Purpose :To determine DO in a water sample
Theory :
In an alkaline solution, dissolved oxygen will oxidize manganese(II) to the trivalent state.
![]()
The analysis is completed by titrating the iodine produced from potassium iodide by manganese(III)
hydroxide.
![]()
Sodium thiosulphate is used as the titrant.
Success of the method is critically dependent upon the manner in which the sample is manipulated.
At all stages, every method must be made to assure that oxygen is neither introduced to nor lost from
the sample. Furthermore, the sample must be free of any solutes that will oxidize iodide or reduce iodine.
Chemicals:
Manganese(II) sulphate solution - prepared by dissolving 48g of
![]() in
in
water to give 100 ml solution;
Alkaline potassium iodide solution - prepared by dissolving 15g of KI in about 25 ml of
water, adding 66 ml of 50% NaOH, and diluting to 100 ml ;
Concentrated sulphuric(VI) acid; 0.0125M sodium thiosulphate solution;
Starch solution (freshly prepared)
Apparatus: 250 ml volumetric flask, 250 ml conical flask; titration apparatus; measuring cylinders;
magnetic stirrer
Procedure:
1. Use a 250 ml volumetric flask to collect a water sample. Fill the flask completely with water
without trapping any air bubbles.
2. Add 1 ml of manganese(II) sulphate solution to the sample using a pipette. Discharge the solution well below the surface (some overflow will occur).
3. Similarly introduce 1 ml of alkaline potassium iodide solution. Be sure that no air becomes entrapped. Invert the bottle to distribute the precipitate uniformly. [Hazard Warning: Care should be taken to avoid exposure to any overflow, as the solution is quite alkaline.]
4. When the precipitate has settled at least 3 cm below the stopper, introduce 1 ml of concentrated sulphuric acid well below the surface. A magnetic stirrer is helpful here.
5. Allow the mixture to stand for 5 minutes and then withdraw 100 ml of the acidified sample into a 250 ml conical flask.
6. Titrate with 0.0125M sodium thiosulphate until the iodine colour becomes faint. Then add 1 ml of starch solution, and continue adding the thiosulphate solution until the blue colour disappears.
7. Record the volume of thiosulphate solution used and calculate the dissolved oxygen content in the sample in mg dm-3 .
Remarks:
1. If the water sample has a low DO value, it is recommended to withdraw 200 ml of the acidified
sample into a 500 ml flask for titration described in step (5).
2. This experiment can be further developed into a project to study the extent of water pollution.
(a) The water sample under investigation is divided into two portions. One portion of the sample is immediately analyzed for dissolved oxygen using Winkler method. The other portion is stored in the dark for 5 days.
(b) Repeat the analysis with the water sample that has two measurements is the 5-days biochemical oxygen demand (BOD ), measured in mg.dm-3 .
Experiment
2
Title : Investigation some of the properties of a pair of cis-trans isomers
Introduction:
Maleic acid and fumaric acid are geometrical isomers of butanedioic acid. Each of these isomers has its own distinctive properties such as melting point, solubility, density and stability.
In this experiment some maleic acid is converted to fumaric acid by heating an aqueous solution of maleic acid in the presence of hydrochloric acid. The hydrochloric acid serves merely as a catalyst of the reaction. The properties of these two isomers are then compared.
Chemicals:
Maleic acid, magnesium ribbon, conc. HCl, bromine
water, pH paper,
![]()
Apparatus: 100 cm3 and 500 cm3 beakers, watch glass, apparatus for suction filtration,
melting point apparatus, 25 cm3 measuring cylinder.
Procedure:
[Hazard Warning: Maleic acid is irritant, conc. HCl is corrosive and bromine water is harmful.]
A.
Conversion of maleic acid to fumaric acid
1. Weigh out about 4g of maleic acid in a clean dry 100 cm3 beaker. Add 10 cm3 of deionized water
and warm slightly to dissolve the acid.
2. Add 10 cm3 of concentrated hydrochloric acid, and cover the beaker with a watch glass. Place the beaker inside a 250 cm3 beaker which is about one third full of water. Heat this water bath to
boiling for about 5 minutes or until a solid material forms in the small beaker.
3. Cool the solution to room temperature by placing the small beaker with its contents in a cold
water bath or in an ice bath.
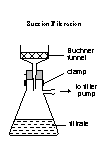
4. Filter the reaction mixture by suction using the following set-up of apparatus:
|
5. Stop suction, either by lifting the funnel or by disconnecting the tubing, and soak the residue in
about 1 cm3 of cold water. (If you turn off the tap, you may get a ‘suck-back’ of water.)
6. Resume suction and dry the crystals by drawing air through them for a few minutes.
7. Transfer the crystals into a weighed watch glass and dry in an oven at about 120o C for 10 minutes.
8. Weigh the dried crystals of fumaric acid.
B.
Comparison of properties of the two isomers
1. Solubility in water - place about 1 g of each isomer into 10 cm3 of water in separate test tubes,
shake to help dissolving. See which isomer is more soluble.
2. Melting point - using the electrical melting point apparatus, measure the m.p. of the two isomers.
3. Acid strength - for each of the two isomers, prepare a solution by dissolving about 0.1 g of the compound in about 20 cm3 of distilled water. Divide the solution into 3 proportions, one for each of the following tests;
(a) Measure the pH of the solution.
(b) Add a 3 cm strip of magnesium ribbon.
(c) Add a small amount of solid sodium carbonate.
4. Reaction with bromine water - suspend about 0.1 g of the acid in about 5 cm3 of water. Add 3
drops of bromine water to the resulting solution/suspension.
