
SERIES EDITOR
Sid Gilman, MD, FRCP
William J. Herdman Distinguished University Professor of Neurology University of Michigan
Contemporary Neurology Series
53 | SLEEP MEDICINE | 69 | PALLIATIVE CARE IN NEUROLOGY |
Michael S. Aldrich, MD | Raymond Voltz, MD, | ||
54 | BRAIN TUMORS | James L. Bernat, MD, | |
Harry S. Greenberg, MD, | Gian Domenico Borasio, MD, | ||
William F. Chandler, MD, and | DipPallMed, | ||
Howard M. Sandler, MD | Ian Maddocks, MD, | ||
56 | MYASTHENIA GRAVIS AND | David Oliver, FRCGP, | |
MYASTHENIC DISORDERS | and Russell K. | ||
Andrew G. Engel, MD, Editor | Portenoy, MD | ||
57 | NEUROGENETICS | 70 | THE NEUROLOGY OF EYE |
Stefan-M. Pulst, MD, Dr. Med., Editor | MOVEMENTS, | ||
58 | DISEASES OF THE SPINE AND | Fourth Edition | |
SPINAL CORD | R. John Leigh, MD, FRCP and | ||
Thomas N. Byrne, MD, | David S. Zee, MD | ||
Edward C. Benzel, MD, and | 71 | PLUM AND POSNER’S DIAGNOSIS OF | |
Stephen G. Waxman, MD, PhD | STUPOR AND COMA, | ||
59 | DIAGNOSIS AND MANAGEMENT | Fourth Edition | |
OF PERIPHERAL NERVE | Jerome B. Posner, MD, | ||
DISORDERS | Clifford B. Saper, MD, PhD, | ||
Jerry R. Mendell, MD, John T. Kissel, MD, | Nicholas D. Schiff, MD, and | ||
and David R. Cornblath, MD | Fred Plum, MD | ||
60 | THE NEUROLOGY OF VISION | 72 | PRINCIPLES OF DRUG THERAPY IN |
Jonathan D. Trobe, MD | NEUROLOGY, | ||
61 | HIV NEUROLOGY | Second Edition | |
Bruce James Brew, MBBS, MD, FRACP | Michael V. Johnston, MD and | ||
62 | ISCHEMIC CEREBROVASCULAR | Robert A. Gross, MD, PhD, Editors | |
DISEASE | 73 | NEUROLOGIC COMPLICATIONS | |
Harold P. Adams, Jr., MD, | OF CANCER, | ||
Vladimir Hachinski, MD, and | Second Edition | ||
John W. Norris, MD | Lisa M. DeAngelis, MD and | ||
65 | MIGRAINE: MANIFESTATIONS, | Jerome B. Posner, MD | |
PATHOGENESIS, AND | 74 | NEUROLOGIC COMPLICATIONS | |
MANAGEMENT, | OF CRITICAL ILLNESS, | ||
Second Edition | Third Edition | ||
Robert A. Davidoff, MD | Eelco F.M. Wijdicks, MD, PhD, FACP | ||
67 | THE CLINICAL SCIENCE | 75 | CLINICAL NEUROPHYSIOLOGY, |
OF NEUROLOGIC | THIRD EDITION | ||
REHABILITATION, | Jasper R. Daube, MD and | ||
Second Edition | Devon I Rubin, MD, Editors | ||
Bruce H. Dobkin, MD | 76 | PERIPHERAL NEUROPATHIES IN | |
68 | NEUROLOGY OF COGNITIVE AND | CLINICAL PRACTICE | |
BEHAVIORAL DISORDERS | Steven Herskovitz, MD, | ||
Orrin Devinsky, MD and | Stephen N. Scelsa, MD, and | ||
Mark D’Esposito, MD | Herbert H. Schaumburg, MD |
CLINICAL NEUROPHYSIOLOGY OF THE VESTIBULAR SYSTEM
Fourth Edition
Robert W. Baloh, MD, FAAN
Department of Neurology and Surgery (Head and Neck)
Reed Neurological Research Center UCLA School of Medicine
Los Angeles, CA
Kevin A. Kerber, MD
Department of Neurology
University of Michigan Health Center Ann Arbor, MI
1
2011
1
Oxford University Press, Inc., publishes works that further Oxford University’s objective of excellence
in research, scholarship, and education.
Oxford New York
Auckland Cape Town Dar es Salaam Hong Kong Karachi Kuala Lumpur Madrid Melbourne Mexico City Nairobi New Delhi Shanghai Taipei Toronto
With offices in
Argentina Austria Brazil Chile Czech Republic France Greece Guatemala Hungary Italy Japan Poland Portugal Singapore South Korea Switzerland Thailand Turkey Ukraine Vietnam
Copyright © 2011 by Oxford University Press, Inc.
Published by Oxford University Press, Inc.
198 Madison Avenue, New York, New York 10016 www.oup.com
Oxford is a registered trademark of Oxford University Press
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, or otherwise,
without the prior permission of Oxford University Press. Library of Congress Cataloging-in-Publication Data
Baloh, Robert W. (Robert William), 1942-
Clinical neurophysiology of the vestibular system / Robert W. Baloh, Kevin A. Kerber. — 4th ed.
; cm. — (Contemporary neurology series ; 77) Includes bibliographical references and index. ISBN 978-0-19-538783-4
Vestibular apparatus. 2. Vestibular function tests. 3. Neurophysiology. I. Kerber, Kevin A. II. Title. III. Series: Contemporary neurology series, 77. 0069-9446 ;
[DNLM: 1. Vestibular Diseases—physiopathology. 2. Vestibular Function Tests. 3. Vestibule, Labyrinth—physiology. W1 CO769N v.77 2011 / WV 255 B195c 2011]
QP471.B34 2011 612.8’58—dc22
2010002663
The science of medicine is a rapidly changing field. As new research and clinical experience broaden our knowledge, changes in treatment and drug therapy occur. The author and publisher of this work have checked with sources believed to be reliable in their efforts to provide information that is accurate and complete, and in accordance with the standards accepted at the time of publication. However, in light of the possibility of human error or changes in the practice of medicine, neither the author, nor the publisher, nor any other party who has been involved in the preparation or publication of this work warrants that the information contained herein is in every respect accurate or complete. Readers are encouraged to confirm the information contained herein with other reliable sources, and are strongly advised to check the product information sheet provided by the pharmaceutical company for each drug they plan to administer.
9 8 7 6 5 4 3 2 1
Printed in the United States of America on acid-free paper
This book is dedicated to our families.
This page intentionally left blank
Foreword
Even the most experienced clinical neurologist may need to take a deep breath before attempting to obtain a clear, crisp history from a patient whose chief complaint is “dizziness”. It is no secret that people with neurological symptoms have widely varying conceptions of the meaning of this word. In some patients, even providing hints or clues cannot induce the patient to express precisely the feeling experienced. It may help to suggest key words such as “off-balance”, “spinning sensa- tion”, “light-headedness”, and “faintness”. I have actually had the experience of running through a long series of words to help guide the patient to express his symptoms precisely when, in response to a hint from me, the patient informed me that he meant that he lost his vision briefly! Taking a clear, precise history in a patient such as this is absolutely essential in order to determine whether the problem might be peripheral or central. This will help greatly in focusing the neurological examination and determining which diagnostic studies to request and how to manage the problem. Even when the clinician determines that the problem is either peripheral or central, the list of neurological disorders that might be responsible can be daunting. With all this in mind, it is a plea- sure to welcome a new contribution to this interesting and challenging field in the fourth edition of the classic book, Clinical Neurophysiology of the Vestibular System. Dr. Robert Baloh, a senior clinician and renowned investigator famous for his seminal work on the interface between clinical neurology and vestibular physiology, has been an author of all of the previous volumes. He is joined in this new version of the book by Dr. Kevin Kerber, a brilliant young clinical neurologist trained in both neurology and in clinical vestibular neurophysiology.
This edition of the book is divided into four parts: 1. Anatomy and Physiology of the Nervous System, 2. Evaluation of the Dizzy Patient, 3. Diagnosis and Management of Common Neurotologic Disorders, and 4. Symptomatic Treatment of Vertigo. The current volume has been completely reorganized and expanded to cover advances over the past decade. This book includes newly described molecular mechanisms of peripheral and central processing within the vestibular system. There is a lucid, clinically practical review of the key features to assess in the clinical evaluation of the patient to determine the site of the lesion. The discussion of the differential diagnosis of dizzi- ness is clear and complete, and I found the description of bedside tests of vestibular function to be practical and helpful. The clinical sections have been completely updated and expanded with an emphasis on evidence-based medicine, but the book is informative even for the clinical scenarios that are lacking in high-level evidence. The chapter on benign paroxysmal positional vertigo con- tains guides to the latest treatment maneuvers. This book also contains a strategy for deciding on which drugs to use for symptomatic control of vertigo and for designing a vestibular exercise pro- gram. This extremely valuable contribution will be useful to clinical neurologists, otolaryngologists, physiatrists, and general and emergency medicine physicians in practice as well as residents and fellows in these specialties. This book is also a comprehensive basic science source for professionals and trainees in vestibular neuroscience.
Sid Gilman, MD, FRCP William J. Herdman Distinguished University Professor of Neurology
Director, Michigan Alzheimer’s Disease Research Center
Department of Neurology University of Michigan
Ann Arbor, MI
vii
This page intentionally left blank
Preface
The purpose of this book is to provide a framework for understanding the pathophysiology of dis- eases involving the vestibular system. The book is divided into four parts: 1. Anatomy and physiol- ogy of the nervous system, 2. Evaluation of the dizzy patient, 3. Diagnosis and management of common neurotologic disorders, and 4. Symptomatic treatment of vertigo. Part 1 reviews the anatomy and physiology of the vestibular system with emphasis on clinically relevant material. Part 2 outlines the important features in the patient’s history, examination, and laboratory evalua- tion that determine the probable site of lesion. Part 3 covers the differential diagnostic points that help the clinician decide on the cause and treatment of the patient’s problem. Part 4 describes the commonly used antivertiginous and antiemetic drugs and the rationale for vestibular exercises.
This completely reorganized and expanded fourth edition covers the rapid advances that have occurred in the basic and clinical vestibular sciences in the past 10 years. Recent breakthroughs in our understanding of the molecular mechanisms of peripheral transduction and central processing within the vestibular system are reviewed. We discuss the differential diagnosis of dizziness of both vestibular and nonvestibular etiology and demonstrate bedside tests of vestibular function. Videos showing tests and important clinical findings are available online. The chapter on the laboratory diagnosis of vestibular dysfunction has been expanded to include videonystagmography (VNG) and vestibular evoked myogenic potentials (VEMPs). In Part 3, the chapter on benign paroxysmal posi- tional vertigo includes all the latest treatment maneuvers. We emphasize controlled treatment trials whenever available. In Part 4 we provide a strategy for deciding on which drugs to use for symptomatic control of vertigo and for designing a vestibular exercise program for patients with different types of vestibular lesions.
We believe that this book will be useful to all physicians who treat patients complaining of dizzi- ness. It should be particularly helpful for those in the field of family practice, internal medicine, neurology, head and neck surgery, and neurosurgery. We hope that it will encourage students (in both the clinical and basic sciences) to choose neurotology as their field of study, or at least help clinicians to enjoy the evaluation and management of patients with dizziness. Finally, we hope that the information in this book can contribute to efforts to optimize the care of patients.
K. A. K.
R. W. B.
ix
This page intentionally left blank
Acknowledgments
Our students and colleagues in Neurology and Head and Neck Surgery provided inspiration. We are grateful to the chairmen of our departments, John C. Mazziotta and David J. Fink, and the major sponsors of our research (National Institutes of Health and Agency for Healthcare Research and Quality) for their continued support. We would also like to thank Krister Brantberg, who pro- vided helpful suggestions for the chapter on the clinical evaluation of hearing.
