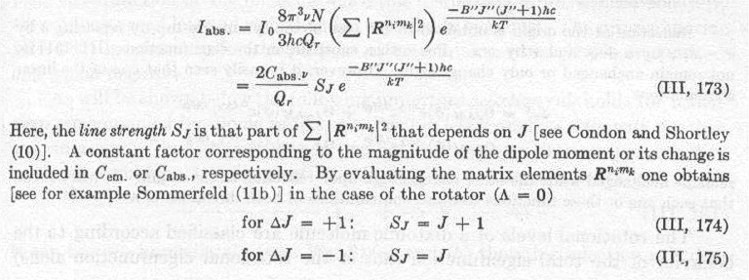
Chromium Dimer
Mike Geusic, Michael Geusic, M. E. Geusic
III. SPECTROSCOPIC INVESTIGATIONS OF TRANSITION METAL DIATOMICS (CHROMIUM DIMER)
A. Introduction
The gas phase spectroscopic investigations of transition metal diatomics have lagged behind as compared to similar studies of other elements within the periodic table. This is primarily due to the extremely high boiling points (approximately 2000-6000 K) of these metals, making their spectroscopic studies extremely difficult, if not impossible, with the techniques used for the alkali and coinage metals. Although about one half of all transition metal diatomics have been studied in the condensed phase by matrix isolation27, some of the answers to important scientific questions have been elusive mainly because of the inability of this technique to permit investigation of high resolution rotational spectra. One of the most important and also controversial questions surrounding the transition metal diatomics has been the nature of the metal-metal bond, or more specifically, the role of d-orbitals in the bonding of these molecules.
In an effort to obtain experimental data pertaining to this question, the cluster source previously described was used to produce and cool (Vib <125 K, Rot <10 K) beams of these transition metal clusters. The diatomics could then be spectroscopically probed using high resolution resonant two-photon ionization.
At the start of these studies, it was apparent that chromium dimer had become a central issue in the controversy over the nature of multiple metal-metal bonding. The ground state of the chromium atom is a half-filled 3d5 4s1 configuration in a high spin 7S state. One might argue that this would lead to chromium dimer having a very tightly bound ground state with a bond order of perhaps as high as six. This would be accomplished through a single 4sσ bond and five d-d bonds involving 3dσ , π and δ orbitals. If this argument were accepted, all the valence electrons would be paired making the ground state electronic symmetry that of 1Σ, and most likely 1Σg+. With six bonds one would expect an exceedingly short bond length, high vibrational frequency and dissociation energy. Up until the time of our experiments, the experimental gas phase data pertaining to chromium dimer consisted of two studies. The first, done by Kant37 used mass spectroscopic measurements of the Cr/Cr2 equilibrium ratio in a Knudsen effusion cell to determine the binding energy (De = 1.56 + .03 eV) of Cr2. The other experiment relevant to the bonding in chromium dimer was reported by Efremov et al.38 in 1974. In a flash photolysis study of chromium hexacarbonyl vapor, a transient absorption was observed in the 4600 A region. Rotational analysis of this spectrum indicated a bond length of r = 1.68 A, but the analysis could not rule out the possibility of this spectrum being due to CrO2 or CrC2.
Since then, the idea of a sextuplet bond has been the subject of extensive theoretical calculations.30-36 Some of these calculations supported the idea; others suggested an effective bond order of considerably less than six. The most recent calculation (at the time of this re-investigation), done by Goodgame and Goddard36 using a self-consistent 6000 configuration wave function corresponding to spin-optimized generalized valence bond plus inter-pair correlations and van der Waals interactions, suggests that chromium dimer is not best described as a sextuplet bond, but rather as an anti-ferromagnetic dimer. This calculation yielded a ground state bond length of re = 3.0 A and a binding energy of De = .03 eV. In this same paper, they also recalculated the binding energy obtained by Kant arguing that the value may have been too high because of the failure to consider substantial population of the low-lying high spin electronic states, and also that effusive flow was not maintained during the experiment.
Because of the lack of firmly established experimental data and unrestrained theoretical calculations, we felt that re-investigation39 of the 4600 A band of Efremov et al.38 was necessary in order to establish whether the carrier was chromium dimer. We also hoped to obtain more extensive data in our re-investigations. This work will be presented in full. Similar investigations of other transition metal diatomics will be discussed briefly and presented in tabular form.