Results:
A. Conversion of maleic acid to fumaric acid
|
Mass of fumaric acid + watch glass |
g |
|
Mass of watch glass |
g |
|
Mass of fumaric acid |
g |
|
Percentage yield |
% |
B. Comparison
of properties of the two isomers
1. Water solubility :_____________________ acid is more soluble.
2. Melting point of maleic acid :__________C
Melting point of fumaric acid :___________C
3. Acid strength
|
Test |
Maleic acid |
Fumaric acid |
|
pH of solution |
|
|
|
Reaction with Mg |
|
|
|
Reaction with
|
|
|
4. Reaction with bromine water
Maleic acid: _____________________________________________________________
Fumaric acid:_____________________________________________________________
Discussion:
1. Assuming that equilibrium concentration was achieved in procedure A, which isomer would you
classify as the more stable with respect to transformation of one into the other?
2. What do each of the following tests contribute your knowledge of the structure of each isomer?
(a) The reaction with magnesium and sodium carbonate.
(b) The pH value.
(c) The melting point.
3. Considering the structures of the two isomers, try to account for the observed differences in
solubility and melting point.
4. One of the isomers can lose a molecule of water from each molecule of acid when its carboxyl
groups react to form an anhydride. Which geometrical isomer, cis- or trans-, do you predict it is?
Title : Rates of hydrolysis of some halogeno-compounds
Introduction:
Consider the nature of C-Hal bonds, the C-F bond is very strong: compare its bond energy of
485 KJ.mol-1 with 327 KJ.mol-1 for C-Cl. Consequently, fluoro-carbons are extremely inert. The C-Cl bond is more reactive than C-F but nevertheless highly chlorinated compounds such as CCl4 and CHCl3 are fairly inert. The C-Br bond in 1-bromobutane is covalent and contain no Br- ions, so it does not produce a precipitate of silver bromide with silver nitrate. The slow appearance of a precipitate of silver bromide suggests that Br- ions are slowly produced. This is an example of nucleophilic substitution. In this experiment, you may also compare the rates of hydrolysis with iodo-alkane, bromobenzene....., and hence deduce the nature of C-Hal bonds in halogeno-alkanes and in aromatic compounds.[Ref. Chemistry in Context pp.504 - 508]
Chemicals:
Ethanol, 0.1M
![]() , 1-chlorobutane, 1-bromobutane, 1-iodobutane, bromobenzene
, 1-chlorobutane, 1-bromobutane, 1-iodobutane, bromobenzene
Apparatus: Test tubes, -10-110 o C thermometer, 10 cm3 measuring cylinder, teat pipette, 250 cm3
beaker
Procedure:
[Hazard Warning: Ethanol and 1-chlorobutane are flammable, 1-bromobutane and 1-iodobutane are harmful, and bromobenzene is irritant.]
A. To compare the rates of hydrolysis of chloro-, bromo-, and iodo- alkanes
1. To three separate test tubes add 2 cm3 of ethanol each and place them in a beaker of water kept at about 60 C.
2. Add 1 cm3 of 0.1M silver nitrate(V) solution to each test tube.
3. Using separate teat pipettes, add 5 drops of 1-chlorobutane to the first test tube, 5 drops of 1-bromobutane to the second and 5 drops of 1-iodobutane to the third.
4. Shake the test tubes and observe the order in which the precipitates appear. Note the colour of the precipitate formed in each case.
B. To compare the rate of hydrolysis of aliphatic and aromatic halogeno-compounds
Repeat part A first at room temperature and then in hot water (at 60 C) with 1-bromobutane
and bromobenzene.
(Take care that there is enough ethanol present to dissolve the aromatic halogeno-compounds.
A slight turbidity on mixing may be due to an emulsion of the organic compound with water. If
this happens, add a few drops of ethanol and shake until the solution is clear.)
Discussion:
1. What are the precipitates observed?
2. Write equations for the reactions occurred.
3. What effect does the halo group have on the rate of the hydrolysis reaction? Explain your answer.
4. What effect does the phenyl group have on the rate of the hydrolysis reaction? Explain your answer.
Title : Reaction of –OH group in butan-1-ol by Br- / Cl- : A competitive reaction
Introduction:
This experiment is a modification of the one on the preparation of haloalkanes such as
1-bromobutane. However, instead of treating an experiment as a preparation exercise for its
own sake, it is modified as shown here to illustrate a competitive nucleophilic substitution
reaction (Sn1 / Sn2 ).
It is very important that the heating rate must not be too high, otherwise little or no product would
be obtained. The equation for the reaction is
Br- / Cl-
CH3CH2CH2CH2OH -------------> CH3CH2CH2CH2Br + CH3CH2CH2CH2Cl
c. H2SO4
Chemicals: Ammonium bromide, ammonium chloride, conc. sulphuric acid, butan-1-ol,
anhydrous calcium chloride, 5% aqueous sodium bicarbonate, crushed ice, filter paper.
Apparatus: Quick fit, electronic balance, bunsen burner, condenser, separating funnel,
conical flask, filter funnel.
Procedure:
1. Carefully pour 50 ml of c. H2SO4 over 60 g of crushed ice in a 250 ml boiling flask. Mix the solution by swirling.
2. Add 13.5g (0.25 mole) of ammonium chloride, 24.5g (0.25 mole) of ammonium bromide and some boiling chips to the flask.
3. Attach the flask to a reflux condenser fitted with a trap for acidic gases, and warm the mixture until the solids are completely dissolved (swirling would help).
4. Add in 14.8g (0.20 mole) of butan-1-ol through the top of the condenser and reflux the resulting solution for about 90 minutes. 【IMPORTANT: the heating rates must be controlled so that no organic materials are carried through the top of the condenser.】
5. Allow the reaction mixture to cool and transfer it to a separating funnel. Discard the aqueous layer and wash the crude halide twice with about 100 ml of 5% aqueous sodium bicarbonate.
6. Transfer the organic material to a 50 ml conical flask containing 2-3g of anhydrous calcium chloride. Stopper the flask and swirl the contents for a few minutes. If the supernatant liquid is clear, decant it into a small, dry conical flask. If a suspension of drying agent remains, separate the solid by gravity filtration and collect the filtrate in a small flask.