xi
This page intentionally left blank
PART 1 ANATOMY AND PHYSIOLOGY OF THE
OVERVIEW OF VESTIBULAR ANATOMY AND PHYSIOLOGY 3 PERIPHERAL VESTIBULAR RECEPTORS 4
Hair Cells The Macules The Cristae Basis of Stimulus Specificity of the Inner Ear Receptor Organs
CENTRAL VESTIBULAR PATHWAYS 11
Vestibular Nuclei
Horizontal Canal-Ocular Reflex Nystagmus Translational Vestibulo-Ocular Reflexes
The Ocular Tilt Reflex Vestibulospinal Reflexes Vestibulo-Autonomic Reflexes
MOTION PERCEPTION AND ORIENTATION 19 PATHOPHYSIOLOGY OF VESTIBULAR SYMPTOMS 20 CENTRAL COMPENSATION FOR VESTIBULAR LESIONS 21 SUMMARY 22
THE PERIPHERAL VESTIBULAR SYSTEM 25 TEMPORAL BONE 25
Tympanic Membrane Middle Ear Facial Nerve
Phylogeny Structure Fluid Dynamics Fluid Chemistry Blood Supply Innervation
Embryonic Development
Morphologic Characteristics Sequence of Hair Cell Activation Relationship between the Direction of Force and Hair Cell Activation Mechanism of Hair Cell Activation Hair Cell Influence on Afferent Nerve Activity Signal Processing at the Hair Cell/Afferent Nerve Junction
THE INNER EAR VESTIBULAR RECEPTORS 43
Anatomy of the Semicircular Canals Physiology of the Semicircular Canals Anatomy of Otolith Organs Physiology of the Otolith Organs
xiii
Anatomy of Primary Neurons Physiology of Primary Neurons
THE CENTRAL VESTIBULAR SYSTEM 63 VESTIBULAR NUCLEI 63
Phylogeny Anatomy Neurotransmitters Physiology
Overview Rotational Vestibulo-Ocular Reflexes Translational Vestibulo-Ocular Reflexes
Ocular Counterrolling Semicircular Canal-Otolith Interaction
Anatomic and Physiologic Basis Characteristics of Neck-Induced Eye Movements
VISUAL–VESTIBULAR INTERACTION 89
Visual Tracking Eye Movements Organization of Visually Guided Tracking Eye
Movements Comparison of Vestibular- and Visual-Induced Eye Movements Visuo-Vestibulo- Ocular Connections Model of Visual-Vestibular Interaction Adaptive Modification of the Vestibulo-Ocular Reflex with Vision Cellular Basis for Visual Vestibular Interaction
Comparison of Ocular and Spinal Vestibular Reflexes Vestibulospinal Connections
Cerebellar–Vestibular Interaction Vestibulo-Collic Reflexes Cellular Mechanisms
SUBJECTIVE VESTIBULAR SENSATION 102
Vestibulothalamocortical Connections Response Properties of Thalamic Relay Neurons Response Properties of Vestibular Cortex Neurons Functional Brain Imaging in Normal Human Subjects Lesions of the Vestibulocortical Pathways in Patients Psychophysical Studies
EPIDEMIOLOGY OF DIZZINESS 121 SPECIFIC DISORDERS 123
BURDEN ON PATIENTS 124 HEALTH CARE UTILIZATION 125 SUMMARY 125
THE HISTORY OF THE DIZZY PATIENT 127
Central versus Peripheral Causes Time Course Precipitating Factors Associated
Symptoms Compensation Predisposing Factors Family History Diagnosis and Management
Orthostatic Hypotension Postural Tachycardia Syndrome (POTS) Vasovagal Attacks Hyperventilation
PSYCHOPHYSIOLOGIC DIZZINESS (CHRONIC SUBJECTIVE DIZZINESS) 134
Panic Disorder Phobic Dizziness Chronic Anxiety Pathophysiology Diagnosis and Management
Common Causes Gait Disorders in Older People Falls in Older People
Diagnosis and Management
Common Causes Oscillopsia Management
Management
Motion Sickness Space Sickness Height Vertigo Mal de Debarquement Syndrome
SUMMARY: DISTINGUISHING BETWEEN VESTIBULAR AND NONVESTIBULAR TYPES OF DIZZINESS 144
BEDSIDE EXAMINATION OF THE VESTIBULAR SYSTEM 149 EXAMINATION OF THE EAR 149
Fistula Test
TESTS OF VESTIBULOSPINAL REFLEXES 151
Pastpointing Static Posture Walking Tests
TESTS OF VESTIBULO-OCULAR REFLEXES 153
Doll’s Eye Test (Oculocephalic Response) Head-Thrust Test Dynamic Visual Acuity
Cold Caloric Test Rotational Testing
TESTS FOR PATHOLOGIC NYSTAGMUS 156
Methods of Examination
TYPES OF PATHOLOGIC NYSTAGMUS 158
Spontaneous Nystagmus Gaze-Evoked Nystagmus Positional Nystagmus
Vibration-Induced Nystagmus Head-Shaking Nystagmus Hyperventilation-Induced Nystagmus
Dissociated Spontaneous Nystagmus Voluntary Ocular Oscillations (Voluntary Nystagmus) Convergence Retraction Nystagmus Saccadic Intrusions Ocular Bobbing Palato-Ocular Myoclonus
LABORATORY EXAMINATION OF THE VESTIBULAR SYSTEM 171 NYSTAGMOGRAPHY 171
Methods of Recording Eye Movements Interpreting the Recording Recording Pathologic Nystagmus Bithermal Caloric Test Tests of Visual-Ocular Control
ROTATIONAL TESTING OF VESTIBULO-OCULAR REFLEXES 189
Relationship between Stimulus and Response Results in Normal Subjects Results in Patients
VISUAL-VESTIBULAR INTERACTION 204
Methodology Results in Normal Subjects Results in Patients
TESTS OF OTOLITH-OCULAR REFLEXES 207
Ocular Counterrolling Eccentric Rotation Off-Vertical Rotation Linear Acceleration
Static-Force Platforms Moving-Platform Posturography
VESTIBULAR-EVOKED POTENTIALS 209
Brain Stem and Cortical Vestibular Evoked Myogenic Potentials (VEMPs)
CLINICAL EVALUATION OF HEARING 219 TYPES OF HEARING DISORDERS 219
Conductive Sensorineural Central Hearing Disorders
BEDSIDE TESTS OF HEARING 220 BEHAVIORAL AUDIOMETRY 221
The Audiogram Speech Recognition Tests Stenger Test
The Acoustic Reflex
Electrocochleography Brainstem Auditory-Evoked Response
CENTRAL AUDITORY SPEECH TESTS 228 SUMMARY OF AUDITORY TEST RESULTS 229
PART 3 DIAGNOSIS AND MANAGEMENT OF COMMON NEUROTOLOGIC DISORDERS
ACUTE OTITIS MEDIA AND OTOMASTOIDITIS 233
Diagnosis and Management
CHRONIC MASTOIDITIS AND CHOLESTEATOMA 235
Diagnosis Management
Diagnosis Management
Diagnosis Management
INTRACRANIAL EXTENSION OF EAR INFECTIONS 238
Routes of Spread Meningitis Epidural Abscess Lateral Sinus Thrombophlebitis
Brain Abscess Otitic Hydrocephalus Diagnosis Management
Diagnosis Management
VIRAL INFECTIONS OF THE INNER EAR 242
Clinical Syndromes Diagnosis Viral versus Other Causes of Peripheral Cochleovestibular Loss Management
SYPHILITIC INFECTIONS OF THE EAR 248
Diagnosis Management
TUBERCULOSIS AND MYCOTIC INFECTIONS OF THE INNER EAR 249
Tuberculous Mastoiditis Mycotic Mastoiditis Basilar Meningitis
BENIGN POSITIONAL VERTIGO 255 HISTORICAL BACKGROUND 255
CAUSES OF BENIGN POSITIONAL VERTIGO 257
POSTERIOR CANAL VARIANT OF BENIGN POSITIONAL VERTIGO 258
Clinical Features Pathophysiology Diagnosis Management
OTHER VARIANTS OF BENIGN POSITIONAL VERTIGO 265
Horizontal Canal Benign Positional Vertigo Anterior Canal Benign Positional Vertigo
Mimics of Benign Positional Vertigo
ENDOLYMPHATIC HYDROPS (MENIERE’S SYNDROME) 273 BACKGROUND 273
Genetics Migraine and Meniere’s Syndrome Infection/Autoimmune
Audiometric Testing Vestibular Testing Imaging
Medical Managment Surgical Managment
Migraine without Aura Migraine with Aura Migrainous Vertigo Basilar Migraine
Migraine and Meniere’s Syndrome Migraine Equivalents
Genetics Spreading Wave of Depression Vasomotor Abnormalities
Migraine without Aura Migraine with Aura Migraine Aura without Headache Basilar Migraine Migrainous Vertigo
Symptomatic and Abortive Treatment Prophylactic Treatment
IMMUNE-MEDIATED DISEASES 303 AUTOIMMUNE INNER EAR DISEASE 303
Background Pathophysiology Clinical Features Diagnosis Management
PARANEOPLASTIC IMMUNE DISORDERS 309
Background Pathophysiology Clinical Features Diagnosis Management
Background Pathophysiology Clinical Features Diagnosis Management
VASCULAR DISORDERS 319 VERTEBROBASILAR ISCHEMIA 319
Pathophysiology Transient Ischemic Attacks (TIAs) Stroke Syndromes Diagnosis Treatment
INTRALABYRINTHINE HEMORRHAGE 332
Diagnosis and Management
HEMORRHAGE INTO THE BRAIN STEM AND CEREBELLUM 332
Diagnosis and Management
VASCULAR COMPRESSION SYNDROMES 334
Vertebrobasilar Doliochoectasia Vascular Compression by Normal Vessels (Vestibular Paroxysmia) Rotational Vertebral Artery Syndrome
TUMORS OF THE MIDDLE EAR AND TEMPORAL BONE 339
Malignant Tumors Glomus Body Tumors (Paragangliomas) Diagnosis Management
TUMORS OF THE INTERNAL AUDITORY CANAL AND CEREBELLOPONTINE ANGLE 341
Schwannomas Meningiomas Epidermoid Cysts (Primary Cholesteatomas) Cholesterol Granulomas Metastatic Tumors Diagnosis Management
Brain Stem Fourth Ventricle Cerebellum Diagnosis and Management
TRAUMA TO THE TEMPORAL BONE 353
Fracture Labyrinthine Concussion Posttraumatic Positional Vertigo Diagnosis Management
Pathophysiology Diagnosis Management
SEMICIRCULAR CANAL DEHISCENCE SYNDROME 358
Pathophysiology Diagnosis Management
Intracranial Complications Associated with Temporal Bone Fractures Dizziness Due to Brainstem Trauma Postconcussion Syndrome Whiplash Injuries Diagnosis Management
DIZZINESS AND SYSTEMIC METABOLIC DISORDERS 367
Diabetes Mellitus Uremia Hypothyroidism Alcohol and Thiamine Deficiency Management
METABOLIC DISORDERS OF THE TEMPORAL BONE 371
Otosclerosis Paget’s Disease Other Disorders Diagnosis Management
Aminoglycosides “Loop” Diuretics Anti-inflammatory Drugs Platinum Compounds Diagnosis Management
Heavy Metals Organic Solvents Diagnosis Management
DEVELOPMENTAL AND GENETIC DISORDERS 383 THE INNER EAR 383
Acquired Disorders Hereditary Disorders Pathology Diagnosis Management
DISORDERS OF THE CRANIAL VERTEBRAL JUNCTION 390
Basilar Impression Bony Fusions Atlantoaxial Dislocation Chiari Malformation Syringobulbia Diagnosis Management
INHERITED SPINOCEREBELLAR ATAXIA SYNDROMES 393
Autosomal Dominant Spinocerebellar Ataxia Syndromes Autosomal Recessive Spinocerebellar Ataxia Syndromes Episodic Ataxia and Vertigo Syndromes Diagnosis Management
ANTIEMETIC AND ANTIVERTIGO DRUGS 405 VESTIBULAR SUPPRESSANTS 407
How Do They Work? How to Use Them Indications Precautions What to Tell the Patient
How Do They Work? How to Use Them Precautions What to Tell the Patient
Scopolamine (Transderm Sc−o p) Buclizine Hydrochloride (Bucladin-S) Diphenhydramine Hydrochloride (Benadryl) Meclizine (Antivert, Bonine) Dimenhydrinate
(Dramamine) Promethazine Hydrochloride (Phenergan) Betahistine (Serc) Metaclopramide (Reglan) Benzquinamide Hydrochloride (Emete-con) Trimethobenzamide Hydrochloride (Tigan) Diazepam (Valium) Droperidol (Inapsine) Diphenidol (Vontrol) Prochlorperazine (Compazine) Dronabinol (Marinol)
ADAPTIVE CONTROL OF NORMAL VESTIBULAR REFLEXES 420 MECHANISMS FOR COMPENSATION AFTER VESTIBULAR LOSS 420 SPECIAL CIRCUMSTANCES 421
Vestibular Loss in Children Vestibular Loss in the Elderly Failure of Compensation
CONTROLLED TRIALS OF VESTIBULAR EXERCISES 422 STRATEGY FOR DESIGNING VESTIBULAR EXERCISES 423
Unilateral Vestibular Lesions Bilateral Vestibular Lesions Central Vestibular Lesions
APPENDIX 20-1. SAMPLE HOME EXERCISE PROGRAM 428
Head-Turning Practice Walking Practice Other Exercises Dizziness Exercises
This page intentionally left blank
![]()
Anatomy and Physiology of the Nervous
![]()
This page intentionally left blank
![]()
Overview of Vestibular Anatomy and
PERIPHERAL VESTIBULAR RECEPTORS
Hair Cells The Macules The Cristae
Basis of Stimulus Specificity of the Inner Ear Receptor Organs
CENTRAL VESTIBULAR PATHWAYS
Vestibular Nuclei
VESTIBULAR REFLEXES
Horizontal Canal-Ocular Reflex Nystagmus
Translational Vestibulo-Ocular Reflexes The Ocular Tilt Reflex
Vestibulospinal Reflexes Vestibulo-Autonomic Reflexes MOTION PERCEPTION AND
ORIENTATION PATHOPHYSIOLOGY OF VESTIBULAR
SYMPTOMS
CENTRAL COMPENSATION FOR VESTIBULAR LESIONS
SUMMARY
The vestibular system like other sensory systems (i.e, auditory, visual, olfactory, gusta- tory, and somatosensory) serves the basic func- tion of translating environmental information into biological signals. However, unlike other sensory systems there is usually no conscious awareness of it during routine activities when the system is functioning normally. In fact, the inner ear vestibular receptors were not even recognized until the seminal work of Prosper Meniere in the mid 1800s.1 Meniere worked in a deaf-mute institute and noticed that many of his patients with hearing loss also had vertigo. Prior to Meniere, vertigo—the most common symptom of vestibular dysfunction—was con- sidered a cerebral symptom, similar to epilep- tic seizures. The semicircular canals had been identified but were considered to be part of the hearing apparatus. Meniere’s notion that ver- tigo could result from damage to the inner ear was met with great scepticism. The vestibular system continues to be underappreciated in
most comprehensive clinical and basic science medical textbooks.
The vestibular system has a “behind the scenes” role of maintaining spatial orientation and driving reflexes that stabilize vision and balance. To do this, it transforms forces associ- ated with head acceleration and gravity into biological signals that travel directly to motor centers for postural and ocular stability and to the cortex to aid in orientation. When the sys- tem functions normally, you have no awareness of these ongoing activities. Unlike the ability to appreciate visual, olfactory, or auditory stimuli, you do not appreciate the function of the ves- tibular system until something goes awry.
This is not to say that you cannot perceive motion. The vestibular system projects to many areas of the cerebral cortex but unlike other sensory systems there is no primary vestibular cortex that only receives vestibular signals. All cortical neurons that receive vestibular signals also receive other sensory signals, particularly
3
visual and somatosensory. It is not possible to determine which signal is responsible for the perceived motion.
An acute malfunction of the vestibular system causes a profound inability to function, leaving one completely disabled because of severe spatial disorientation, imbalance, nau- sea, and vomiting during the most intense peri- ods. These are some of the most bizarre and incapacitating symptoms in all of medicine. The patient simply cannot navigate the envi- ronment because, to the patient, the world is moving as though he is on an unremitting car- nival ride. Interestingly, however, a chronic lesion—even a bilateral loss of function—leads to relatively little disability in most patients affected by it. In fact, many patients with a bilateral vestibular loss probably go undiag- nosed because of few or mild symptoms that either do not lead to a medical evaluation or are not recognized by physicians. As opposed to gradual hearing loss or visual loss, a gradual vestibular loss can go virtually unnoticed.
Vestibular symptoms pose a great deal of difficulty in clinical medicine. First, patients suffering vestibular symptoms often have diffi- culty describing the symptoms. Many patients with a vestibular disturbance will simply report “dizziness”—a nonspecific term that can refer to symptoms stemming from cardiac distur- bances, a psychological disorder, medication side effects, or many other disturbances. Second, there is much overlap among the symptoms and signs of vestibular disorders, and discriminating among vestibular lesions depends on appreciat- ing rather subtle differences in how the eyes are moving. In fact, most of the examination of the vestibular system involves observing eye move- ments since the vestibular structures cannot be visualized at the bedside and the most recogniz- able functions of the vestibular system are manifest by the vestibulo-ocular reflex. Most physicians can recognize nystagmus—a hall- mark movement of the eyes reflecting vestibu- lar function – but most physicians do not appre- ciate that characterizing the pattern of nystagmus can discriminate a benign disorder from a life-threatening disorder. Physicians typ- ically have little training in the basic science and clinical evaluation of the vestibular system, and this in turn results in overuse of tests, misdiag- nosis, and underuse of effective treatments.
The purpose of this book, then, is to provide the basic science and clinical training needed
to diagnose and treat vestibular system disorders. In this overview chapter, we provide the most salient information regarding the vestibular system. An overview chapter is important because it rapidly and succinctly presents the essential elements that can be periodically revisited. Whenever appropriate the reader is referred to later sections where the material is described in more detail.