B. Experimental
Chromium clusters were produced within a supersonic molecular beam using the laser vaporization technique previously described. In this experiment, a pulsed solenoid nozzle is used to produce a 200-400 us pulse of helium (FWHM). As the helium flows over a chrome-plated steel target (.06 cm diameter, plating depth of .013 cm),a Q-switched Nd:YAG laser (second harmonic ) focused to a 0.15 cm diameter spot, is fired at a chosen time within the helium pulse. The laser pulse vaporizes the target material and produces a plasma which is entrained in the helium carrier gas. An exit channel (.2 cm diameter, .32 cm length) enables sufficient three-body collisions to produce clusters prior to expansion. The supersonic free jet (He + chromium clusters) then expands proceeding downstream during which time it is skimmed twice at positions 32 and 65 cm away from the nozzle before entering the ionization region of a time of flight mass spectrometer (TOFMS). The resonant two-photon ionization with mass-selective detection is obtained using a Nd:YAG pumped dye laser as the excitation with a KrF excimer laser (4.98 eV) providing the ionization photon. For high resolution scans, the dye laser line width is narrowed to approximately 0. 05 cm-1 by the insertion of an inter-cavity etalon and then pressure-tuned using either SF6 or N2. Both lasers are overlapped temporally and spatially in the ionization region. The ionization laser is chosen such that it is incapable of one-photon ionizing the cluster of interest but can directly ionize an excited state produced by absorption of a photon from the excitation (dye) laser. This provides a resonant enhancement when the dye laser is tuned to an absorbing transition. Mass-selective detection is accomplished using a time of fight mass spectrometer to monitor the resulting photoion signal. The signal is then multiplied and digitized. The intensity of each photoion can then be averaged and recorded by a lab minicomputer ( MIK RT-11/2 ). In this way, the spectrum of chromium dimer can be determined simultaneously (but independently) for the various isotopic species.
C. Selection Rules
In these experiments the information pertaining to the bonding in transition metal (chromium dimer) diatomics was obtained through spectroscopically probing rovibronic transitions. The probability that a transition between two different electronic states will be induced by the oscillating electric field of light is proportional to the square of the transition moment Integral40 ,
M = <Y' l d l Y"> = ∫ Y'*NY"N [ ∫ Y'*el del Y"el dtel ] dtN
where, the single prime indicates the upper of the electronic states. From this integral the overall selection rules for a one-photon electric dipole transition for diatomic molecules (with an emphasis on a 1Σ-1Σ transition) are seen to be :41
1) The spin multiplicity cannot change.
ΔS=0
Singlet<---------->Singlet
Triplet <--------->Triplet
Singlet <---l----->Triplet
2) The electronic orbital angular momentum can change at most by one quantum.
ΔΛ=0,+ 1
3)For homonuclear diatomics, even electronic states combine only with odd electronic states.
gerade<--------->ungerade
gerade<----l---->gerade
ungerade<----l---->ungerade
4) Σ + states cannot combine with Σ - states.
Σ+<--------->Σ+
Σ-<--------->Σ-
Σ+<----l---->Σ-
5) The vibrational quantum number may change by
ΔV = 0, +1, +2, +3...etc.
6) The rotational quantum number may change by at most one.
ΔJ = 0, +1
ΔJ = +1 for transition Σ - Σ
Along with these selection rules, there are factors which modulate the intensity of individual vibrational and rotational lines within an electronic transition. For the vibrational bands, it is seen that the square of the integral,
∫ Y'vibY"vibdtn
is proportional to the intensity of the band. Therefore, the magnitude of the overlap between the two vibrational wave functions is the determining factor of the strength of a particular vibrational band within the electronic transition. The square of this integral is called the Franck-Condon factor. For rotational lines within a particular vibrational band, there is also a modulation in intensity. These intensities can be shown to be proportional to the Honl London factors41 whose formulae are given below for a ΔΛ=O transition:
SRJ = (J'+Λ')(J'-Λ')
J' emission
= (J''+1+Λ'')(J''+1-Λ'')
J''+1 absorption
SQJ = (2J'+1)Λ'2
J' (J'+1) emission
= (2J''+1)Λ''2
J''(J''+1) absorption
SPJ= (J'+1+Λ')(J'+1-Λ)
J'+1 emission
= (J''+Λ'')(J''-Λ'')
J'' absorption
Of the two forms the first is more useful for emission, and the second for absorption since the Boltzman factor contains J' and J'' respectively.