Given the densities of proposed products:
1-chlorobutane : 0.89 g cm-3
1-bromobutane : 1.28 g cm-3
Questions
for discussion:
1. Estimate the % yield of the products obtained.
2. Why do we have to start out with the same number of moles of ammonium chloride and ammonium bromide?
3. Why do we need to wash the crude product with various solutions?
4. Why is one type of haloalkane formed more than the other?
5. Why is a trap for acidic gases necessary?
Title : Identification of a carbonyl compound by preparing its derivatives
Introduction:
Crystalline derivatives of many carbonyl compounds can be formed by condensation reactions with compounds such as phenylhydrazine and 2,4-dinitrophenylhydrazine. These derivatives can usually be isolated in relatively pure forms which have well defined melting points. Phenylhydrazine forms derivatives (phenylhydrazones) readily with aromatic aldehydes, but in general
2,4-dinitrophenylhydrazine is preferred because its derivatives (2,4-dinitrophenylhydrazones) have higher melting points and are less soluble. These derivatives are useful in identification of carbonyl compounds.
Chemicals: Ethanol, methanol, dilute sulphuric acid, 2,4-dinitrophenylhydrazine in methanol,
carbonyl compound labelled X (different groups of students may work on different
carbonyl compounds)
Apparatus: Beaker, 100 cm3 measuring cylinder, apparatus for melting point determination,
apparatus for suction filtration, rubber rings (cut from a rubber tubing of appropriate
diameter), ice bath
Procedure:
[Hazard Warning: Methanol solution of 2,4-dinitrophenylhydrazine is flammable and toxic, methanol, ethanol and many carbonyl compounds are flammable, and bench dilute sulphuric(VI) acid is corrosive.]
A. Preparation of 2,4-dinitrophenylhydrazone of compound X
1. Add 10 drops of compound X to 5 cm3 of 2,4-dinitrophenylhydrazine solution in a 50 cm3 beaker. If crystals are not formed, added about 1 cm3 of dilute sulphuric(VI) acid. If they are still not formed, warm the mixture gently, then cool with scratching in ice water.
2. Filter off the crystals by suction filtration. While still on suction, wash the crystals with 1 cm3 of methanol.
3. Recrystallize the crystals from hot ethanol as follows:
(a) Transfer the crystals to a 100 cm3 beaker standing on a steam bath (or in a 250 cm3 beaker of
hot water).
(b) Dissolve the crystals in the minimum amount of hot ethanol.
(c) When the crystals have dissolved, cool the solution in an ice-water mixture until crystals reappear.
(d) Filter the crystals by suction. If necessary, rinse the beaker with the filtrate (not the extra solvent) to complete the transfer. Finally, wash the crystals with a few drops of cold ethanol. Dry the crystals by drawing air through them for a few minutes.
4. Spread the crystals on a dry watch glass and leave overnight in a desiccator for drying.
B. Determination of the melting point of the 2,4-dinitrophenylhydrazone of compound X
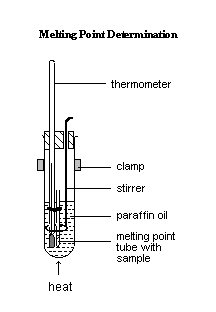
1. Introduce a small amount of the crystals into a melting point tube until a total length of about 0.5
cm is compacted at the bottom of the tube.
2. Attach the prepared melting point tube to the thermometer, as shown in Figure 1.
3. Half-filled a boiling tube with paraffin oil, and position the thermometer with attached tube and the stirrer as shown in Figure 2.
4. Position the apparatus over a low Bunsen flame and gauze and gently heat the apparatus, stirring the paraffin oil all the time by moving the stirrer up and down.
5. Note the temperatures when the crystals start to melt and when the melting is completed.
6. Compare the melting point of the crystals with the values given in the following table and thus identify compound X.
 |
Discussion:
1. What soluble impurities may be present in your product before recrystallization?
How can they be removed in the recrystallization process?
2. What factors should be considered in selecting a suitable solvent in the recrystallization step?
3. In the recrystallization procedure, why were the crystals dissolved in only the minimum amount of hot ethanol?
4. If the sample contains insoluble impurities such as pieces of filter paper, cork, etc., suggest how they can be removed.
5. If the melting point of the 2,4-dinitrophenylhydrazine is 156 ℃, suggest how you can confirm whether compound X is propanal or pentan-3-one.
Title : Preparation and Reaction of amines and diazonium salt
Note: Use IUPAC naming for amines
Common name: aniline, anilinium ion, 2,4,6-tribromoaniline
IUPAC name: benzenamine, benzenammonium ion, 2,4,6-tribromobenzenamine
Introduction:
A.
Preparation
of aniline (Demonstration)
(a) Reduction of nitrobenzene
Place 4ml of nitrobenzene and 10g of tin in a 250ml round-bottomed flask fitted with a
reflux condenser. Add excess concentrated hydrochloric acid, by adding it to the flask a
few ml at a time, until a total of 24ml has been added. It is most convenient to pour the acid
down the condenser, and it is advisable to shake the flask between each addition. When all
the acid has been added heat the reaction mixture on a water-bath for 15 minutes.
(b) Steam distillation
Cool the flask and add sodium hydroxide solution until the solution is strongly alkaline. The
hydroxide of tin, which is precipitated first, should be dissolved - about 50 ml of 30%
sodium hydroxide solution should be sufficient.
Set up the apparatus for steam distillation and proceed until oily droplets of aniline are no
longer visible in the distillate coming over.
(c) Ether extraction
Add concentrated hydrochloric acid to the distillate, until the solution is acidic - about 3 or 4
ml of acid should be needed - and, when cool, transfer the solution to the separating funnel.