PERIPHERAL VESTIBULAR RECEPTORS
The role of the inner ear vestibular receptors in maintaining orientation has remained the same from the earliest organisms in the animal king- dom.2 A primitive gravity-detection organ, the statocyst, appeared more than 600 million years ago in some bygastrulated animals such as jel- lyfish, allowing the animal to regulate its static position in space (see Fig. 2–5 in Chapter 2). With the advent of modern fish (about 100 mil- lion years ago), the vestibular labyrinth reached its peak of development, and relatively little change has taken place since that time. The basic structure of the three semicircular canals, the utricle, and the saccule is similar in all higher vertebrates. The membranous laby- rinths of modern fish lie in the bony chamber of the skull directly behind the orbits. In its subsequent evolution in amphibians, birds, and mammals, the membranous labyrinth is com- pletely surrounded by a bony labyrinth enclos- ing the periotic space. This space is filled with perilymphatic fluid and suspensory connective tissue acting as a shock absorber. The relative positions of the planes of the three semicircu- lar canals vary from species to species, although in primates they are approximately orthogonal to each other. The shape of each semicircular canal also varies considerably from that of a tri- angle in reptiles to an ellipse in birds to an almost true circle in mammals.2
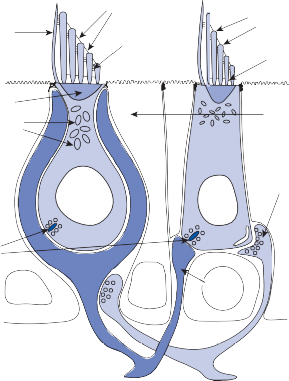
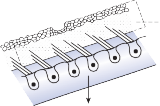
Hair Cells
The basic element of the labyrinthine receptor organs that transduces mechanical force to nerve action potentials is the hair cell. Already developed in the statocysts of invertebrates, this specialized sensory cell becomes more
sophisticated in mammals.2 Transducer cells are surrounded by supporting cells in special- ized areas in the walls of the sensory epithe- lium. Two types of hair cells occur in birds and mammals (Fig. 1–1). Type II cells are cylindri- cal, with multiple nerve terminals at their base, whereas type I are globular or flask shaped, with a single large, chalice-like nerve terminal surrounding the base. A bundle of nonmobile stereocilia protrudes from the cuticular plate on the apical end of each hair cell. The height
of the stereocilia increases stepwise from one side to the other, and next to the tallest stereo- cilia a thicker, longer cilia, the kinocilia, protrudes from the cell’s cytoplasm through a segment of the cell membrane lacking a cuticu- lar plate. The tips of the cilia are connected by tip-links that open and close mechanosensory channels (Fig. 1–1; also see Fig. 2–12 in Chapter 2).3
IINNHIBITION
IINNHIBITION
The adequate stimulus for hair cell activation is a force acting parallel to the top of the cell,
Kinocilium
Cuticular plate Mitochondria
EXCIITTAATTIIOON
EXCIITTAATTIIOON
Tip links
Shaft links
Cilia
Ankle Links
Supporting cell
I II
Efferent bouton
Ribbon Synapses
Calyx
Afferent bouton
Figure 1–1. Schematic drawing of the two types of hair cells. Inset illustrates relationship between the direction of force and maximum hair cell activation.
resulting in bending of the cilia (a shearing force).4 Force applied perpendicular to the cell surface (a compressional force) is ineffective in stimulating the hair cell. The stimulus is maxi- mum when the force is directed along an axis that bisects the bundle of stereocilia and goes through the kinocilium (Fig. 1–1, insert). Deflection of the cilia toward the kinocilium opens the mechanosensory channels at the tips causing an influx of potassium and depolariza- tion of the resting membrane potential.5 This opens voltage-gated calcium channels at the base and releases neurotransmitter (mostly glu- tamate) activating the afferent nerve terminals. Bending of the cilia in the opposite direction produces the reverse effect (closing of the channels and hyperpolarization of the hair cells).
Much of the basic information regarding the physiological properties of hair cells and their afferent nerves has been obtained through the study of hair cell systems in nonmammalian spe- cies. Analysis of the lateral line organs of amphib- ians and fish has been particularly informative.6 The organs consist of groups of hair cells, the neuromasts, aligned in longitudinal rows on the side of the animal’s body and head. A free- standing gelatinous cupula covering the cilia transmits the force associated with water dis- placement into hair cell deflection, which in turn results in change in firing rate of the affer- ent nerve. A key observation that has been confirmed in all other hair cell systems is a con- tinuous spontaneous activity of the afferent nerves.7 A small percentage of the mechano- sensory channels remains open at rest, leading to the spontaneous afferent nerve discharge. Depolarization and hyperpolarization of the hair cells’ membrane potential result in modulation of this spontaneous activity (Fig. 1–2). Bending of the cilia toward the kinocilium increases the spontaneous activity, and bending of the cilia away from the kinocilium results in a decrease. The spontaneous firing rate varies among differ- ent animal species and among different sensory receptors. It is thought to be highest in the afferent neurons of the semicircular canals of mammals (up to 90 spikes per second).8
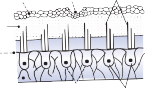
The Macules
The membranous labyrinth forms two globular cavities within the vestibule: the utricle and
the saccule. Each cavity contains a separate macule.9 In the saccule, the macule is located on the medial wall in the sagittal plane; in the utricle, the macule is mostly in the horizontal plane next to the opening of the horizontal semicircular canal (Fig. 1–3C). The surfaces of the utricular and saccular macules are covered by the otolithic membrane, a structure consist- ing of a mesh of fibers embedded in a gel with a superficial layer of calcium carbonate crys- tals, the otoconia (Fig. 1–3A).10 The stereocilia of the macular hair cells protrude into the oto- lithic membrane. The striola, a distinctive curved zone running through the center, divides each macule into two parts. The hair cells on each side of the striola are oriented so that the kinocilia are in opposite directions (as indicated by the arrows in Fig. 1–3C). In the utricle, the kinocilia face the striola, and in the saccule, they face away from it. Because of the different orientation, displacement of the otolithic membrane has an opposite effect on the set of hair cells on each side of the striola.
The density of the otolithic membrane over- lying the hair cells of the macules is much greater than that of the surrounding endo- lymph, owing to the presence of the calcium carbonate crystals. The weight of this mem- brane produces a shearing force on the under- lying hair cells that is proportional to the sine of the angle between the line of gravitational force and a line perpendicular to the plane of the macule (Fig. 1–3B). During linear head accel- eration tangential to the surface of the recep- tor, the force acting on the hair cells is the result of the two forces: one in the opposite direction of the head displacement and the other in the direction of gravitational pull. Recordings of afferent neuronal activity from the macules of primates confirm that the utric- ular and saccular macules are responsive to static tilt and dynamic linear acceleration forces (see Fig. 3–6 in Chapter 3).11
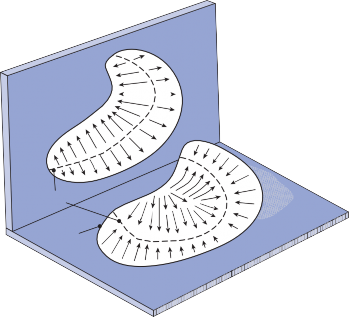
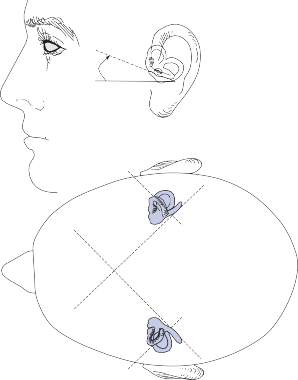
The Cristae
The cristae are the receptor organs of the semi- circular canals. The semicircular canals are aligned to form a coordinate system.12 The hor- izontal canal makes a 30-degree angle with the horizontal plane, and the vertical canals make 45-degree angles with the frontal plane (Fig. 1–4C). At the anterior opening of the
II
I
Efferent
Afferent
PRIMARY AFFERENT
FIRING RATE
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
nerve nerves

![]()
100 msec
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Figure 1–2. Hair cell modulation of spontaneous afferent nerve firing rate. Bending of the stereocilia toward the kinocilium depolarizes the hair cell and increases the firing rate, and bending away from the kinocilium hyperpolarizes the hair cell and decreases the firing rate. Kc – kinocilium.
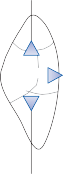
horizontal and anterior semicircular canals and the inferior opening of the posterior canal, each tube enlarges to form the ampulla. The crista, the sensory epithelium composed of hair cells and supporting cells, crosses each ampulla in a direction perpendicular to the longitudinal axis of the canal (Fig. 1–4A).9 Hair cells are located on the surface of the crista, with the cilia pro- truding into the cupula, a gelatinous mass that extends from the surface of the crista to the ceiling of the ampulla, forming a watertight seal.
The hair cells in each crista are oriented with their kinocilia in the same direction. In the horizontal canal, however, the kinocilia are directed toward the utricular side of the ampulla, whereas in the vertical canals the kinocilia are directed toward the canal side of the ampulla. This difference in morphological polarization explains the difference in direc- tional sensitivity between horizontal and verti- cal canals.13 The afferent nerve fibers of the horizontal canals increase their baseline firing when endolymph moves toward the utricle and
Otoconia Gelatin layer
Reticular membrane
Supporting cells
Striola
Kc
Hair cells
Static tilt Otolith
displacement
Gravitational force
Dorsal
Dorsal
(c)
Saccular macule
Anterior
Anterior
Striola
Lateral
Lateral
Utricular macule
Figure 1–3. The macule: (a) anatomy, (b) mechanism of hair cell activation with static tilt, and (c) orientation of saccular and utricular macules. Arrows indicate the direction that the kinocilia point toward. (Adapted from Barber HO, Stockwell CW. Manual of Electronystagmography. CV Mosby, St. Louis, 1976.)
ampulla (ampullopetal flow), but the afferent nerves of the vertical canals increase their base- line firing rate with endolymph flow away from the ampulla (ampullofugal flow).
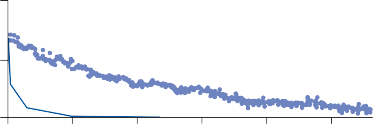
Since the cupula has the same specific grav- ity as the surrounding endolymph, it is not subject to displacement by changes in the line of gravitational force. The forces associated with angular head acceleration displace the cupula and bend the hair cells of the crista, however. The motion of the cupula can be lik- ened to that of a pendulum in a viscous medium.14,15 Sudden displacement of the cup- ula by a step change in angular velocity is fol- lowed by a gradual exponential return of the cupula to its baseline position (Fig. 1–5). The
rate of return (time constant, Tc) is determined by the ratio of the viscous drag coefficient of
the endolymph to the elasticity coefficient of
the cupula according to the pendulum model
(see Chapter 2).
Precise measurements of primary afferent nerve activity originating from the cristae of animals during physiological rotatory stimula- tion reveal that the change in frequency of action potentials is approximately proportional to the deviation of the cupula as predicted by the pendulum model.8 For example, during sinusoidal head rotation in the plane of a semi- circular canal, a sinusoidal change in firing fre- quency is superimposed on the rather high rest- ing discharge (about 90 spikes per second in the squirrel monkey). The peak firing rate occurs at the time of maximum cupular displacement, which occurs at the time of peak angular head velocity. With small-amplitude sinusoidal rota- tion, the modulation is almost symmetrical about the baseline firing rate. For larger

Utricular sac of macule
Cupula
Ampulla
Cupula
displacement Relative
endolymph flow
Angular acceleration
Supporting cells
Hair cells
Semicircular canal
![]()
Left and
right HC
30
Right AC
Left PC
Right PC
Left AC
Figure 1–4. The crista: (a) anatomy, (b) mechanism of hair cell activation with angular acceleration, and (c) orientation of the semicircular canals within the head. AC, anterior canal; HC, horizontal canal; PC, posterior canal.
Slow phase velocity (deg/sec)
Slow phase velocity (deg/sec)
100
50
0
0 10 20 30
Time (seconds)
40 50
Figure 1–5. Rate of return of the cupula to its initial position after a step change in angular velocity (thin solid line) and rate of decay in nystagmus slow phase velocity after the same step change in angular velocity (each blue dot represents a single beat of nystagmus). Note that the nystagmus outlasts the cupular deviation (and afferent nerve activity) due to central velocity storage.
Basis of Stimulus Specificity of the Inner Ear Receptor Organs
The inner ear receptors all work on the same basic principal: activation of hair cells by an applied external force. The density of the oto- lithic membrane overlying the hair cells of the macule is greater than that of the surrounding endolymph. The hair cell cilia are embedded in the otolithic membrane and, when displaced, produce a shearing force (Ft) on the underlying
hair cells that is proportional to the sine of the
angle between the line of resulting gravitational vector and a line perpendicular to the plane of the macule. Each macule is bisected by a distinctive curved zone, the striola. Hair cells are oriented in opposite directions on each side of the striola so that displacement of the oto- lithic membrane has an opposite effect on the set of hair cells on each side of the striola (see Fig. 1–3C).
The hair cell cilia in the cristae of the semi- circular canals are embedded in the cupula, a jelly-like substance of the same specific gravity as that of the surrounding fluids. The cupula, therefore, does not exert a force on the under- lying crista and is not subject to displacement by changes in the line of gravitational force. The forces associated with angular head accel- eration, however, do result in a displacement of the cupula that stimulates the hair cells of the crista in the same way that displacement of the otoliths stimulates the macular hair cells (Fig. 1–4B). However, in the cristae, all the hair cells are oriented in the same direction in the crista surface. All hair cells are either excited or inhibited by motion of the fluid in the canal, but the orientation is different in dif- ferent semicircular canals.
In the cochlea, the hair cells are mounted on the flexible basilar membrane in the organ
of Corti. Covering the organ of Corti and rest- ing over the hair cells is the tectorial mem- brane, a relatively rigid structure attached to the wall of the cochlea. A small, acoustically induced pressure difference across the basilar membrane causes the organ of Corti and hair cells to vibrate at the frequency of sound. The motion of the basilar membrane has a different effect on the outer hair cells than on the inner. Outer hair cells have their cilia embedded in the tectorial membrane and are directly stimu- lated as the cilia are displaced in relation to the relatively fixed tectorial membrane, which acts as a hinge.16 In contrast, the inner hair cell cilia are not embedded in the tectorial mem- brane but are instead surrounded by endo- lymph. Their stimulation is produced by the dragging viscous force of the fluid on the cilia. Intracellular recordings in mammalian cochlear hair cells show a difference of phase between the receptor potentials of the inner and outer hair cells as predicted by the difference in the coupling of the cilia to the tectorial membrane.17 The outer hair cells respond to position and the inner hair cells respond to the velocity of the basilar membrane motion.
In all cases, the effective stimulus to the sen- sory cells is the relative displacement of the cilia produced by application of mechanical force to their surroundings. Since the mechan- ical properties of the “supporting and coupling” structures are different, the frequency ranges at which the cilia can be moved by the applied force are different. Because of the great flexi- bility of the basilar membrane, the range of sound frequencies to which the hair cells in the cochlea are sensitive varies from 20 to 20,000 Hz. In the macules, the otoconia are maximally displaced during constant accelera- tions such as those associated with steady head displacement. Owing to the characteristics of the restraining viscoelastic forces holding the otoliths to the macule, their motion rapidly diminishes if the linear acceleration changes at a frequency >0.5 Hz.18 The semicircular canals also respond maximally to constant angular acceleration, but they can respond to changes in angular acceleration as high as 40 to 50 Hz.19 This frequency limitation is due to the inertial and viscous forces restraining the displacement of fluid and cupula in the narrow semicircular canals.
