1/(2U) dqij
dove u
ij = ½( d
i u
j + d
j u
i ) e`
il tensore di deformazione.
L e
U non sono indipendenti ma sono
legati da una equazione di stato.
Se l'equazione di stato afferma che
U e` la costante di Lame`,
la precedente relazione viene chiamata equazione di Prandtl-Reuss.
Se l'equazione di stato e` 2LU=1 essa diviene du
ij = d(Lq
ij)
quindi il tensore di deformazione e` proporzionale al tensore
deviatore degli sforzi.
Poiche` qij aij = 0 le deformazioni preservano il volume,
duij aij = 0.
L'equilibrio statico di un mezzo plastico e` determinato dalle due precedenti
equazioni e dalla condizione
dj sij = Fi
Le incognite sono le tre componenti di s
ij, le due componenti di
u
i e
L (funzione ausiliaria). Il sistema deve essere integrato
tenendo conto delle condizioni al contorno.
Per la dinamica di un mezzo plastico quest'ultima relazione ha anche il
termine di accelerazione,
k ( vi di vj + dvj/dt ) =
Fj - di sij
e la legge di Mises riguarda solo il deviatore degli sforzi plastici
(cioe` senza quello degli sforzi viscosi),
(qij + 2 U nij) (qij + 2 U nij) = 2 h2
dove
U e` il coefficiente di viscosita` e
n
ij = ½(d
i u
j + d
j u
i) la velocita`
di deformazione,
qij = - 2 (M+U) nij
U e` costante, ma in genere
M non lo e`.
Infine c'e` l'equazione d
i u
i = 0.
In tutto sono quattro equazioni in quattro incognite (vi, M
e p) che debbono essere integrate tenendo conto delle condizioni al
contorno.
Come esempio consideriamo la soluzione stazionaria per un ghiacciaio
su un pendio inclinato (con inclinazione percentuale pari a a =
tan(t) ).
Il ghiacciaio e` un mezzo deformabile che si estende nel semipiano
y + a x>0.
La velocita` normale si annulla sulla superficie y+ax=0.
Supponiamo inoltre che la pressione sia data, p=p0(y+ax),
e cerchiamo una soluzione in cui le componenti della velocita`
dipendono solo da y+ax e M sia costante.
Dalla equazione di continuita` risulta
v'y = -
a v'x
Quindi le componenti della velocita` di deformazione sono
vxx = a v'
vxy = (1 - a2)/2 v'
vyy = - a v'
L'equazione di Mises conduce a v' = cos
2(t) h/M, e le componenti
della velocita` sono (qui
z = cos(t) y + sin(t) x )
vx = (h/M) cos(t) z + vox
vy = - (h/M) sin(t) z + voy
Poiche` la velocita` normale si annulla sulla superficie (z=0), deve essere
voy - a vox.
Il ghiacciaio scorre parallelamente al pendio con velocita` che aumenta
con la distanza da questo.
L'equazione della dinamica diventa una equazione omogenea per il deviatore
degli sforzi, d
i q
ij = 0. Inoltre c'e` la relazione che lega
vij a
qij.
Il deviatore degli sforzi risulta proporzionale a
| cos2(t) |
- cos(t) sin(t)
|
| - cos(t) sin(t) |
sin2(t)
|
che rappresenta uno sforzo normale diretto lungo il pendio: il ghiaccio
scorre sotto la spinta del proprio peso.
marco corvi - Mon Nov 19 11:34:13 2007
Prev Up Top Next Contents
This work is licensed under a Creative Commons
Attribution-NonCommercial-ShareAlike 2.0 Italy License.