Extract with two separate 5 ml portions of ether. After shaking the separating funnel invert
it and open the tap to release the ether vapour. Discard the ether layer in each case (run it
into the ether residues bottle).
Transfer the aqueous layer to a beaker and add sodium hydroxide pellets, one at a time, until
the solution is alkaline.
Shake this alkaline solution, in a separating funnel, with three 5ml portions of ether. Retain
the ether layer from this separation.
Dry the ethereal extract, by standing it over potassium hydroxide pellets in a stoppered flask
until the next practical session.
(d) Ether distillation
Heat a water bath to about 70C and, after extinguishing all flames, decant the ethereal
solution into a 50 ml round-bottomed flask fitted with a stillhead adaptor and water
condenser. Heat the flask in the water bath until no more ether distills.
Replace the water condenser by air condenser, fit a thermometer pocket and 250C
thermometer to the stillhead adaptor and heat the flask over a gauze using a Bunsen burner.
Collect the fraction which distills between 180 and 185C and record the yield.
1. In an alternative procedure the reflux condenser is removed and the reaction mixture is heated
on a water bath prior to making the solution alkaline for the steam distillation. The later, ether
extraction of the acid solution is then omitted. Explain this procedure.
2. Why must the solution be made alkaline before steam distillation?
3. The aniline formed by reduction exists as anilinium chloride and also as aniliniumhexachlorostannate(IV). Write formulae for these two compounds.
4. Why is the reaction mixture cooled before the addition of sodium hydroxide in part (b)?
5. Would you expect a mixture of aniline and water to boil at a temperature above, or below, the boiling point of water? Explain your answer.
6. Why is hydrochloric acid added to the distillate before the first ether extraction, and why is this ethereal solution discarded?
7. Why is sodium hydroxide added before the second ether extraction and why is this ethereal solution retained?
8. Why are two separate 5 ml portions of ether used in part ( c), rather than one 10 ml portion?
9. Why is potassium hydroxide used for drying aniline, rather than the cheaper calcium chloride?
B.
Properties
of aniline
(a) Shake a few drops of aniline with water in a test tube, and add a piece of red litmus paper to
the solution.
Add a few drops of aniline to 2ml of dilute hydrochloric acid in a test tube.
Describe your observations and explain the difference in solubility of aniline in the two
solutions.
10. Would you expect aniline to be more, or less, basic than methylamine?
(b) To five drops of aniline add concentrated hydrochloric acid until the aniline dissolves.
Add bromine water and note the rapid reaction producing 2,4,6-tribromoaniline.
(c) To three drops of aniline add 5ml of 10% sodium hydroxide solution followed by five drops
benzoyl chloride. Stopper the test tube and shake until a solid product is formed. Isolate the
product and if time permits, recrystallize from hot ethanol determine the melting point of
the benzanilide.
(d) To three drops of aniline in a test tube add concentrated hydrochloric acid and a little water
until the solid formed dissolves. Cool the tube in ice and then add a few crystals of sodium
nitrite. Keep half of the solution in ice and heat the other portion gently over a Bunsen
burner.
11. What do you observe in each of the two samples? What can you smell in the tube that has
been heated?
12. Compare the reaction of aniline in (d), with the behaviour of aliphatic amines on treatment
with sodium nitrite and hydrochloric acid.
13. Why is a mixture of hydrochloric acid and sodium nitrite used in these reactions rather than
nitrous acid?
Note:
1. Cut the quantities by 1/4 times.
2. Part(b): Add NaOH solution to the pear-shape flask using a filter funnel and make sure that no alkali has adhered to the inner part of the neck of the flask.
(Reason: strong alkali will attack glass especially on heating. The pear-shape flask and the
stillhead adaptor will be fused together after experiment and cannot separated.)
3. During distillation,
-check the water level in the safety-tube regularly,
-check that the rubber-tubing is not blocked by condensed water,
-check that the rubber tubing is not close to the burners.
4. Check for the use of air condenser and water condenser.
C.
Small
scale preparation and reactions of benzenediazonium chloride solution
(a)
Preparation
of benzenediazonium chloride:
To 1.5 ml of phenylamine and 5 ml of water in a boiling tube, add 4 ml of concentrated
hydrochloric acid. Cork and shake the tube until the amine has dissolved.
Cool the solution in a beaker containing ice to about 5C, and add a solution of sodium nitrite (1.5g in 4 ml of water), previously cooled to 5C. Make sure that the temperature of the mixture does not rise above 10C. (Warning: the temperature overshoots very readily, and sodium nitrite solution should be added slowly.)
(b)
Reactions
of benzenediazonium chloride solution:
Replacement reactions
1. Boil 2 ml of the diazonium solution. Note the odour of phenol which separates as an oily liquid.(Note: Phenol will often occur as side product in these reactions, and may influence the observation of other reactions.)
2. To 2 ml of the diazonium solution at 5C, add, drop by drop, 1 ml of a 10% solution of potassium iodide, previously cooled to 5C. Allow to stand for 5 minutes and then gently boil. Observe oily drops of iodobenzene formed.(Iodobenzene:colourless liquid)
Coupling reactions
3. To 4 ml of benzenediazonium chloride solution in a test tube, add phenylamine (cool to below 5C) dropwise.
4. Dissolve 2 or 3 crystals of phenol in 2 ml of dilute sodium hydroxide solution. Cool the solution in ice, and add the diazonium solution drop by drop. A yellow precipitate of a dye, the sodium salt of 4-hydroxyazobenzene, is obtained.
5. Repeat part (4) using naphthalen-2-ol instead of phenol.
![]()
Title : Reactions of aminoacids (glycine)
Introduction: Glycine is one of the simplest aminoacids H O
The structure of glycine is: H - N - C - C - OH
. H H
Procedure:
Reactions of glycine
1. Describe the appearance of glycine (aminoethanoic acid).
2. Test the solubility of aminoethanoic acid in -water; -ethanol; -ether
3. Test the pH of aminoethanoic acid in water.
Properties
of the amino-group
4. Dissolve a small amount of aminoethanoic acid in the minimum amount of concentrated hydrochloric acid .Cool the mixture and observe whether a white crystalline solid is formed.