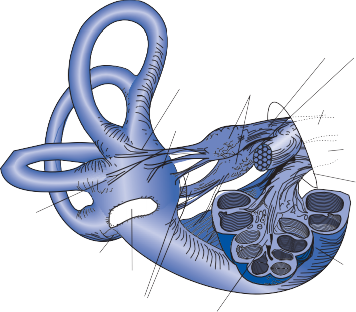
Parallel to the separation of receptor organs, afferent nerve fibers differentiate into bundles that maintain independent identity in the inter- nal auditory canal and at the entrance to the brain stem.20 The afferent nerve from the utricle and horizontal and anterior semicircular canals and some of the nerve fibers from the saccule form the superior division of the vestibular nerve; most nerve fibers from the saccule and the nerve from the posterior semicircular canal contribute to the inferior branch (Fig. 1–6). The afferent fibers from the auditory organ form a separate nerve anterior and inferior to the ves- tibular nerve to innervate the organ of Corti, the auditory receptor organ. Together these two nerves constitute the eighth cranial nerve and, within them, a system of efferent fibers from the central nervous system (CNS) gates or mod- ulates the activity of the peripheral organs.21–23 Phylogenetically, this neural feedback system is already present in gastropods, in which action potentials directed from the brain to the recep- tors have been recorded.24
In comparison with the vestibular sensory organs, central vestibular connections become progressively more complex in higher
vertebrates.25,26 This complexity accompanies the development of other afferent systems for the maintenance of equilibrium (vision, prop- rioception) and pathways for interaction of these systems with the vestibular system.
Vestibular Nuclei
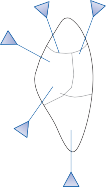
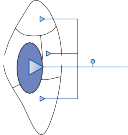
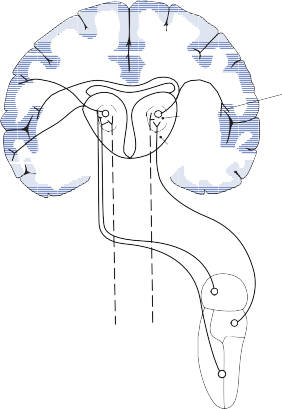
The central processes of the primary vestibular neurons divide into an ascending and descend- ing branch after entering the brain stem at the inner aspect of the restiform body (see Fig. 3–1 in Chapter 3). The ascending branch ends either in the rostral end of the vestibular nuclei or in the cerebellum, and the descending branch ends in the caudal vestibular nuclei. None of the primary afferents cross the mid- line. Four distinct anatomical groups of neu- rons have traditionally been identified: medial, lateral, superior, and inferior nuclei (Fig. 1–7).27 Canal and otolith signals converge on most sec- ondary neurons that receive primary afferent input (Fig. 1–7C). Major connections run to and from the cerebellum, particularly the so- called vestibulocerebellum (uvula, nodulus, and flocculonodular lobes) (Fig. 1–7E). The two sides are connected by reciprocal commissural
Nerve from horizontal canal
Nerve from posterior canal
Round window
Nerve from anterior canal
Utricular nerve
Scarpa’s ganglion
Vestibular nerve superior division inferior division
Facial nerve
Auditory nerve
Internal auditory canal
Cochlea
Figure 1–6. Innervation of the labyrinth.
Saccular
nerves Spiral ganglion
Afferent inputs to the vestibular nuclei
(B)
Efferent outputs from the vestibular nuclei
Visual inputs
Thalamus/Hypothalamus Cerebellum
Hippocampus
![]()
VN Vestibular nerve afferent inputs
Commissural projections to contralateral
side
VN
![]()
Extraocular motoneurons
Proprioceptive spinal inputs
(C)
(C)
Labyrinthine nerve afferent projections
(D)
Neuronal integrator
(Ncl. prepositus hypoglossi)
Vestibular commissural projections
![]()
(E)
Spinal cord
Afferent and efferent cerebellar projections
SVN
Semicircular
SVN
SVN
Flocculus Uvula / Nodulus SVN
afferent
afferent
canal/otolith
MVN MVN
MVN
LVN
DVN
nerve fibers
LVN MVN LVN LVN
DVN DVN
DVN
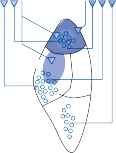
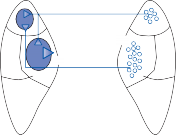
Figure 1–7. Main afferent and efferent connections of the vestibular nuclei (A and B). The vestibular nuclei (VN) receive afferent signals related to head motion in space (A) and project to target areas involved in stabilization of gaze and posture as well as in vegetative and cognitive functions (B). The MVN is the major relay station for vestibular signals related to gaze and postural stabilization. A large area of the MVN receives afferent labyrinthine inputs from the semicircular canal and otolith organs (C). The MVN is the largest source and target area for reciprocal commissural pathways (D). The MVN is the major vestibular nucleus for signals from and to the flocculus (E). DVN, LVN, MVN, SVN, for descending, lateral, medial and superior vestibular nucleus. (From Straka H, Vibert N, Vidal PP, Moore LE, Dutia MB. Intrinsic membrane properties of vertebrate vestibular neurons: function, development and plasticity. Prog Neurobiol. 2005;76:349, with permission.)
pathways most of which originate in the medial nucleus (Fig. 1–7D). Secondary vestibular neu- rons project to target areas involved in stabiliza- tion of gaze and posture, vegetative regulation, and higher cognitive function (Fig. 1–7B).
Vestibular nucleus neurons receive afferent visual and proprioceptive signals in addition to primary vestibular signals (Fig. 1–7A).28 For example, visual and proprioceptive signals are organized such that movement of the visual surround in one direction excites and inhibits the same neurons that are excited and inhibited by movement of the head and neck in the opposite direction. The vestibular nucleus is
therefore not simply a relay station for vestibu- lar signals but rather an important sensorimo- tor interaction center.
The basic elements of a simple vestibular reflex arc are a hair cell, an afferent bipolar neuron, an interneuron, and an effector neuron (Fig. 1–8).29 This simple three-neuron reflex arc is already developed in the phylum Mollusca, among which the class Cephalopoda

Interneuron
Motoneuron
Figure 1–8. Three neuronal reflex arc. The primary affer- ent neuron carries signals generated by the hair cells to interneurons in the brainstem which in turn activate motoneurons that initiate the motor response.
has contributed to many classic anatomic and physiologic studies of gravitational reflexes.30 Vestibular reflexes have developed further in vertebrates and mammals with the addition of multiple neuronal pathways.29
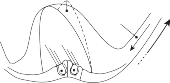
The terminal fibers of the afferent neuron make synaptic contact with the hair cell and transmit nerve signals to neuronal sensory pools on the same side of the CNS (the vestibular nuclei) that contain both excitatory and inhibi- tory neurons. Besides receiving signals from excitatory first-order neurons from the ipsilat- eral ear, the excitatory neurons also receive signals from the inhibitory neurons of the con- tralateral side by way of crossed neural path- ways. The output of the excitatory vestibular nuclei interneurons is transmitted to the effec- tor motor pools, which consequently reflect the activity of both ears. The effector neuron, in turn, controls the activity in an appropriate muscle to coordinate orienting behavior.
In 1947, Sherrington noted, “The simple reflex is probably a purely abstract conception
because all parts of the nervous system are con- nected together and no part of it is probably ever capable of reaction without affecting and being affected by various other parts….”31 The maintenance of body equilibrium and posture in everyday life is a complex function involving multiple receptor organs and neural centers in addition to the labyrinths. Visual and proprio- ceptive reflexes in particular must be integrated with vestibular reflexes to ensure postural sta- bility. The prominent role of sensory interac- tion in orientation can already be appreciated in the behavior of gastropods. The invertebrate Hermissenda has only rudimentary vestibular and visual receptors, yet the two systems fully interact to control behavior.32 Afferent signals from photoreceptors in the eye and from hair cells in the statocyst converge on interneurons in the cerebroplural ganglia, which control a putative motor neuron in each pedal ganglion. Excitation of the motor neuron produces turn- ing of the animal’s foot in the ipsilateral direc- tion, consistent with the animal’s turning behavior toward light. In humans, during most natural head movements, gaze stabilization is achieved by a combination of vestibular, neck proprioceptive, and visual inputs; the interac- tion can be synergistic or antagonistic. For example, when the vestibular induced eye movements lie in a direction opposite to that required to maintain the desired gaze position, the visual reflexes override the vestibular reflex. The kind of head rotation that would produce compensatory eye movement in the dark does not do so in the light if the subject fixates on a target moving in phase with the head (Fig. 1–9). In this simple example, failure to override the vestibular signal leads to disorientation.
Horizontal Canal-Ocular Reflex
The direct pathways from the horizontal canals to the horizontal extraocular muscles deserve particular attention, since the horizontal ves- tibulo-ocular reflex is the focus of most clinical vestibular testing (Fig. 1–10).33,34 The second- ary vestibular neurons lie in the medial and lat- eral vestibular nuclei. The more medial group of excitatory neurons projects to the contralat- eral abducens nucleus, while the more laterally located excitatory neurons (in the medial part of the lateral nucleus) project to ipsilateral
60°/sec 0
60°/sec
60°/sec 0
60°/sec
Eye movements in the dark
Eye movements in the dark
5 sec
15°
Eye movements with fixation
Right
Left
5 sec
15°
Eye movements with fixation
Right
Left
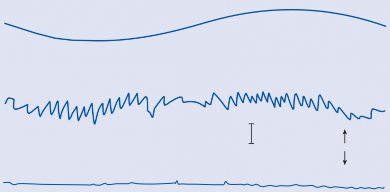
Figure 1–9. Eye movement induced in a normal human subject by sinusoidal angular acceleration (0.05 Hz, maximum velocity 60°/sec) in the dark and in the light with a target moving in phase with the subject.
LR MR MR LR
Oculomotor IR nucleus (III) IO
SR
MR
Trochlear ATD
nucleus (IV)
MLF
Abducens nucleus (VI)
S L
SG
M I
Vestibular nucleus
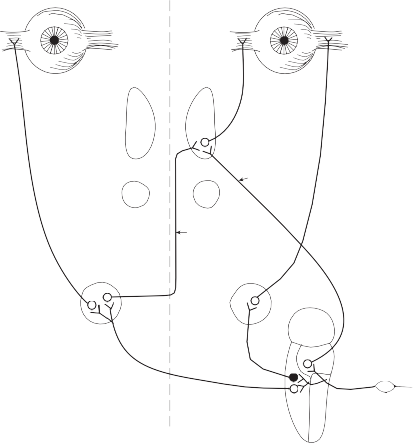
Figure 1–10. Direct pathways of the horizontal semicircular canal-ocular reflex. The darkened cell body indicates an inhibitory secondary vestibular neuron. SG – Scarpa ganglion, S – superior nucleus, L – lateral nucleus, M – medial nucleus, I – inferior (descending) nucleus, MLF – medial longitudinal fasciculus, ATD – ascending tract of Deiters, IR – inferior rectus, IO – inferior oblique, SR – superior rectus, MR – medial rectus, LR – lateral rectus.
medial rectus motoneurons via the ascending tract of Deiters (ATD). The ipsilateral medial rectus neurons also receive a strong excitatory input via the medial longitudinal fasciculus (MLF) from interneurons in the contralateral abducens nucleus. These interneurons are
CLOCKWISE
![]()
Right
ROTATION
Left
excited by the same secondary vestibular neu- rons that excite the abducens motoneurons.35 The relative contributions to the horizontal vestibulo-ocular reflex of the ATD and MLF
horizontal canal
horizontal canal
excitatory pathways is not entirely clear, but the MLF pathway seems more important since the eyes cannot adduct past the midline if
R+ L+
R–
the MLF is sectioned.36 Inhibitory secondary neurons in the rostral part of the medial ves- tibular nucleus run directly to the ipsilateral abducens nucleus. Contralateral medial rectus
Vestibular nucleus
Vestibular
L– nucleus
motoneurons apparently do not receive disyn- aptic inhibition from the horizontal semicircular canals.
In addition to the direct and indirect con- nection between secondary vestibular neurons and oculomotor neurons, commissural connec- tions between the two vestibular nuclei play an important role in controlling the rotational ves- tibulo-ocular reflex.37 Through GABAnergic interneurons, secondary vestibular neurons
R+ L– R– L+
motoneurons
motoneurons
motoneurons
motoneurons
![]()
![]()
Agonist
Agonist
Antagonist
Antagonist
Increased Decreased
![]()
on one side inhibit their counterparts of the Excitatory
opposite side (see Fig. 3–4 in Chapter 3). As
Inhibitory
will be seen later, the commissural connections are particularly important after unilateral loss of vestibular function since they provide a mechanism for a single labyrinth to control the vestibular nuclei on both sides, thus maintain- ing a functional vestibulo-ocular reflex.38
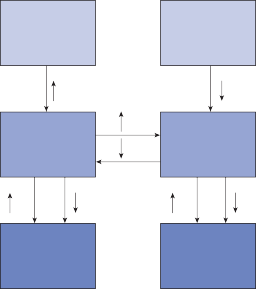
Because physiological stimuli activate both labyrinths, the horizontal vestibulo-ocular reflex is controlled by a four-way push-pull mechanism (Fig. 1–11). For example, physio- logical stimulation of the crista of the right horizontal semicircular canal excites the left lateral rectus and the right medial rectus and inhibits the right lateral rectus. Because of the symmetry between the labyrinths, the same receptor in the other ear simultaneously diminishes its afferent output, thereby disfacili- tating the left medial rectus and right lateral rectus and disinhibiting the left lateral rectus. The end result is contraction of the left lateral and right medial rectus muscles and relaxation of the left medial and right lateral rectus muscles.
Figure 1–11. Organization of the horizontal semicircular canal–ocular reflex. R – right, L – left.
Nystagmus
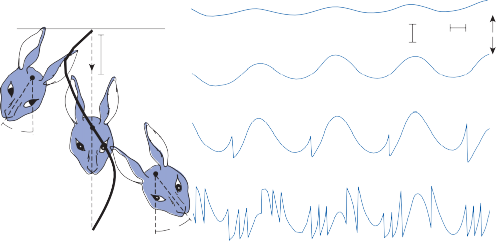
When the head is rotated back and forth in the dark in the plane of the horizontal semicircular canals, compensatory eye movements are pro- duced, with eye velocity approximately equal and opposite to the head velocity. This is easily demonstrated in lower animals such as the rab- bit, who have few spontaneous eye movements (Fig. 1–12A,B). If the angle of rotation is large, such that it cannot be compensated for by the motion of the eye in the orbit, the slow com- pensatory vestibular-induced eye movement is interrupted by quick movements in the oppo- site direction. This combination of rhythmic slow and fast eye movements is called nystag- mus.39 Because of the fast components, the tra- jectory of the eye motion during the slow components effectively compensates for head rotation as if the eye had unlimited range
q = 3
EYE MOVEMENT RECORDINGS
a.
Rt. Lt.
q = 6 4
1sec
rt.
lt.