---
Below is information not originally contained in this thesis but has been added for clarification, see page 127 "Spectra of Diatomic Molecules" Herzberg.
The line intensity in most branches of electronic bands varies in essentially the same way as in the branches of the rotation-vibration bands. For a 1Σ - 1Σ transition there is only a P and R branch and the intensity relation is given below. Of course, in the electronic bands one of the bands usually forms a head. However, this does not alter the intensities of the lines. For Absorption:
---
When dealing with homonuclear diatomic molecules with Λ=O, one must also consider the effects of nuclear spin40 because of the fact that either the even or odd rotational levels will have a greater statistical weight. The ratio of the intensity between the even and odd levels can be shown to be ( I+1/ I ). In the case of a homonuclear diatomic with zero nuclear spin, it can be seen that either the odd or even rotational levels will be non-existent.
D. Results
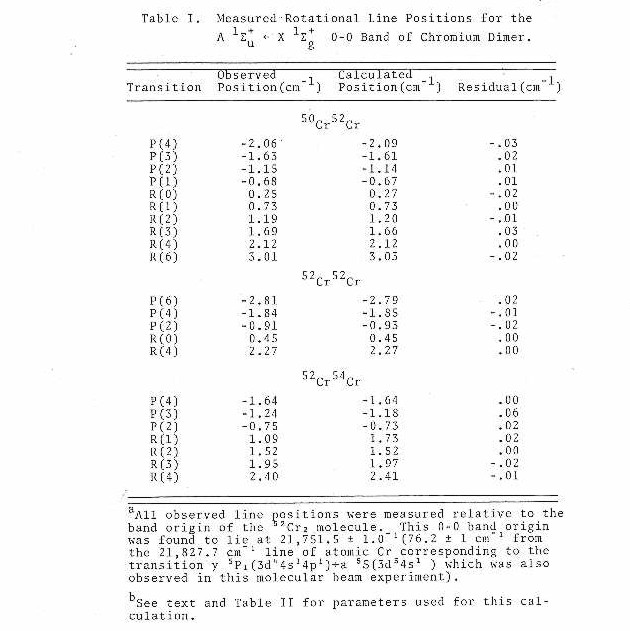
The supersonic beam version of the chromium dimer absorption band in the 4600 A region as reported by Efremov et al.38 is shown under low resolution to consist of a single vibronic transition (see Figure 11). In this figure, the resonant two-photon ionization signal corresponds to a certain isotopic species (52-52) of chromium dimer versus the frequency of the scanning dye laser.
Figure 11
In trying to understand the appearance of the low resolution scan as consisting of a single band assigned as the (0-0) transition due to the small isotope shift, two possible explanations are conceivable. The first is that the transition was between two nearly identical states that is at least near the minimum of their potential energy wells. If this were the case, one might expect to observe a v=0 progression. The second explanation is that the higher vibrational levels of the excited state are predissociative. Measurement of the lifetime shows the v=0 level of the excited state to be 30 ns which is supportive of the predissociative argument. Further evidence however is needed before either or both possibilities may be ruled out.