5. To about 1 ml of an ice-cool solution of sodium nitrite in a test tube, add 1 ml of dilute hydrochloric acid. Some decomposition of the nitrous acid formed will occur. When the effervescence has subsided, introduce a few crystals (or a few drops of a concentrated aqueous solution) of aminoethanoic acid. Effervescence occurs again as nitrogen is evolved.
This indicates the presence of an amino-group:
![]()
Properties
of carboxyl group:
6. To a solution of aminoethanoic acid in water, add some solid sodium hydrogencarbonate.
Evolution of carbon dioxide indicates the presence of an acid group:
![]()
7. To an aqueous solution of aminoethanoic acid add a few drops of copper(II) sulphate solution.Observe the deep blue colour of the copper salt of the amino-acid.
8. Identification of aminoethanoic acid: Mixture of amino-acids can be separated by chromatography, and then compare the R values of the spots with literature values.Location of spots: spray the chromatography paper sparingly with 0.02M ninhydrin solution and then heat in an oven at about 110C for 10 minutes. (+)result: give purple spots.
![]()
Title : Reactions of group II elements and their compounds
Introduction:
The s-block of the periodic table contains the most reactive and, in chemical terms, the most typically metallic elements. All the elements in group I are highly reactive, but those in group II are slightly less so and show a rather more obvious trend in reactivity. In this practical you will study some of the properties of the elements of group II and their compounds. The members of the group are shown in table 1.
|
Elements of gp II |
Beryllium Magnesium Calcium Strontium Barium Radium |
|
Symbols |
Be Mg Ca Sr Ba Ra |
We will concentrate on the three elements: magnesium, calcium and barium. Beryllium will not be studied because its compounds are extremely toxic and very expensive.
Some physical data concerning the elements of group II are given in table 2.
|
Element |
Be |
Mg |
Ca |
Sr |
Ba |
|
Electronic structure |
(He)2s2 |
(Ne)3s2 |
(Ar)4s2 |
(Kr)5s2 |
(Xe)6s2 |
|
1st ionization energy (kJ.mol-1) |
900 |
740 |
590 |
550 |
500 |
|
2nd ionization energy (kJ.mol-1) |
1800 |
1450 |
1150 |
1060 |
970 |
|
3rd ionization energy (kJ.mol-1) |
14800 |
7700 |
4900 |
4200 |
---0 |
|
Atomic (metallic) radius (nm) |
0.11 |
0.16 |
0.20 |
0.21 |
0.22 |
|
Ionic radius (M2+ ) (nm) |
0.030 |
0.065 |
0.094 |
0.110 |
0.134 |
|
Hydration energy(M2+) (kJ.mol-1) |
---- |
-1891 |
-1561 |
-1414 |
-1273 |
|
Standard electrode potential E [M2+(aq) | M2+(s) (volts) |
-1.85 |
-2.37 |
-2.87 |
-2.89 |
-2.91 |
1. What type of ion is formed by group II elements when they react?
2. Why do ionization energies decrease as you go down group II?
3. Why do atomic radii increase as you go down group II?
4. Which of the various sets of data in table 1 gives the most accurate indication of the likely reactivity trend within the group?
Chemicals: Universal indicator solution, indicator paper, filter paper, splints, magnesium ribbon,
magnesium powder, calcium granules and turnings, barium metal in small pieces,
magnesium oxide, calcium hydroxide, barium hydroxide, hydrated magnesium chloride,
hydrated calcium chloride, hydrated barium chloride, magnesium carbonate, lime water,
barium carbonate.
0.1M solutions of Mg2+ (aq), Ca2+ (aq), Ba2+ (aq)
1.0M
solutions of NaOH,
![]() and
and
![]()
Apparatus: Hard-glass test tubes (8), Angled glass bend with bung to fit test tube, 400ml beaker, funnel.
Procedure:
A.
Reaction of elements with water
Put a very small piece of calcium metal into a large beaker of cold water. Observe the reaction and identify the products. Repeat, using a small piece of clean magnesium ribbon and then a small piece of barium metal.
Care: Barium and its compounds are poisonous. Handle with care and wash your hands after practical. Eye protection must be worn. Your teacher may prefer to demonstrate the reaction of barium with water.
1. Write equations for any reactions which occur.
2. How do the metals differ in the vigor with which they react with water?
3. In all their reactions the group II metals behave as reducing agents. What is being reduced in this reaction?
4. Explain the reactivity trend among the three metals in this experiment using some of the data in table 2.
5.
What other factor, not listed in the table, may affect the relative
reactivity of the metals with water? The reaction of
magnesium with water was probably very slow. You can
investigate this reaction further by setting up the
experiment shown in figure 1(in the mannual only ).Leave the experiment for half
an hour or so, then test the products.
B.
Acid-base character
Place a very small quantity (0.01g) of magnesium oxide, calcium hydroxide and barium hydroxide in three separate test tubes. Add 10ml distilled water to each tube and shake. Add 2 drops of universal indicator solution to each tube and mix. Record the pH values indicated for the three tubes.
1. How does the acid-base character of the hydroxides vary within the group?
2. Write a general ionic equation to represent the equilibrium between the undissolved solid hydroxide and its aqueous ions.
3. Why is it valid to use magnesium oxide instead of magnesium hydroxide in this experiment?
4. What is milk of magnesia and what is it used for?
5. Look at the values for ionic radius in table 2. Which group II ion will have the strongest attraction for OH- ions? What effect will this have on the basic strength of its hydroxide?
C.