1 sec
b.
q = 12
q c.
q = 24
d.
q
Figure 1–12. Compensatory eye movements in the rabbit that are produced by sinusoidal angular acceleration of the head (0.2 Hz) at four different peak angular displacements ().
of motion. If the fast components were removed from the tracings in Figure 1–12C,D and the slow components joined end to end, the result- ing sinusoidal eye movement would be approx- imately equal and opposite in direction to the sinusoidal head movement just as in Figure 1–12A,B. Thus, the quick component of nys- tagmus is a strategy developed in the brain to increase the functional capabilities of the reflex.
Spontaneous nystagmus occurs after lesions of the labyrinth, the vestibular nerve or the central vestibulo-ocular neurons and intercon- necting pathways. The driving force of the spontaneous nystagmus is an imbalance of tonic signals within the vestibulo-ocular pathways. Damage to one labyrinth or its vestibular nerve results in spontaneous nystagmus, with the slow phase directed toward the damaged side; the tonic input from the intact side is no longer balanced by tonic input from the damaged side. This spontaneous nystagmus is similar to nys- tagmus produced by physiological stimulation of the horizontal semicircular canals (Fig. 1–13; also see Video 6–4). The direction of nystag- mus associated with lesions of the brain stem is less predictable, depending on the location and extent of the lesion.40 Central spontaneous nystagmus can be purely vertical or torsional, since tonic signals for vertical and torsional eye movements run in different tracts from the
vestibular nuclei to the oculomotor neurons. By contrast, peripheral spontaneous nystagmus aligns with the planes of the semicircular canals, producing a combination of torsional and linear components. After a complete unilateral peripheral vestibular loss, the nystagmus is horizontal/torsional because the vertical com- ponents from the loss of vertical canal input cancel out.
Groups of neurons in the paramedian pon- tine reticular formation (PPRF) adjacent to the abducens nuclei fire in short bursts just before the onset of horizontal fast components.41 Pathways interconnect neurons in the vestibu- lar nuclei with neurons in this region of the PPRF, and these neurons project directly to oculomotoneurons and interneurons in the abducens nucleus. Neurons in the PPRF moni- tor vestibulo-ocular signals and intermittently trigger bursts of firing in the opposite direction mainly based on the eye position in the orbit. During angular rotation, the fast components of the initial beats of nystagmus are larger in amplitude than the preceding slow components so that the eyes deviate in the direction of the fast components. The apparent advantage of this strategy is that the eyes are ready to follow new targets arriving in the field of vision and fixation can be maintained during the subse- quent slow component. Unilateral lesions of the PPRF impair ipsilateral rapid eye movements
PHYSIOLOGIC NYSTAGMUS SPONTANEOUS NYSTAGMUS


![]()
AC Damaged
PC
HC
Ampulla
Utricle
PRIMARY AFFERENT FIRING RATE
![]()
100 msec
Figure 1–13. Primary afferent nerve activity associated with rotation-induced physiological nystagmus and spontaneous nystagmus resulting from a lesion of one labyrinth. The thin straight arrows indicate the direction of the slow components; the thick straight arrows indicate the direction of fast components; curved arrows show the direction of endolymph flow in the horizontal semicircular canals. AC – anterior canal, PC – posterior canal, HC – horizontal canal.
(both voluntary and involuntary), and the eyes deviate to the contralateral side of the orbit.42 Stimuli that normally would produce nystagmus with ipsilateral fast components simply cause a tonic contralateral deviation of the eyes.
Translational Vestibulo-Ocular Reflexes
Natural head movements consist of a combina- tion of rotation and translation. For images to remain stable on the retina, vestibular reflexes must compensate for both types of movement. Translational movements are sensed by the otolith organs of the inner ear, and compensa- tory eye movements are generated by the oto- lith ocular reflexes. Although the rotational vestibulo-ocular reflexes are highly conserved throughout evolution, translational reflexes develop later in frontal-eyed animals with foveal vision. Unlike the rotational vestibulo- ocular reflexes where an equal and opposite eye movement suffices regardless of target dis- tance, the translational vestibulo-ocular reflexes must be scaled to viewing distance to compen- sate for the fact that the size of the required
compensatory eye movement increases as the target moves closer, the so-called motion paral- lax (see Chapter 3).43,44
Furthermore, unlike the rotational vestib- ulo-ocular reflexes that stabilize images on the entire retina, the translational vestibulo-ocular reflexes only stabilize images on one spatial location in the visual field, usually the fovea. Not surprisingly, there is a close functional relationship between the translational vestib- ulo-ocular reflexes and the other foveal stabi- lizing systems, the smooth pursuit and the vergence systems.45
The Ocular Tilt Reflex
If a subject is tilted in the roll plane (about the nasal occipital axis), there is a reflex counter- rolling and skewing of the eyes to maintain gaze stabilization (see Fig. 6–7 in Chapter 6). This represents an utriculo-ocular reflex primarily mediated by excitation of the utricle of the dependent ear with synapses in the ipsilateral vestibular nucleus and in the contralateral ocul- omotor complex in the rostral brain stem.46,47 Unlike the translational vestibulo-ocular reflex,
it develops early in evolution being particularly prominent in lateral-eyed animals. It is a rudi- mentary reflex in humans since the amount of ocular counterrolling is only about 10% of the angle of head tilt.48 Attempts to use this reflex as a clinical test of otolith function have been largely abandoned because of the large vari- ability in normal subjects and the lack of consis- tent asymmetry after unilateral lesions. However, understanding the connections of this otolith-ocular reflex is critical for localizing peripheral and central lesions that cause double vision due to skew deviation (see Chapter 6).47
Vestibulospinal Reflexes
At least three major functional roles for vestibu- lospinal reflexes can be identified.49,50 The first is to maintain posture, namely, the upright posi- tion in relation to the earth vertical. Vestibular reflexes of this kind induce muscle contractions that produce negative geotropic movement or forces that compensate for steady changes in the direction of the force of gravity. If the pull of gravity on the body were unopposed by forces developed in the muscles, the body would col- lapse. Reflexes in this category in humans are dependent on the function of the otolith organs but not on that of the semicircular canals. The second role is to produce “kinetic,” or transi- tory, contractions of muscles for maintenance of equilibrium during movement. This category
includes reflexes arising from both the semicir- cular canals during angular acceleration and the otolithic organs during linear acceleration.51 Most natural head movements contain both types of acceleration, and the vestibular reflexes act in combination to maintain equilibrium. A third role of vestibular reflex activity is to help maintain muscular tone, a role in which both the macules50 and cristae participate.52 The labyrinthine contribution to skeletal- muscle tone can be demonstrated by the change in posture that follows unilateral laby- rinthectomy in normal animals.53 Tone is increased in the extensor muscles of the con- tralateral extremities and decreased in the ipsi- lateral extensor muscles. An even more striking demonstration of the vestibular role in mainte- nance of muscle tone is the removal of decerebrate rigidity after sectioning of both vestibular nerves or destruction of the vestibu- lar nuclei (see later discussion).54,55 The exten- sor rigidity that results from transection of the nervous system at the caudal end of the mesencephalon is markedly decreased when the tonic labyrinthine input is removed.
The anterior horn cells of the antigravity muscles (extensors of the neck, trunk, and extremities) are under the combined excitatory and inhibitory influence of multiple supraspi- nal neural centers (Fig. 1–14).54 At least in the cat, one finds two main facilitatory centers (the lateral vestibular nucleus and rostral retic- ular formation) and four inhibitory centers

1
–
2 –
–
3
–
–
–
+ 5 6 +
+
4 –
Figure 1–14. Facilitatory (+) and inhibitory (−) pathways influencing the myotatic spinal reflex in the cat. Inhibitory path- ways are (1) corticobulboreticular, (2) caudatospinal, (3) cerebelloreticular, and (4) reticulospinal. Facilitatory pathways are
reticulospinal and (6) vestibulospinal. (From Lindsley DB, Schreiner LH, Magoun HW. An electromyographic study of spasticity. J Neurophysiol. 1949;12: 197, with permission.)
(the pericruciate cortex, basal ganglia, cerebel- lum, and caudal reticular formation). The bal- ance of input from these different centers determines the degree of tone in the antigrav- ity muscles. If one removes the inhibitory influ- ence of the frontal cortex and basal ganglia by sectioning the animal’s midbrain, a characteris- tic state of contraction in the antigravity mus- cles results—so-called decerebrate rigidity. The extensor muscles increase their resistance to lengthening and the deep tendon reflexes become hyperactive. As noted earlier, the ves- tibular system contributes largely to this increased extensor tone since there is a marked decrease upon bilateral destruction of the laby- rinths.56 Unilateral destruction of the labyrinth or the lateral vestibular nucleus results in an ipsilateral decrease in tone, indicating that the main excitatory input to the anterior horn cells arrives from the ipsilateral lateral vestibulospi- nal tract.55
In a decerebrate animal with normal laby- rinths, the intensity of the extensor tone can be modulated in a specific way by changing the position of the head in space.31,49,50 The tone is maximal when the animal is in the supine posi- tion with the angle of the mouth 45 degrees above horizontal and minimal when the animal is prone with the angle of the mouth 45 degrees below horizontal. Intermediate positions of rotation of the animal’s body about the trans- verse or longitudinal axis result in intermediate degrees of extensor tone. If the head of the upright animal is tilted upward (without neck extension), extensor tone in the forelegs increases; downward tilting of the head causes decreased extensor tone and flexion of the fore- legs. Lateral tilt produces extension of the extremities on the opposite side. These tonic labyrinthine reflexes, mediated by way of the otoliths, seldom occur in intact animals or human subjects because of the inhibitory influ- ence of the higher cortical and subcortical cen- ters; however, they can be demonstrated in premature infants.57
Vestibulo-Autonomic Reflexes
The strong connections between vestibular and vegetative centers are apparent based on the prominent vegetative symptoms that accom- pany vestibular lesions. Nausea and vomiting, diarrhea, perfuse sweating, and fainting can be
the predominant presenting symptoms of a ves- tibular lesion. Animal and human studies have shown that electrical or physiological stimula- tion of the vestibular receptors alters the activ- ity of sympathetic efferents.58,59 Neurons in the caudal vestibular nuclei project to medullary regions known to participate in regulation of blood pressure, heart rate, and breathing; lesions in this region abolish cardiovascular and respiratory responses to stimulation of vestibu- lar afferents. Loss of vestibulocardiac and ves- tibulovascular reflexes may explain the fainting and near fainting often associated with vestibu- lar lesions. There are also connections from the vestibular nuclei to the locus coeruleus, area postrema, and more centrally to the hypothala- mus, amygdale, and limbic cortex that could explain the motion sickness and symptoms of fear and panic that commonly accompany vertigo.60
MOTION PERCEPTION AND ORIENTATION
Several important clinical observations support the existence of a specific vestibular sensation. Probably the most convincing is that patients without vestibular function (either on an acquired or congenital basis) do not experience a turning sensation when rotated in the dark if visual and tactile cues are eliminated.61 In contrast, in patients with the sensation of move- ment, it is not dependent on vision or associ- ated nystagmus, since blind subjects and patients with complete oculomotor paralysis experience a spinning sensation comparable to that of normal subjects when their vestibular end organs are stimulated. Focal cortical lesions can interfere with spatial orientation and the performance of three-dimensional construc- tion tasks, and epileptic discharges from many different areas of the cortex can be associated with a subjective illusion of movement (usually spinning). These observations imply a cerebro- cortical representation for vestibular sensation. The vestibulocortical pathway via the thala- mus is concerned with the control of body posi- tion and orientation in space (Fig. 1–15).63,64 Thalamic and cortical units that receive ves- tibular signals are also activated by propriocep- tion and visual stimuli. Most units respond in a similar way to rotation in the dark, or to moving
Cerebral cortex
IPL
VPL
PIVC
STG
Thalamus
Muscle and cutaneous afferents
S
L
M
M
Vestibular
I nucleus
Figure 1–15. Vestibulothalamocortical projections. I, inferior nucleus; IPL, intra-parietal lobe; L, lateral nucleus; M, medial nucleus; PIVC, parieto-insular vestibular cortex; S, superior nucleus; STG, superior temporal gyrus; VPL, nucleus ventralisposterior lateralis.
visual fields, indicating that they play a role in relaying information about self-motion. From a functional point of view, the vestibulothalamo- cortical projections appear to integrate vestibu- lar, proprioceptive, and visual signals to provide one with a “conscious awareness” of body ori- entation. Beginning at the vestibular nuclei, a stepwise integration of body-orienting signals occurs, reaching its maximum at the level of the cortex.
PATHOPHYSIOLOGY OF VESTIBULAR SYMPTOMS
Much of our knowledge of labyrinthine func- tion was accumulated at the turn of the twenti- eth century from clinical and experimental observations in humans and animals with uni- lateral and bilateral lesions of the peripheral labyrinth.65–67 At that time, a controversy existed
concerning whether the symptoms associated with acute unilateral labyrinthine damage was due to irritation or paralysis of the affected labyrinth. The subsequent discovery of the continuous flow of action potentials in the vestibular nerve at baseline led to the present concept that symptoms are usually caused by an imbalance of the normal resting state activity—that is, by a unilateral decrease in activity.
Symptoms and signs after labyrinthine lesions can largely be traced to asymmetric tone or loss of function within the vestibular reflex pathways (Table 1–1). The magnitude of symptoms and signs depends on (1) whether the lesion is unilateral or bilateral, (2) the rapidity with which the functional loss occurs, and (3) the extent of the lesion. In most experimental animals, simultaneous removal of both labyrinths does not produce severe abnormalities, although vestibular reflex activ- ity is lost and ocular and postural stability is
Table 1–1 Symptoms and signs after labyrinthine lesions result from asymmetric tone and/or loss of function within vestibular reflex pathways
![]()

Pathway Asymmetric Tone Loss of Function
Vestibulo-ocular Spontaneous nystagmus
Ocular roll & skew
Vestibulo-spinal Head tilt Lateropulsion
Vestibulo-autonomic Nausea, vomiting, fainting,
fear, anxiety Vestibulo-cortical Illusion of movement
Tilt of subjective vertical
Head movement dependent oscillopsia Imbalance worse with eyes closed Resistant to motion sickness
Decreased motion perception, visual dependency
![]()
impaired. Similarly, patients who lose vesti- bular function bilaterally (e.g., secondary to gentamicin treatment) usually do not com- plain of vertigo, but they do report visual blur- ring or oscillopsia with head movements and instability when walking at night (due to loss of vestibulo-ocular and vestibulospinal reflex activity).
In contrast, animals and humans develop severe symptoms and signs following acute unilateral labyrinthectomy. Lower mammals are initially unable to walk and develop head tilt and decreased ipsilateral muscle tone. Nystagmus is prominent, with the slow compo- nent directed toward the damaged side and the fast component toward the intact side. These signs abate with time but may persist for months after surgery.