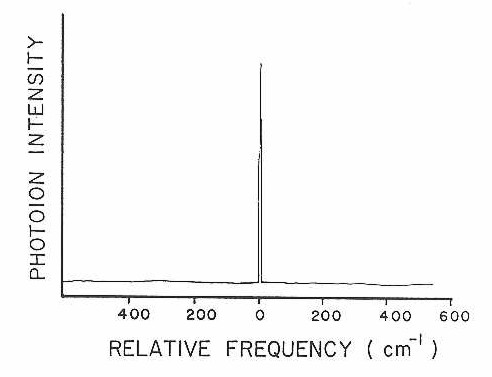
Higher resolution scans of the (0-0) band are shown in Figures 12 and 13 corresponding to the 52-52 and 50-52 isotopic species, respectively. From the well-resolved rotational structure, it is quite apparent that a Q branch is not present. This type of rotational structure, consisting of a P and R branch, is what would be expected for a Σ-Σ rovibronic transition. Since the 52-52 isotopic species has zero nuclear spin, one also expects that alternating rotational lines would be absent in the spectrum, due to nuclear spin statistics. This is easily confirmed by examining the band gap (spacing between the inner most lines of the P and R branches) in Figure 12. When all lines are present the ratio of the spacing of the band gap to any two consecutive lines in either the P or R branch will be two. If only alternating lines are present this ratio will be three-halves. Also since the 50-52 isotopic species has no symmetry restrictions on the existence of its rotational levels, a quick comparison of Figure 13 to Figure 12 would further confirm the absence of alternating lines in the 52-52 rotational structure. High resolution spectra of the 52-54 isotopic species has also been obtained showing a similar rotational structure as that for the 50-52 isotopic species as would be expected.
Using the selection rules for a Σ-Σ transition and the Honl London factors presented in the last section, symmetries of the states involved can be narrowed to either
Σ+u<--Σ+g or Σ-g<--Σ-u
The first of these is more likely and therefore, the transition is assigned as 1Σ+u <-- 1Σ+g.
Figure 12
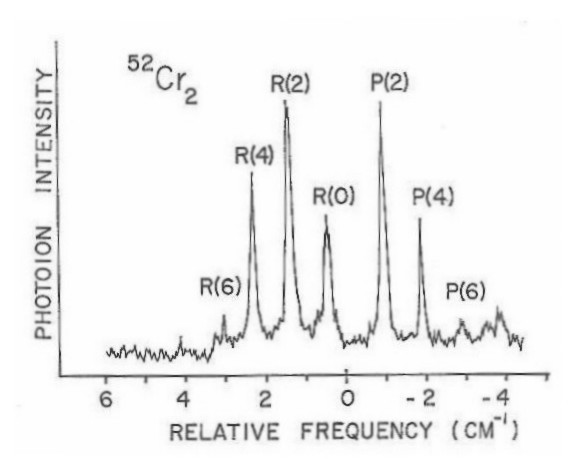
Figure 13
Figure 14

The assignments and measured positions of all observed rotational lines are listed (see Table I) for each of the three isotopic species mentioned above. These line positions were then fit simultaneously to the formula41
ν + = F' (J"+l) -F"(J")
where the + and -refer to the R(ΔJ=+l) and P(ΔJ=-l) branches of the rovibronic transition, respectively, where
F'(J) = ν00 + 1/2ρ ωe' + ρ2β0' J(J+l)
F"(J) = 1/2ρ ωe" + ρ2β0" J(J+l)
and
ρ =( μ/μ i )1/2
μ = the reduced mass of the 52-52 isotopic species
μi= the reduced mass of the other isotopic species of chromium dimer being measured.
Results of the fit are listed in Table I. The parameters are presented in Table II and compared to the results of Efremov et al.38
As shown in Table I, the fit of the measured data to the suggested model is excellent. It should be pointed out that due to the extensive rotational cooling (approximately 4 K which has been determined by computer simulation of a rovibronic band) in the supersonic molecular beam, only the low rotational levels are populated. This leads to a spectrum consisting of a limited number of lines in both the P and R branches. The accuracy of the data is limited but is still sufficient to determine that the ground state and excited state bond lengths for v=0 lie within the range of 1.67 to 1.69 A.

Similar gas phase studies have been published by both Bondybey42 and Riley.43 Bondybey has reported data concerning the ground state vibrational frequency which he estimated to be approximately 460 cm-1. In both of these studies, information regarding the predissociation of the v=0 level is presented and shown to have a strong rotational dependence.