Hydrolysis of chlorides
Ionic chlorides dissolve in water forming simple hydrated ions. Many covalent and partly covalent chlorides, however, are hydrolysed, giving hydrogen chloride and the oxide or hydroxide. For example, aluminium chloride reacts vigorously with water as follows:
![]()
The extent of hydrolysis of the group II chlorides can be estimated by heating the hydrated chloride and testing for hydrogen chloride gas. Working in a fume cupboard, strongly heat about 1 cm depth of each of the hydrated chlorides of magnesium, calcium and barium in separate, dry hard-glass test tubes. Test for the evolution of hydrogen chloride.
1. Are any of the chlorides hydrolysed? Is there a trend in the tendency towards hydrolysis?
2. Which of the chlorides shows the greatest covalent character? Explain this tendency towards covalency in terms of some of the data in table 2.
D.
Thermal stability of the carbonates
Strongly heat about 1 cm depth of each of the dry carbonates of magnesium, calcium and barium separately in the apparatus shown in figure 2. Continue heating strongly for several minutes. Note how rapidly gas is evolved, and the extent to which the lime-water becomes milky. Remember to remove the tube from the lime-water as soon as heating is stopped.
1. Write equations for any reaction which occur.
2. What trend do you detect in the thermal stability of the carbonates of the elements in gp II?
E.
Solubility of some compounds of group 2 elements
To investigate the solubility of group II compounds, solution containing the appropriate anions and cations are mixed. If the compound is insoluble, a precipitate will form.
Put 2 ml of a 0.1M solution of each of the group II cations under investigation (Mg2+, Ca2+, Ba2+) in separate test tubes. Add an equal volume of a 1.0M solution of hydroxide ions, and mix. Note whether a precipitate is formed, and if so, how dense it is. Repeat the experiment twice, using first a 1.0M solution of sulphate ions and then a 1.0M solution of carbonate ions, instead of the hydroxide ions. Tabulate your results.
1. What trends do you notice in the solubility of
(a) hydroxides, (b) sulphates, (c ) carbonates ?
If you have time, you could extend this experiment by investigating the solubility of some other compounds - say chromates and oxalates. You might then be able to formulate a more general rule concerning solubility trends within the group.
2.. Using the results and conclusions you have derived from this practical, predict the following properties of (a) beryllium, (b) strontium, and their compounds:
(i) reaction with water
(ii) acid-base character of the hydroxide
(iii) tendency of the chloride to hydrolyse
(iv) thermal stability of the carbonate
(v) solubility of the hydroxide
(vi) solubility of the carbonate
(vii) solubility of the sulphate
![]()
Title : Reactions of group VII elements and their compounds
Procedure:
A.
The elements
Make a comparison of the appearance of samples of chlorine, bromine and iodine. Record your results in the form of a table, adding any further data you wish.
Discover the colour of solutions of chlorine, bromine and iodine in trichloromethane (or tetrachloromethane). Use this information to establish what happens when chlorine water is added to an aqueous solution of (a) potassium iodide, (b) potassium bromide, with trichloromethane present in each case.
1. Why is it necessary to use chloroform to establish whether bromine or iodine is produced?
2. Which of these three elements is the strongest oxidizing agent?
3. Look up the electrode potentials. Do these confirm your results?
E-values for
![]() Cl = +1.36V;
Br=+1.07V; I=+0.53V
Cl = +1.36V;
Br=+1.07V; I=+0.53V
4. What would you expect to see if bromine water were added to an aqueous solution of
(i) potassium chloride, (ii) potassium iodide, with trichloromethane present in each case.
Use the electrode potentials to arrive at your prediction.
B.
The hydrogen halide
Add syrupy phosphoric acid to a small sample of each of the solid sodium or potassium halide and warm. Record your observations.
5. Does heating cause decomposition of any of the hydrogen halide? If so, which is most readily decomposed?
6. Does the syrupy phosphoric acid produce the hydrogen halide because it is a stronger acid than the hydrogen halide, or because it is less volatile?
7. What would you predict to happen if concentrated hydrochloric acid were added to anhydrous sodium phosphate?
Add concentrated sulfuric acid to a small sample of each of the solid sodium or potassium halide. Note the appearance and (caution!) the smell of the products. Suggest the identity of the products of these reactions.
8. Why does concentrated sulphuric acid produce a different reaction from that with phosphoric acid?
9. What does this suggest about the relative ease of oxidation of these hydrogen halides?
C.
The silver halide
Place about 1 ml of silver nitrate solution in each of 3 test tubes and add an equal volume of potassium chloride, potassium bromide and potassium iodide to each of the 3 tubes. Mix the contents of each tube.
Divide the solutions into two portions. Expose the first portion to sunlight for a few minutes, and add excess concentrated ammonia solution to the second tube.
10. Record your observations and attempt to explain the solubility in the presence of ammonia solution, using the relative solubilities of the silver halides.
D.
sodium hypochlorite
Investigate the effect of dilute acid on sodium hypochlorite solution.
11. Record your observations, including identification of any gases evolved, and interpret the reactions.
E.
Oxidizing power of hypochlorite, chlorate and perchlorate
List of reduction potentials:
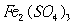
12. Using the above electrode
potentials in acid solutions to predict the reaction between
(i) hypochlorite and iron(II) ion;
(ii) hypochlorite and iodide.
Carry out reactions to confirm your predictions above.
(i) hypochlorite and iron(II) ion;
(ii) hypochlorite and iodide.
F.
An investigation of the thermal stability of chlorate
Heat small sample of potassium chlorate. (Take care in heating the solid!)
13. Record your observations, including identification of any gases evolved. Write suitable equation for the reaction occurred.
![]()
Title : Reactions of nitrogen and sulphur compounds
Introduction:
A.
Reactions of sulphurous acid
(Sulphurous acid is a solution of sulphur dioxide in water. In redox reaction, sulphur would be oxidized from +4 to +6 oxidation state)
1. Pour about 2 ml of sulphur dioxide solution into a test tube. To the test tube pour in 1 ml of chlorine water. Observe the colour change. (Sulphate ion and chloride ion should be formed, but they are difficult to be tested for. Why?)