A sudden unilateral loss of labyrinthine function in humans is a dramatic event.68 The patient complains of severe vertigo and nausea, is pale and perspiring, and usually vomits repeatedly. The patient prefers to lie motion- less but can walk if forced to (deviating toward the side of the lesion). Head and ocular tilt and changes in extremity tone occur but less fre- quently than in lower animals. A brisk, sponta- neous nystagmus interferes with vision. These symptoms and signs are temporary, and the process of compensation starts almost immedi- ately. Within 1 week of the occurrence of the labyrinthine lesion, a young patient can walk without difficulty and, with fixation, can inhibit the spontaneous nystagmus. Within 1 month, most patients return to work with few, if any, residual symptoms. If a patient slowly loses vestibular function unilaterally over a period of months or years (e.g., with a vestibu- lar schwannoma), symptoms and signs may be absent.
CENTRAL COMPENSATION FOR VESTIBULAR LESIONS
In animals immediately after a labyrinthec- tomy, ipsilateral secondary vestibular neurons lose their afferent input, become silent, and do not respond to ipsilateral angular rotation.69–71 At the same time, contralateral healthy second- ary neurons lose their inhibitory contralateral input, and their spontaneous activity increases in comparison to normal levels. An imbalance in ocular and skeletal muscle tone takes place, resulting in the clinical signs of labyrinthec- tomy—nystagmus and disequilibrium. A few days after the labyrinthectomy, the previously silent secondary neurons on the damaged side recover their spontaneous activity and begin to respond to physiologic stimulation of the con- tralateral labyrinth, the result of their connec- tions through the commissural pathways. Although the responses of secondary neurons on the damaged side are not as intense as those on the normal side, they are qualitatively simi- lar. The recovery of sensitivity in the ipsilateral secondary neurons after a labyrinthectomy parallels the time course of recovery in clinical symptoms and signs.
The genesis of the renewed tonic input to ipsilateral secondary neurons several days after a complete labyrinthectomy is not entirely known. It does not come from the healthy side, since afferent activity on that side does not change. It probably results from changes in ion channels expressed in the cell membrane, from the sprouting of axons from other sources (e.g., neck proprioceptive), and from up and down regulation of synaptic receptors (particularly GABA receptors) (see Chapter 3).72–75 In ani- mal studies, the course of compensation is affected by exercise,76 visual experience,77 and
drugs (as a rule, stimulants accelerate and sed- atives slow compensation).78 If a second laby- rinthectomy is performed after compensation for the first occurs, the animal again develops signs of acute unilateral vestibular loss with nystagmus directed toward the previously operated ear (Bechterew’s compensatory nys- tagmus),79 as if the first labyrinthectomy had not taken place. Compensation after the sec- ond labyrinthectomy is slightly faster than after the first, but it still requires several days.
The vestibular system transduces the forces associated with head acceleration and gravity into a biologic signal. The control centers in the brain use this signal to develop a subjective awareness of head position in relation to the environment and to produce motor reflexes for
avenues of research in the study of vestibular function in health and in disease.
REFERENCES
Baloh RW. Prosper Ménière and his disease. Arch Neurol. 2001;58:1151.
Gray O. A brief survey of the phylogenesis of the laby- rinth. J Laryngol. 1955;69:151.
Vollrath MA, Kwan KY, Corey DP. The micromachin- ery of mechanotransduction in hair cells. Annu Rev Neurosci. 2007;30:339.
Hudspeth AJ, Corey DP. Sensitivity, polarity, and conductance change in the response of vertebrate hair cells to controlled mechanical stimuli. Proc Natl Acad Sci USA. 1977;74:2407.
Hudspeth AJ. How the ear’s works work: mechano- electrical transduction and amplification by hair cells. C R Biol. 2005;328:155.
Lowenstein O, Wersall J. A functional interpretation of the electron microscopic structure of the sensory hairs in the cristae of the elasmobranch Raja clavata in terms of directional sensitivity. Nature. 1959;184:1807.
sh.
equilibrium, relating these experiences to those of other sensory systems during locomotion. The vestibular system, by means of its recep- tors for the perception of linear and angular acceleration, plays a central role in orientation. Inertial guidance systems that control the trajectory of space vehicles include the same basic components: a monitor of displacement based on sensors for linear and angular accel- eration, and a central processor that integrates this information, computing the coordinates of the space position. The central processor also maintains a memory of the trajectory and can therefore make appropriate adjustments in course when necessary.80 Here the similarities of vestibular organs to space vehicle guidance systems end, for they do not explain the com- plex operational capabilities of the brain in support of the sensory function of orientation. The performance of space vehicles is based upon preprogrammed strategies while the brain can resolve even the most unexpected conflicts. For example, the direction of the ves- tibulo-ocular reflex can be reversed (i.e., the eyes will move in the same direction as that of the head instead of in the opposite direction) if one wears glasses with reversing prisms for sev- eral days or even hours.81 Patients with vestibu- lar system disorders can adapt rapidly to per- turbed disequilibrium. The neuroanatomic and
Hoagland H. Impulses from sensory nerves of catfi
Proc Natl Acad Sci USA. 1932;18:701.
Goldberg JM, Fernandez C. Physiology of peripheral neurons innervating semicircular canals of the squirrel monkey. 1. Resting discharge and response to constant angular accelerations. J Neurophysiol. 1971;34:635.
Hunter-Duvar IM, Hinojosa R. Vestibule: sen- sory epithelia. In: Friedmann I, Ballantyne J, eds. Ultrastructural Atlas of the Inner Ear. London: Butterworths; 1984.
Lundberg YW, Zhao X, Yamoah EN. Assembly of the otoconia complex to the macular sensory epithelium of the vestibule. Brain Res. 2006;1091(1):47.
Fernández C, Goldberg JM. Physiology of periph- eral neurons innervating otolith organs of the squirrel monkey. I. Response to static tilts and to long-duration centrifugal force. J Neurophysiol. 1976;39:970.
Blanks RHI, Curthoys IS, Markham CH. Planar relationships of the semicircular canals in man. Acta Otolaryngol (Stockh). 1975;80:185.
Lowenstein O, Wersäll J. A functional interpretation of the electron-microscopic structure of the sensory hairs in the cristae of the elasmobranch raja clavata in terms of directional sensitivity. Nature. 1959;184:1807.
Steinhausen W. Über Sichtbarmachung and Funktionsprüfung der Cupula terminalis in den Bogengangs-ampullen der Labyrinths. Arch Ges Physiol. 1927;217:747.
Dohlman GF. Some practical and theoretical points of labyrinthology. Proc R Soc Med. 1935;28:1371.
Von Békésy G. Experimental models of cochlea with and without nerve supply. In:Rasmussen GL, Windle WF, (eds). Neural Mechanisms of the Auditory and Vestibular System. Springfield, IL: Charles C Thomas; 1960.
Dallos P. Membrane potential and response changes in mammalian cochlear hair cellsduring intracellular
physiologic substrates for this capability are becoming better understood, opening new
recording. J Neurosci. 1985;5:1609.
De Vries H. The mechanics of labyrinth
Otolaryngol (Stockh). 1950;38:262.
otoliths. Acta
Egmond AAJV, Groen JJ, Jongkees LBW. The mecha- nism of the semicircular canal. J Physiol. 1949;110:1.
Lorente de Nó R. Anatomy of the eighth nerve. The central projection of the nerve endings of the internal ear. Laryngoscope. 1933;43:1.
Rasmussen G. The olivary peduncle and other fiber projections of the superior olivary complex. J Comp Neurol. 1946;84:141.
Warr WB. Olivocochlear and vestibulocochlear effer- ent neurons of the feline brain stem: their location, morphology and number determined by retrograde axonal transport and acetylcholinesterase histochem- istry. J Comp Neurol. 1975;161:159.
Brown MC. Morphology of labeled efferent fibers in the guinea pig cochlea. J Comp Neurol. 1987;260:605.
Wolff HG. Efferente Aktivatät in den Statonerven eini- ger Landpulmonaten (Gastropoda). Z Vergl Physiol. 1970;70:401.
Fristsch B. Evolution of the vestibulo-ocular system.
Otolaryngol Head Neck Surg. 1998;119:182.
Graf W, Brunken WJ. Elasmobranch oculomotor organization: anatomical and theoretical aspects of the phylogenetic development of vestibulo-ocular connec- tivity. J Comp Neurol. 1984;227:569.
Straka H, Vibert N, Vidal PP, Moore LE, Dutia MB. Intrinsic membrane properties of vertebrate vestibular neurons: function, development and plasticity. Prog Neurobiol. 2005;76(6):349.
Angelaki DE, Cullen KE. Vestibular system: the many facets of a multimodal sense. Annu Rev Neurosci. 2008;31:125.
Lorente De Nó R. Vestibulo-ocular reflex arc. Arch Neurol Psychiatory. 1933;30:245.
Budelmann BU. Morphological diversity of equi- librium receptor systems in aquatic invertebrates. In: Atema J, Fay RR, Popper AN, Tavolga WN, eds. Sensory Biology of Aquatic Animals. New York: Springer-Verlag; 1988.
Sherrington C. The Integrative Action of the Nervous System. New Haven, CT: Yale University Press; 1906.
Goh Y, Alkon DL. Sensory, interneuronal, and motor interactions within Hermissenda visual pathway. J Neurophysiol. 1984;52:156.
Buttner-Ennever JA. Vestibular oculomotor organi- zation. In: Fuchs AF, Becker W, eds. The Control of Eye Movements. Amsterdam, Netherlands: Elsevier; 1981.
McCrea RA, Strassman A, May E, Highstein SM. Anatomical and physiological characteristics of ves- tibular neurons mediating the horizontal vestibulo- ocular reflex of the squirrel monkey. J Comp Neurol. 1987;264:547.
Baker R, Highstein SM. Vestibular projections to the medial rectus subdivision of oculomotor nucleus. J Neurophysiol. 1978;41:1629.
Evinger LC, Fuchs AF, Baker R. Bilateral lesions of the medial longitudinal fasciculus in monkeys: effects on the horizontal and vertical components of volun- tary and vestibular induced eye movements. Exp Brain Res. 1977;28:1.
Shimazu H, Precht W. Inhibition of central vestibular neurons from the contralateral labyrinth and its medi- ating pathway. J Neurophysiol. 1966;29:467.
Gliddon CM, Darlington CL, Smith PF. GABAergic systems in the vestibular nucleus and their contribution
to vestibular compensation. Prog Neurobiol. 2005; 75:53.
Markham CH. How does the brain generate horizon- tal vestibular nystagmus? In: Baloh RW, Halmagyi GM, eds. Disorders of the Vestibular System. New York: Oxford University Press; 1996: 48.
Uemura T, Cohen B. Effects of vestibular nuclei lesions on vestibulo-ocular reflexes and posture in monkeys. Acta Otolaryngol Suppl (Stockh). 1973;315:1.
Henn V, Hepp K, Buttner-Ennever JA. The pri- mate oculomotor system. II. Premotor system. Hum Neurobiol. 1982;1:87.
Henn V, Lang W, Hepp K, Reisine H. Experimental gaze palsies in monkeys and their relation to human pathology. Brain. 1984;107:619.
Paige GD, Tomko DL. Eye movement responses to linear head motion in the squirrel monkey. II. Visual- vestibular interactions and kinematic considerations. J Neurophysiol. 1991;65:1183.
Angelaki DE. Eyes on target: what neurons must do for the vestibuloocular reflex during linear motion. J Neurophysiol. 2004;92(1):20.
Angelaki DE, Hess BJM. Direction of heading and vestibular control of binocular eye movements. Vision Res. 2001;41:3215.
Westheimer G, Blair M. The ocular tilt reaction— a brain stem oculomotor routine. Invest Ophthalmol. 1975;14:833.
Brandt T, Dieterich M. Pathological eye head coordi- nation in roll: tonic ocular lilt reaction in mesenceph- alic and medullary lesions. Brain. 1987;1(10):649.
Miller EF, II. Counterrolling of the human eye produced by head tilt with respect to gravity. Acta Otolaryngol (Stockh). 1962;54:479.
Magnus R. Some results of studies in the physiology of posture. I. Lancet. 1926;2:531.
Magnus R. Some results of studies in the physiology of posture. II. Lancet. 1926;2:585.
Uchino Y, Sasaki M, Sato H, Bai R, Kawamoto E. Otolith and canal integration on single vestibular neu- rons in cats. Exp Brain Res. 2005;164(3):271.
Mair IWS, Fernandez C. Pathological and functional changes following hemisection of the lateral ampullary nerve. Acta Otolaryngol (Stockh). 1966;62:513.
Dow RS. The effects of unilateral and bilateral laby- rinthectomy in monkey, baboon and chimpanzee. Am J Physiol. 1938;121:392.
Bard P. Postural coordination and locomotion and their central control. In: Bard P, ed. Medical Physiology. 11th ed. Philadelphia: CV Mosby; 1961.
Fulton JF, Liddell EGT, Rioch D. The influence of unilateral destruction of the vestibular nuclei upon posture and knee jerk. Brain. 1930;53:327.
Bach LMN, Magoun HW. The vestibular nuclei as an excitatory mechanism for the cord. J Neurophysiol. 1947;10:331.
Mandich M, Simons CJ, Ritchie S, Schmidt D, Mullett
Motor development, infantile reactions and pos- tural responses of preterm, at-risk infants. Dev Med Child Neurol. 1994;36(5):397.
Wilson TD, Cotter LA, Draper JA et al. Vestibular inputs elicit patterned changes in limb blood flow in conscious cats. J Physiol. 2006;575(pt 2):671.
Kaufmann H, Biaggioni I, Voustianiouk A, et al. Vestibular control of sympathetic activity. An
otolith-sympathetic reflex in humans. Exp Brain Res. 2002;143(4):463.
Balaban CD. Projections from the parabrachial nucleus to the vestibular nuclei: potential substrates for auto- nomic and limbic influences on vestibular responses. Brain Res. 2004;996:126.
Guedry FT. Psychophysics of vestibular sensation. In: Kornhuber HH, ed. Handbook of Sensory Physiology, The Vestibular System, Vol VI, Part 2. New York: Springer-Verlag; 1974.
Walsh EG. Role of the vestibular apparatus in the perception of motion on a parallel swing. J Physiol. 1961;155:506.
Angelica DE, Shaikh AG, Green AM, Dickman JD. Neurons compute internal models of the physical laws of motion. Nature. 2004;430:560.
Britten KH. Mechanisms of self-motion perception.
Annu Rev Neurosci. 2008;31:389.
Ewald J. Physiolgisshe Untersuchungen über das Endorgan des Nervus Octavus. Wiesbaden, Germany: Bergmann; 1892.
Bárány R. Physiologie und Pathologie des Bogengangsapparates beim Menschen. Vienna, Austria: Deuticke; 1907.
Magnus R. Körperstellung. Berlin, Germany: Springer- Verlag; 1924.
Baloh RW. Vestibular neuritis. N Engl J Med. 2003;348:1027.
Curthoys IS. Vestibular compensation and substitu- tion. Curr Opin Neurol. 2000;13:27.
Dieringer N. Activity-related postlesional vestibular reorganization. Ann NY Acad Sci. 2003;1004:50.
Dutia MB. Mechanisms of vestibular compensation. In: Luxon, Davies R, eds. Handbook of Vestibular Rehabilitation. London: Whurr Press; 2005.
Vibert N, Babalian A, Serafin M, Gasc JP, Mühlethaler M, Vidal PP. Plastic changes underlying vestibular
compensation in the guinea-pig persist in isolated, in vitro whole brain preparations. Neuroscience. 1999;93:413.