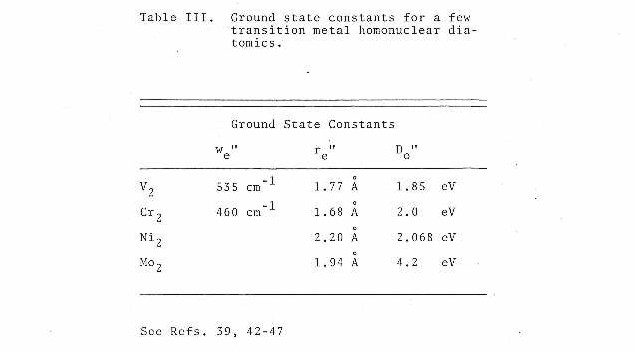
Independent studies on nickel dimer44, vanadium dimer45,and molybdenum dimer46,47 have also been carried out in our laboratory. The measurement of bond lengths and some vibrational frequencies for these species were obtained and are presented in tabular form in Table III. According to this data the contribution of the d-orbital to bonding in transition metal diatomics can be interpreted as a function of position within the periodic table.
E. Conclusion
In examining the data presented one can see that our results are in excellent agreement with those of Efremov et al. This firmly establishes the carrier of the 4600 A vibronic band to be chromium dimer. From the bond length measurement and estimated ground state vibrational frequency, one may reasonably conclude that the d-orbitals play a significant role in the bonding. The extremely short bond length of 1.68 A and high vibrational frequency (~460 cm-1 ) suggest a quintuplet or sextuplet bond.
Recent calculations using a modified generalized valence bond method48 and local spin density formalisms49 have both yielded results predicting a tightly bound dimer with an equilibrium bond length of 1.68 A. This is in agreement with the experimental data, although these calculations lead to differences in the actual shape of the ground state potential energy curve and binding energies. The modified generalized valence bond method predicts a double minimum with the inner minimum at 1.68 A which is due to a quintuplet bond involving only the d-orbitals. The outer minimum at 3.06 is suggested to arise from bonding of 4s-4sσg orbital with the d-shells coupled (antiferromagnetically) into a net singlet state. Using the new values for re, we and an electronic degeneracy of 1, the experimental binding energy has been recalculated to be 2.0 + .03 eV. The calculation using the local spin density formalisms is in agreement with the equilibrium bond length, although this calculation yields no double minimum in the ground state potential energy curve. Also, the binding energy was calculated to be .05 to 1 eV higher than the modified generalized valence bond method.
Our results in conjunction with those of Bondybey42 and Riley43 suggest that the presence of the single vibronic band is due to a combination of both Franck-Condon factors and predissociation. The data shows both the ground and excited states to have nearly identical equilibrium bond lengths, while predissociation was also observed in the v=O and v=l levels of the excited state (see Appendix 1 for further spectroscopic studies of Cr2).
Similar studies concerning a few other transition metal diatomics have also been carried out within this lab. Listed in Table III are the bond lengths and vibrational frequencies obtained. From this data, one can suggest that the bonding in vanadium and molybdenum dimer must involve contributions from the d-orbitals. Theoretical calculations50 concerning Mo2 have predicted the bonding contributions to be identical to Cr2.
In contrast, the results from Ni2 suggest little or no contribution from the d-orbitals. When compared to Cu2 whose bonding is accepted as being due to only the 4s-4sσg orbital, one can conclude from its similar bond length that only a single 4s-4sσg bond is present in Ni2. This agrees with the predictions of theoretical calculations51,52.
Spectroscopic studies of the remaining first row transition metal diatomics and triatomics (with the exception of Cu353) have been carried out in the same spectral region with no observation of any excited state features. A possible explanation for these results is that the open d-shell leads to a large density of states which gives rise to a fast subnanosecond nonradiative decay into states inaccessible to detection by resonant two-photon ionization. A plausible solution to this problem is to probe excited states whose potential energy minimum lies below the dissociation energy of the ground state. Results obtained on Ni2 support the explanation that a high density of states exists leading to a fast nonradiative decay. It also shows that these low-lying states can be probed by way of R2PI.
Figure 14 presents bonding information for the transition metal diatomics. This information shows that in some cases the use of tunable infrared sources will , be needed in order to obtain spectral information necessary to examine these low-lying excited states. Such tunable infrared sources are now available, and so the study of the first row diatomics may continue.
In extending similar studies to the second and third row transition metals the same fast nonradiative decay is likely to exist, however, an additional complication may arise. Due to the heavier masses and an expected increase in bond length, an additional requirement of higher resolution lasers than available in our lab would be needed to resolve the rotational structure of these species. At this time a possible solution which involves the use of a pulse amplified Cw ring laser is being set up and tested in our lab.