2. To a test tube of 2 ml of sulphur dioxide solution add in 1 ml of iron(III) chloride solution. Observe any visible change.
3. Repeat (2) using acidified potassium permanganate solution in place of iron(III) solution.
4. Repeat (2) using acidified potassium dichromate solution in place of iron(III) solution.
B.
Reaction of conc. sulphuric acid
(Caution: Take care in handling conc. sulphuric acid, especially during heating and disposal, because it is corrosive.)
1. Carefully pour about 1 ml of conc. sulphuric acid into a test tube. Add in 3 pieces of copper turnings. Warm if necessary. Observe for the colour change of solution and test for the presence of sulphur dioxide evolved.
2. Place a spatula of copper(II) sulphate crystal into a test tube. Add in about 1 ml of conc. sulphuric acid. Observe for the colour change of the crystals.
3. Place a piece of used match into a test tube containing 1 ml of conc. sulphuric acid. Note the appearance of the used match.
4. Repeat step (3) above using sugar instead of the used match.
C.
Complex formation involving ammonia and sodium hydroxide
N.B. Both ammonia and hydroxide ion can form complex with some metal ions, and such complex formation is often used for the identification of metal ion in an unknown.
1.
Add 1 drop of dilute sodium hydroxide solution to a test tube containing
1 ml of zinc chloride solution. Observe for the formation of precipitate. Repeat
the test using
![]()
2. Divide the solution obtained in (1) into two parts. Add to the first part conc. ammonia solution. Note whether the precipitate can be redissolved or not.
3. To the second portion add conc. sodium hydroxide solution. Note whether the precipitate can be redissolved or not.
D.
Reaction3
of nitric acid
(Caution: conc. nitric acid is corrosive and poisonous nitrogen dioxide may be formed)
1. Add 1 ml of conc. nitric acid into a test tube. Add to it copper turnings.Observe any visible changes. Repeat using magnesium ribbon in place of copper turnings.
2. Repeat (1) using moderately concentrated nitric acid (1V conc. acid + 1V water) instead of conc. nitric acid. Compare your observation with part (1).
E.
Reactions of nitrates
Thermal stability of nitrates: heating 3 typical nitrates.
1. Add half spatula of copper(II) nitrate into a test tube. Heat the tube and test for the evolution of oxygen and nitrogen dioxide. (Caution: a poisonous gas!)
2. Add a small amount of silver nitrate into a test tube and heat. Observe for the formation of nitrogen dioxide, oxygen and silver metal.
3. Add half spatula of potassium nitrate into a test tube. Heat the tube strongly and test for the evolution of oxygen.
Brown ring test: the standard test for the presence of nitrate ion.
4. Pour 1 ml of potassium nitrate solution into a test tube. Add to it 1 ml of iron(II) sulphate solution. Mix the solution. Hold the test tube in a slant position steadily and by using a dropper, add in conc. sulphuric acid slowly. Observe for the formation of a brown layer at the interface of the upper aqueous solution and the lower conc. sulphuric acid.
![]()
Title : Investigation of the oxidation states of Vanadium
Introduction:
Vanadium has a range of oxidation states from 0 to +5. The following experiments are designed to identify the colours of vanadium solutions for the various oxidation states, the ease with which the oxidation states can be formed and the stability of the vanadium compounds made.
Chemicals:
Ammonium metavanadate, 0.1M
![]() , 0.1M
, 0.1M
![]() , 0.1M KI, 0.1M KBr,
, 0.1M KI, 0.1M KBr,
0.1M I2 in KI, aqueous SO2, copper powder, zinc powder, bench dilute H2SO4 .
Apparatus: Bunsen burner, test tube and rack, filter funnel, filter paper.
Procedure:
[Hazard warning: Ammonium metavanadate is toxic, iron(III) sulphate(V) is irritant, and bench dilute sulphuric(VI) acid is corrosive.]
A.
Various oxidation states obtained from ammonium metavanadate:
1. Dissolve a little of ammonium metavanadate in dilute sulphuric(VI) acid.
2. Mix solutions of substances as indicated in table 1 and carefully record your observations. In each case add the other solution and centrifuge or filter quickly.
Table
1
|
Substances to be mixed |
Observation |
|
Ammonium metavanadate solution + iron(III) sulphate(VI) |
|
|
Ammonium metavanadate solution + potassium iodide |
|
|
Ammonium metavanadate solution + aqueous sulphur dioxide |
|
|
Ammonium metavanadate solution + copper powder |
|
|
Ammonium metavanadate solution + zinc powder |
|
B.
Identification of colours of vanadium compounds and a study of these compounds
1. Using the following chart of standard electrodes E and your observation made in part A, deduce the colours of the various oxidation states of vanadium and list them in Table 2. (Be careful about the mixing of coloured solutions even without chemical reactions. A blue and yellow solution can give a green one on mixing. If iodine is present, the colour of other products can only be seen if the iodine is converted to colourless iodide ions or removed in a solution of 1,1,1-trichloroethane.)
2. Again using the table of E values, and your deductions in table 2, predict the likely experimental result when substances are mixed ad in table 3. This should cover the expected oxidation state of vanadium in the final solution and the likely observations.