Vibert N, Bantikyan A, Babalian A, Serafin M, Mühlethaler M,Vidal PP. Post-lesional plasticity in the central nervous system of the guinea-pig: a “top-down” adaptation process? Neuroscience. 1999;94:1.
Vibert N, Beraneck M, Bantikyan A, Vidal P. Vestibular compensation modifies the sensitivity of vestibular neurones to inhibitory amino acids. NeuroReport. 2000;11:1921.
Beraneck M, Idoux E, Uno A, Vidal PP, Moore LE, Vibert N. Unilateral labyrinthectomy modifies the membrane properties of contralesional vestibular neu- rons. J Neurophysiol. 2004;92:1668.
Igarashi M, Levy JK, O-Uchi T Reschke MF. Further study of physical exercise and locomotor balance com- pensation after unilateral labyrinthectomy in squirrel monkeys. Acta Otolaryngol (Stockh). 1981;92:101.
Fetter M, Zee DS, Proctor LR. Effect of lack of vision and of occipital lobectomy upon recovery from unilat- eral labyrinthectomy in rhesus monkey. J Neurophysiol. 1988;59:394.
Lacom M, Xerri C. Vestibular compensation: new per- spectives. In: Flohr H, Precht W, eds. Lesion-Induced Neuronal Plasticity in Sensorimotor Systems. Berlin, Germany: Springer-Verlag; 1984: 240.
Bechterew W. Ergebnisse der Durchschneidung des
acusticus, nebst Erörterung der Bedeutung der semicirculären Canäle für das Körpergleichgewicht. Pfuegers Arch Ges Physiol. 1883;30:312.
Barlow JS. Inertial navigation as a basis for animal navigation. J Theor Biol. 1964;6:76.
Gonshor A, Melvill Jones G. Extreme vestibulo-ocular adaptation induced by prolonged optical reversal of vision. J Physiol (Lond). 1976;256:381.
![]()
TEMPORAL BONE
Facial Nerve
INNER EAR (LABYRINTH)
Phylogeny Structure
Fluid Dynamics Fluid Chemistry Blood Supply Innervation
Embryonic Development
THE HAIR CELL
Morphologic Characteristics Sequence of Hair Cell Activation
Relationship between the Direction of Force and Hair Cell Activation
Mechanism of Hair Cell Activation
Hair Cell Influence on Afferent Nerve Activity Signal Processing at the Hair Cell/Afferent
Nerve Junction
THE INNER EAR VESTIBULAR RECEPTORS
Anatomy of the Semicircular Canals Physiology of the Semicircular Canals Anatomy of the Otolith Organs Physiology of the Otolith Organs PRIMARY VESTIBULAR NEURONS
Anatomy of Primary Neurons Physiology of Primary Neurons EFFERENT VESTIBULAR NEURONS
The ear is divided into three anatomic parts: the external, middle, and inner ear. Except for the auricle and soft tissue portion of the external auditory canal, the ear is enclosed within the temporal bone of the skull.
The temporal bone contributes to the base and lateral wall of the skull and forms part of the middle and posterior fossae.1,2,3 It is divided into four parts: the squamous, tympanic, petrous, and mastoid areas. The petrous por- tion, or pyramid, contains the sense organs of the inner ear. The seventh and eighth cranial nerves enter the petrous portion through the internal auditory canal; the facial nerve exits via the stylomastoid foramen of the mastoid por- tion (Fig. 2–1). The internal carotid artery and internal jugular vein enter the skull through the temporal bone, their bony canals forming part of the anteroinferior wall of the middle ear.
The anatomical proximity of these major ves- sels to the inner ear can explain pulsatile tinni- tus in a patient without vascular abnormalities. A cross section of the temporal bone in Figure 2–2 illustrates the relationship between the three functional parts of the ear. Although the external and middle ear are auditory organs with no direct bearing on vestibular function, a knowledge of their structure, particularly those of the middle ear, is important for under- standing diseases involving the inner ear.4 For example, infection arising in the middle ear can spread directly through its medial wall (oval and round windows) into the inner ear, or it can enter the intracranial cavity by breaking through the roof of the epitympanic recess. The aditus ad antrum interconnects the epitympanic recess with the middle ear by means of air cells throughout the mastoid portion of the temporal bone so that infection beginning in the middle ear can spread to the
25
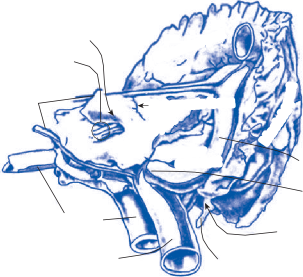
Squamous part
Squamous part
Pore of internal acoustic meatus
Vestibulo acoustic (VIII), facial (VII) nerves
part
part
Mastoid
Mastoid
Subarcuate fossa
Subarcuate fossa
Superior, inferior petrosal sinuses
Petrous part
Petrous part
Vestibular
Vestibular
aqueduct
aqueduct
Transverse sinus
Internal carotid artery
Internal jugular vein
Superior bulb of jugular vein
Stylomastoid foramen
Facial (VII) nerve
Figure 2–1. Medial view of the temporal bone. (From Anson BJ, Donaldson JA. Surgical Anatomy of the Temporal Bone and Ear. WB Saunders, Philadelphia, 1973, with permission.)
vessels and nerves passing through the temporal bone.
Tympanic Membrane
The ear drum, or tympanic membrane, forms a partition between the external and middle ear. The tympanic membrane has a thickness of
0.1 mm and a diameter of 8.5 to 10 mm. It con- sists of three layers, an inner mucosal layer, a middle fibrous layer, and an external epidermal layer. It is attached to the tympanic ring in the external canal at a distance of 2 to 5 mm from the opposite (medial) wall of the middle ear. From the external canal, the tympanic mem- brane appears as a thin, semitransparent disk that normally has a glistening, pearly-gray color

Integument Facial musculature
Squama of temporal bone
Meninges Brain
Auricle
.
.
C
C
.
.
L
L
Cartilages of external ear
Epitympanic recess
Auditory ossicles
Semicircular ducts (in canals)
Endolymphatic duct
(in vestibular aqueduct)
Endolymphatic sac (in dura mater)
Perilymphatic duct
(in cochlear canaliculus) Scala tympani Cochlear duct
Scala vestibuli Mucous membrane
Eustacian tube Levator veli palatini
Secondary tympanic membrane
S
S
E
E
N
N
Vestibule
N
N
I
I
Mastoid air cells
Mastoid process
Tympanic membrane and cavity
Styloid process
Figure 2–2. Cross section of the ear. (From Anson BJ, Donaldson JA. Surgical Anatomy of the Temporal Bone and Ear. WB Saunders, Philadelphia, 1973, with permission.)
The mallear stria (the manubrium shining through the tympanic membrane) passes from slightly inferior and posterior of the center (umbo) toward the superior margin of the tym- panic membrane. Near the superior margin, the mallear prominence is formed by the lat- eral process of the malleus. From the mallear prominence, two folds stretch to the tympanic sulcus of the temporal bone, enclosing the tri- angular area of the pars flaccida, or Shrapnell’s membrane.
Middle Ear
The middle ear, or tympanic cavity, is a flat cleft with a volume of approximately 2.0 cc, containing three tiny bones whose main role is to provide an interface for transmitting to the inner ear the changes in atmospheric pressure produced by sound waves (Fig. 2–3).5 The manubrium is attached, like the radius of a circle, to the inner side of the tympanic mem- brane in a superoanterior direction. Superiorly, the head of the malleus is bound to the incus,
forming the incudomalleal articulation, a type of diarthric joint. The so-called long process of the incus (7 mm), directed down and anteri- orly, is connected to the stapes, the smallest of the three middle ear ossicles. The footplate of the stapes articulates with the walls of the ves- tibule at the oval window to which it is attached by a ring of ligaments. The dimensions of the window are 1.2 by 3 mm, with a total area that is one-seventeenth that of the tympanic mem- brane. Sound-induced displacements of the tympanic membrane and its attached manu- brium are transmitted through the medial arm of the assembly of middle ear bones, acting as a lever to the inner ear; in this fashion the middle ear functions as a mechanical transformer. Additional amplification is produced as the force applied over the surface of the tympanic membrane is funneled into the smaller area of the oval window. The middle ear compensates for the loss of energy—approximately a 99.9% loss—that would occur if sound were transmit- ted directly from air to the fluids of the inner ear.6
The ossicles are suspended by several liga- ments and are dynamically controlled by the action of two muscles. The tensor tympani, innervated by a branch of the trigeminal nerve, is connected by a tendon to the upper part of
Superior malleal ligament
Posterior ligament incus
Lateral malleal ligament
Tympanic membrane pars flaccida (Shrapnell’s membrane)
Tendon tensor tympani muscle
Tympanic mambrane pars tensa
Fabrous annulus
Epitympanic recess
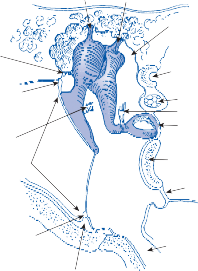
Horizontal semicircular canal
Facial nerve
Tendon stapedius muscle Oval window
Cochlear promintory
Round window
Eustachian tube
Bony annulus
Figure 2–3. Cross section of the middle ear.
the manubrium. Coursing in a lateral direction from the anterior part of the medial wall of the tympanic cavity, this muscle draws the manu- brium medially, tensing the tympanic mem- brane. The stapedius muscle, innervated by the facial nerve, is attached to the posterior wall of the tympanic cavity and is directed anteriorly to anchor in the upper part of the stapes. Contraction of these muscles leads to stiffening of the middle ear system and thus less sound transmitted to the inner ear (i.e., increased impedance) (Fig. 2–3). This is the reason that a lesion of the facial nerve (e.g., Bell’s palsy) can result in hyperacusis on the affected side. Another condition involving these muscles, middle ear myoclonus, results in repetitive clicking sounds in the affected ear.
The medial, or labyrinthine, wall of the mid- dle ear is an irregular surface because of the structures bulging from the inner ear: the promontory of the basal turn of the cochlea and the prominences of the facial canal and horizontal semicircular canal (Fig. 2–3). Beneath the cochlear prominence is the mem- brane of the cochlea or round window, which seals the scala tympani of the cochlea and its fluid from the middle ear. It provides an outlet for equilibrium of pressure in the inner ear whenever sound displaces the stapes. Without this compliance, sound energy could not dis- place the basilar membrane of the cochlea because the endolymph fluid is incompress- ible. The vestibular, or oval, window is located just above the cochlear prominence, where it is closed by the base of the stapes and the annular ligament. These windows between the middle and inner ear can be a route for infection or toxins to spread from the middle to inner ear, or they can rupture, allowing perilymph to leak from the inner ear to the middle ear (perilymph fistula).
Facial Nerve
The facial nerve arises at the inferior border of the pons and proceeds to the internal auditory canal on the superior surface of the cochlear nerve. Within the temporal bone, four portions of the facial nerve can be classified: (1) the canal (meatal) segment (7 to 8 mm), (2) the labyrin-
thine segment (3 to 4 mm), (3) the tympanic
(horizontal) segment (12 to 13 mm), and (4) the mastoid (vertical) segment (15 to 20 mm)
(Fig. 2–4). The canal segment runs in close company in an anterosuperior position with the vestibular and cochlear divisions of the eighth nerve, while in its remaining segments the facial nerve lies separately within a bony canal—the facial or fallopian canal. The laby- rinthine segment runs at nearly a right angle to the petrous pyramid superior to the cochlea and vestibule to reach the geniculate ganglion. At the geniculate ganglion, the nerve takes a sharp turn posteriorly, marking the beginning of the tympanic segment. The horizontal tym- panic segment courses in a posterior direction along the medial wall of the middle ear supe- rior to the oval window and inferior to the hor- izontal semicircular canal (see Fig.2–3). At the sinus tympani, the nerve bends inferiorly, marking the beginning of the vertical, or mas- toid, segment that continues toward the stylo- mastoid foramen. At this level, the facial nerve consists exclusively of motor fibers that inner- vate the muscles of the facial expression after coursing through connective tissue septa in the parotid gland. Three major groups of fibers have been recognized that are directed to (1) the auricular and occipital muscles, (2) the orbicu- laris and muscles of mimetic facial expression, and (3) the buccinator and buccolabial muscles. Three major branches of the facial nerve lie within the temporal bone: (1) the greater superficial petrosal nerve, arising from the geniculate ganglion; (2) the nerve to the stape- dius muscle, arising from the initial part of the mastoid segment; and (3) the chorda tympani, leaving the facial nerve approximately 5 mm above the stylomastoid foramen. The greater superficial petrosal nerve is composed of
(1) parasympathetic efferent fibers originating in the superior salivatory nucleus for innerva- tion of the lacrimal glands and seromucinous glands of the nasal cavity and (2) afferent cuta- neous sensory fibers from parts of the external canal, tympanic membrane, and middle ear, destined for the nucleus of the solitary tract. The nerve to the stapedius muscle and the main facial nerve trunk are motor nerves originating from the facial nucleus in the caudal pons. The chorda tympani, like the greater superficial petrosal, is a mixed nerve containing (1) para- sympathetic efferent fibers from the superior salivatory nucleus, destined for the sublingual glands, and (2) afferent taste fibers from the anterior two-thirds of the tongue, ending in the nucleus of the solitary tract.
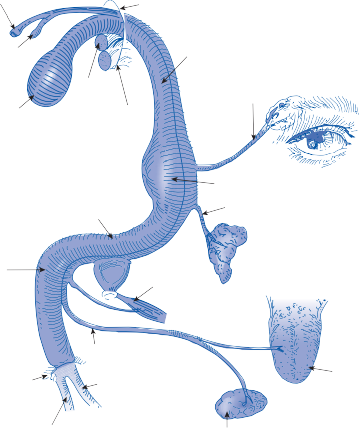
Internal auditory canal
Nucleus of the solitary tract
Facial nucleus
Cochlear nerve
Vestibular nerve
Horizontal (intratympanic segment)
Labyrinthine segment
Greater superficial petrosal nerve for lacrimation
Geniculate ganglion
Lesser superficial petrosal nerve to parotid gland
Lower (posterior) genu
Vertical (mastoid) segment
Stylomastoid foramen
Nerve to stapedius muscle
Chorda tympani Upper division
Tongue
Lower division
Submaxillary salivary gland (salivation)
Figure 2–4. Schematic diagram of the facial nerve within the temporal bone.
Knowledge of the structure and function of each division of the facial nerve allows the cli- nician to localize disease affecting the nerve within the temporal bone.7,8 Lesions in the internal auditory canal commonly involve both the seventh and eighth cranial nerves. Lesions of the labyrinthine segment of the facial nerve above the geniculate ganglion impair ipsilateral
lacrimation (resulting in dry eyes and increased tearing), (2) stapedius reflex activity (resulting in hyperacusis), (3) taste on the ante- rior two-thirds of the tongue, and (4) facial muscular strength. A lesion of the tympanic segment central to the nerve of the stapedius muscle affects only the latter three functions (2–4) listed above, and a lesion of the mastoid segment before the origin of the chorda tympani
affects only the latter two (3, 4). Finally, a lesion at the stylomastoid foramen causes only ipsilat- eral facial muscle weakness or paralysis. Pain is another common symptom particularly with inflammatory disorders (e.g., Bell’s palsy) and is typically postauricular pain with a lesion involving the geniculate ganglion.