3. Test them out by performing the actual experiments. Record the results also in table 3.
Table
3
|
Substances to be mixed |
Prediction |
Experimental Result |
|
V(IV) + V(II) |
|
|
|
V(V) + V(III) |
|
|
|
V(IV) +
|
|
|
|
V(III) +
|
|
|
|
V(IV) + KI |
|
|
|
V(V) + KBr |
|
|
|
V(III) + I2 |
|
|
|
V(IV) + I2 |
|
|
|
V(V) + Cu |
|
|
Discussion:
1. What is the oxidation state of vanadium in ammonium metavanadate?
2. What is the ion present when vanadium has an oxidation number of +4?
3. How did you make your solutions of V(II), V(III) and V(IV) for the experiments in table 3?
![]()
Title : Catalytic effect of d-block ions
Introduction:
This experiment is a study of the catalytic action of d-block ions on the reaction between iodide and persulphate ions. Iodide ions are oxidized by persulphate (peroxodisulphate) ions to iodine and various metal ions will catalyse this reaction:
![]()
A convenient way in which to measure the rate of this reaction is to add a fixed volume of sodium thiosulphate solution to the reaction mixture. This reacts with the iodine formed in the reaction as follows:
![]()
When the thiosulphate has been used up, free iodine is produced, and if some starch solution has also been added, a deep blue colour will be produced. The time that it takes for the blue colour to appear is inversely proportional to the average rate of reaction during this time, and so, whether or not an ion catalyses the reaction can be found by comparing the times for the reaction with and without the metal ion.
Procedure:
Place 10 ml of iodine solution, 10 ml of thiosulphate solution and 5 ml of starch solution in a 150 ml flask, and 20 ml of persulphate solution in a boiling tube. Add the persulphate solution to the flask and start the stopclock. Note the time taken for the blue colour to develop. Repeat the experiment adding three drops of a solution of one of the ions listed below. The ions can be shared out amongst the class so that the whole range is tested.
Cr(III) Mn(II) Fe(II) Co(II) Ni(II) Cu(II)
Cr(VI) Mn(VII) Fe(III)
Compare the times from the various experiments with and without metal ion and so find out if the addition of the metal ion affect the rate of the reaction. In order to compare the times some idea of the errors involved must be known. If two times are
1.
130
![]() 30 seconds, and
30 seconds, and
2.
80
![]() 20 seconds
20 seconds
then although inspection at 80 and 130 seconds might suggest that the reaction in (2) was nearly twice as fast as that in (1), the errors indicate that (1) could be as low as 100 seconds and that (2) could be as high as 100 seconds so the rate in the two experiments may be the same. Comparison of several results will enable you to gain some idea of the errors, and, indeed, an average of several experimental results will give better figures with which to decide whether the rate is being affected.
Questions for discussion:
1. Which ions catalyse the reaction?
2. What sort of reaction is being catalysed? (Precipitation, redox, neutralization, ….etc.)
3. What is being transferred in the reaction?
4. How might the catalyst (an ion of a metal in the d-block) help this reaction?
5. Can you find out if your suggestion is possible by looking up Eo values?
6. Can you test your suggestions practically?
Figure:
Redox potentials at pH=0
E (volt)
![]() .0 -
.0 -
![]()
0.2 -
0.4
-
![]()
0.6 -
0.8 -
![]()
![]()
1.0 -
![]()
1.2
- &
![]()
![]()
1.4
- &
![]()
1.6
- &
![]()
![]()
![]()
1.8 -
![]()
![]()
![]()
![]() 2.0
-
2.0
-
![]()
![]() |
|
| |
|
|
|
| |
|
|
|
| |
|
|
|
| |
|
7 6 5 4 3 2 1 0 -1 -2 Oxidation number
Title : Analysis of Sulphur Dioxide Content in Wine
Introduction:
In the presence of atmospheric oxygen, the alcohol content of wine can be converted to ethanoic acid making the wines sour and unpalatable. Even small amount of air, over a period of time, can adversely affect wines. The problem can be minimized by the introduction of a suitable reductant which will preferentially react with the oxygen. One such reductant is sulphur dioxide but because this substance is, itself, toxic and pungent in odour, limits are set on the amount of ‘free’ sulphur dioxide allowed in wine. Most of the preservative present in wine is ‘fixed’ in the form of sodium hydrogensulphate(IV) (i.e. sodium hydrogensulphite). Although this can act as a source of sulphur dioxide, the actual amount of free sulphur dioxide is quite low.
In this experiment, the amount of
total available sulphur dioxide in the wine, irrespective of its actual form in
the sample is determined. The method involves first the conversion of all
sulphur dioxide into sulphite ion (
![]() ). Acidification of the solution then liberates all sulphur dioxide.
). Acidification of the solution then liberates all sulphur dioxide.
![]()
which is then titrated with iodine solution according to the equation:
![]()
Chemicals: White wine (non-sparking or non-carbonated), 1M NaOH, 2M H2SO4, 0.0050M I2 ,
starch solution (freshly prepared)
Apparatus: Titration apparatus, measuring cylinder, teat pipette
Procedure:
1. Determine, from the label, the volume of wine in a bottle.
2. Using a pipette, transfer 25 ml of white wine into 250 ml conical flask.
3. Add about 12 ml of 1 M sodium hydroxide solution and allow to stand for about 15 minutes.
4. Add about 10 ml of 2 M sulphuric acid to the mixture and titrate with 0.0050 M iodine solution. (Why should this titration be done quickly?)
5. Record the actual molarity of the iodine solution.
6. Record the titre required to produce the first faint but permanent blue colour.
7. Repeat the procedure to get two additional concordant titres.
Results and Discussion:
1.
|
Mean titre (ml) |
|
|
no. of moles of I2 in the titre |
|
|
no. of moles of SO2 in the original aliquot |
|
|
no. of moles of SO2 in the bottle of wine |
|
|
Mass of SO2 in the bottle of wine (mg) |
|
2. Compare your results with the limit of 450 mg dm-3 stated in Hong Kong Preservatives in Food Regulations. Comment upon the comparison.
3. What is the most questionable assumption you have made in arriving at this figure?
4. By the use of oxidation numbers, show that the reaction during the titration was a redox reaction.
5. Suggest an experiment to determine the efficiency of sodium hydrogensulphate(IV) as a wine preservative. If time allows, conduct the experiment and comment on your results.