Phylogeny
The most primitive gravity-detection organ, the statocyst, appeared more than 600 million years ago in the late Precambrian era.9–11 It is
present in some bygastrulated animals with the most developed Coelenterata, such as jellyfish, allowing the animal to orient itself in relation to the horizon by sensing the direction of the gravitational force of the earth. The statocyst is a fluid-filled invagination or sac containing a calcareous particle, the statolith, or multiple particles, the statoconia, of a density greater than that of the fluid (Fig. 2–5a). Attracted by
gravity, the particles rest their weight differen- tially over cilia protruding from specialized sensory neurons in the wall of the cyst. A large central cilia, the kinocilia, is surrounded by rows of smaller cilia. Tethers between the cilia and the kinocilia open and close mechanosen- sory channels that control the firing rate of the sensory neurons, allowing the animal to regu- late its static position in space.
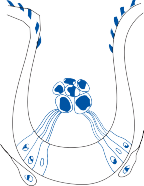
a
S
CB
N N
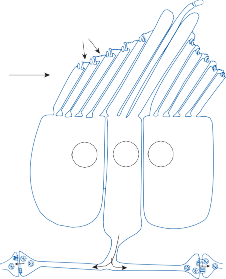
K
b
tl Is
N N N
HC SN HC
Figure 2–5. a: Statocyst of the ctenophore comb jelly Pleurobrachia. Ciliary bundles (CB) of hair cells support the extracel- lular statoliths (S). N, neurons (transverse section). (Adapted from Budelmann BU. Morphological diversity of equilibrium receptor systems in aquatic invertebrates. In: Atema J, et al. (eds). Sensory Biology of Aquatic Animals. Springer-Verlag, New York, 1988, with permission.) b: Hair bundle complex of a sea anemone. Supporting cells located on opposite sides of the sensory neuron (SN) function in a manner comparable that of to hair cells (HC). Large-diameter stereocilia (Is) and the kinocilium (K) of the sensory neuron are also shown. During deflection with a vibratory stimulus, the sensory neuron integrates the input from the supporting cells and then signals other neurons in the nerve net. Note that when the cilia of the hair cell bend toward the kinocilium, the tip links (tl) open ion channels that lead to excitation of the sensory neuron. By contrast, the cilia of the hair cell to the right bend away from the kinocilium and the ion channels remain closed. (Adapted from Watson GM, Mire P. A comparison of hair bundle mechanoreceptors in sea anemones and vertebrate systems. Curr Top Dev Biol. 1999; 43: with permission.)
A primitive receptor organ for generating kinetic reflexes can be found on the tentacles of marine invertebrates, such as sea anemo- nes.12,13 A sensory neuron is coupled to two neighboring hair cells that act as mechanore- ceptors of water pressure waves in their vicinity (Fig. 2–5b). Supporting cells located on oppo- site sides of the sensory neuron function in a manner similar to that of hair cells. During deflection associated with a vibration stimulus, the sensory neuron integrates the input from the supporting cells and signals other neurons in the nerve net. Note that when the cilia of the supporting cell bend toward the kinocilium of the nerve cell, the tethers (tip links) open ion channels whereas when the cilia bend away from the kinocilium the ion channels are closed. Among important signals are vibrations pro- duced by minuscule prey animals. The neuron reaction leads to a “motor” response involving a sensory cell, a surrogate vestibular nucleus, and an effector neuron. The reflex response con- sists of the secretion of a paralyzing substance
by the enmatocyst, the “stinging organelle” of the anemones.
From these simple mechanotransduction receptors to the labyrinth of higher animals, a continuous increment in anatomic complexity occurs that accompanies the phylogenetic evo- lution of the taxa. Next developmentally are the mechanoreceptors of mollusks (e.g., octo- pus, sepia), in which both types of receptors, the static otolith and the kinetic cristae recep- tors are seen.14–17 These new receptors, incor- porated in an invaginated common cavity, accompany the appearance of motor responses to motion, including nystagmus.14 The statocyst cavity, previously open to the outside, is closed and filled by an endogenous secretion (endo- lymph). The otolith/macula system consists of a rounded plate of mechanosensory cells with a compact statolith (Fig. 2–6a). The force exerted by the statolith mass on the cilia of the mecha- nosensory cells depends on the magnitude and direction of any applied linear acceleration including gravity. The cristae/cupula system

(b)
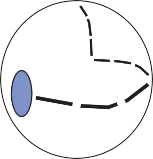
Figure 2–6. Drawing of the octopus statocyst showing the ovoid plate of macula cells and the crista strip which runs around the inside of the statocyst sphere and is divided into nine segments (a). Drawing of an expanded transverse section through one of the crista segments showing the rows of primary sensory hair cells (white), secondary sensory hair cells (light blue) and afferent neurons (dark blue) (b). The direction of movement of the overlying cupula is shown by the arrow. Scale bar in b = 15 micrometers. (From Williamson R, Chrachri A. A model biological neural network: the cephalopod vestibular system. Phil Trans R Soc B. 2007;362:473, with permission.)
Two surviving cyclostomes, the hagfish and the lamprey, demonstrate important steps in the phylogenetic development of the vestibular lab- yrinth. In the hagfish, a simple circular tube is interrupted anteriorly and posteriorly by bul- bous enlargements, the ampullae, each contain- ing a primitive crista (Fig. 2–7a). Between the ampullae, in an intercommunicating channel, lies the macule communis, the forerunner of the utricular and saccular macules. The labyrinth of
the lamprey is more complex, consisting of an anterior and posterior canal communicating with a bilobulated sac containing separate utric- ular and saccular macules (Fig. 2–7b). The pre- decessor of the auditory organs appears after the development of a membranous labyrinth that is divided into two cavities. In the inferior of the two cavities (the saccule), two new receptor areas develop: the lagenar macule and the basi- lar papilla. In crocodiles, however, these recep- tors are contained in a cavity separate from the saccule, while in birds the basilar papilla is a long, uncoiled organ, the predecessor of the coiled cochlea (Fig. 2–7c).18 The basic structure of the three semicircular canals—the utricle, the saccule, and the cochlea—is similar in all mammals (Fig. 2–7d).
Structure
Within the petrous portion of the temporal bone, a series of hollow channels, the bony labyrinth, contain the auditory and vestibular

a
C. post.

b
M. comm.
C. ant.
C. post.
M. negl.
C. ant.
M. utr.
lag. M. sacc.
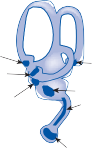

c d
C. ant.
C. post. C. ant.
C. lat.
M. utr.
M. utr.
M. sacc. M. sacc. Pap. bas.
C. post.
M. lag. Cochlea
Figure 2–7. Phylogeny of the labyrinth, (a) myxine; (b) petromyzon; (c) bird; (d) mammal. C. ant., anterior canal; C. lat., lateral or horizontal canal; C. post., posterior canal; M. comm., common macule; M. lag., lagenar macule; M. negl., neglector macule; M. sacc., saccular macule; M. utr., utricular macule; Pap. bas., basilar papilla. (From Wersall DJ, Bagger Sjoback D. Morphology of the vestibular sensor organs. In: Kornhuber, HH (ed). Handbook of Sensory Physiology, Vol VI, Part 2. Springer Verlag, New York, 1974, with permission.)
sensory organs (see Fig. 2–2). The bony laby- rinth consists of an anterior cochlear part and a posterior vestibular part.1 The vestibule is a central chamber (about 4 mm in diameter) marked by the recesses of the utriculus and sacculus (i.e., the macules). The superior and posterolateral walls contain openings for the three semicircular canals, and anteriorly the vestibule is continuous with the scala vestibuli of the snail-shaped cochlea.
Medial to the bony labyrinth is the internal auditory canal, a cul-de-sac housing the sev- enth and eighth cranial nerves and the internal auditory artery. The aperture on the cranial side is located at approximately the center of the posterior face of the pyramid of the tempo- ral bone (see Fig. 2–1). Two other important orifices are in this vicinity. Halfway between the canal and the sigmoid sinus, the slit-like aperture of the vestibular aqueduct contains the endolymphatic sac, a structure important in the exchange of endolymph. The second opening is that of the cochlear aqueduct, at the same level as the auditory canal but on the infe- rior side of the pyramid. The labyrinthine opening of this channel is located in the scala tympani, providing a connection between the
subarachnoid and the perilymphatic spaces. Infection or blood in the cerebrospinal fluid (CSF) can make its way into the inner ear through this channel (see “Fluid Dynamics”).
The membranous labyrinth is enclosed within the channels of the bony labyrinth (Fig. 2–8). A space containing perilymphatic fluid, a sup- portive network of connective tissue, and blood vessels lies between the periostium of the bony labyrinth and the membranous labyrinth; the spaces within the membranous labyrinth contain endolymphatic fluid.
Fluid Dynamics
Perilymph is thought to be a filtrate of CSF and from blood vessels in the ear.19–21As noted pre- viously, the CSF communicates directly with the perilymphatic space through the cochlear aqueduct, a narrow channel 3 to 4 mm long with its inner ear opening at the base of the scala tympani (Fig. 2–8). In most instances, this channel is filled with a loose net of fibrous tis- sue continuous with the arachnoid. The size of the bony canal varies from individual to indi- vidual. Necropsy studies in patients who died

CEREBROSPINAL FLUID
K = 4 mEq/liter
Na = 152 mEq/liter Protein = 20–50 mg%
Endolymphatic sac
CSF
Dura mater
Posterior canal
Horizontal canal
Anterior canal
Cochlear aqueduct Endolymphatic duct
Scala vestibuli PERILYMPH
K+ = 10 mEq/liter Na = 140 mEq/liter
Protein = 200–400 mg% Cochlear duct
Scala tympani
ENDOLYMPH
K+ = 144 mEq/liter Na+ = 5 mEq/liter
Utricle
Saccule Ductus reuniens
Round window
Protein = 126 mg%
Figure 2–8. Cross section of the inner ear.
of subarachnoid hemorrhage or meningitis have revealed free passage of leukocytes and red blood cells into the inner ear in some patients, whereas in others the cells were blocked from passing through the aqueduct.22,23 Blood cells have also been found passing into the internal auditory canal and through the porous canaliculi that contain the vestibular and cochlear nerves, suggesting another route for CSF–perilymph communication. Probably the most important source of perilymph, how- ever, is filtration from blood vessels within the perilymph space, since blocking the cochlear aqueduct does not appear to affect inner ear morphology or function.24,25
The main sites for the production of endolymph are the marginal cells of the stria vascularis of the cochlea and the dark cells of the vestibular labyrinth.19,26,27 Endolymph pro- duction is tightly coupled to K+ secretion.28,29A Na-K-Cl cotransporter expressed in the baso- lateral membrane of marginal and dark cells pumps K+ into these cells to high levels. Potassium channels at the apical surface of the marginal and dark cells allow K+ accumulating in the cells to flow back into the endolymph, thus maintaining the high K+ concentration and the generator potential. In mice, mutations in the genes that code for the Na-K-Cl cotrans- porter protein or the apical K+ channel proteins lead to a failure to produce endolymph and a phenotype of deafness and imbalance.30–32 Cellular water channels, aquaporins, are essen- tial for the fluid regulation of several organs (e.g., kidney, lung, and brain), and aquaporins 1–6 are widely expressed in the inner ear but their role in labyrinthine fluid dynamics is yet to be defined.33
Three theories have been proposed regard- ing the regulation of endolymph volume. The longitudinal, or Guild, theory34 assumes that endolymph is produced in the cochlea and ves- tibular labyrinth and flows toward the endo- lymphatic sac, where it is resorbed. The radial theory assumes a local transverse and active diffusion process between endolymph and per- ilymph.35 The dynamic theory, a combination of the Guild and radial theories, assumes a radial ionic diffusion process and a slow longi- tudinal bulk process.36,37
The pressure of the inner ear fluids has been shown by direct measurements to be different from the atmospheric pressure of the middle ear.38–41 The perilymph and endolymph are
both at an equal positive pressure of approxi-
mately 7 to 10 cm of H2O.38 When the pressure in the intracranial cavity or the labyrinth
increases to above normal, the pressure will tend to equilibrate between the two compart- ments.42 The round window elasticity provides a measure of protection for pressure regulation in the inner ear.43
Destruction of the epithelium lining the endolymphatic sac or occlusion of the duct results in an increase of endolymphatic volume in experimental animals.25,44 The first change is an expansion of cochlear and saccular mem- branes, which may completely fill the perilym- phatic space. The anatomic changes resulting from this experiment are comparable to those found in the temporal bones of patients with Meniere’s syndrome (either idiopathic or sec- ondary to known inflammatory disease).
Fluid Chemistry
The chemical compositions of the fluids filling the inner ear are similar to those of the extra- cellular and intracellular fluids throughout the body. The endolymphatic system contains intracellular-like fluids with a high potassium and low sodium concentration, whereas the perilymphatic fluid resembles the extracellular fluid with a low potassium and high sodium concentration.36,45 Figure 2–8 shows the relationship between electrolytes and protein concentration of the different fluid compart- ments.21,46 The high protein content in the endolymphatic sac, compared with that in the rest of the endolymphatic space, is consistent with the sac’s role in the resorption of endo- lymph. The difference in protein concentration between perilymph and CSF argues against a free communication between the compart- ments of these two fluids and in favor of an active process of perilymph production. The electrolyte composition of the endolymph is critical for normal functioning of the sensory organs bathed in fluid. Rupture of the mem- branous labyrinth in experimental animals causes destruction of the sensory and neural structures at the site of the endolymph– perilymph fistula.47
It is possible to sample the fluid in the vesti- bule by introducing a micropipette through a tiny fistula in the footplate of the stapes.48,49 The fluid obtained normally has the chemical
composition of perilymph given in Figure 2–8. In 29 patients with vestibular schwannomas, the protein content of the perilymph was con- sistently elevated, with an average value of 1800 mg.49 Elevation of perilymph protein can occur when the protein content of CSF is nor- mal or only slightly elevated. The electrolyte composition of perilymph remains normal in such patients. In patients with Meniere’s syn- drome, the markedly dilated sacculus or herni- ated cochlear duct is usually in contact with the footplate, so that endolymph rather than peri- lymph is obtained from tapping the vestibule. The chemical composition of perilymph obtained from other regions of the labyrinth at the time of surgery is normal in patients with Meniere’s syndrome.49
Blood Supply
The labyrinthine artery irrigates the membra- nous labyrinth and its neural structures and does not communicate with arteries in the otic capsule and the tympanic cavity.50 It usually originates from the anteroinferior cerebellar artery (AICA), but occasionally it arises directly from the basilar artery or some of its branches.51 As it enters the temporal bone, it forms branches that irrigate the ganglion cells, nerves, dura, and arachnoidal membranes in the inter- nal auditory canal.52 Shortly after entering the inner ear, the labyrinthine artery divides into two main branches: the common cochlear artery and the anterior vestibular artery (Fig. 2–9A). The common cochlear artery forms two branches: the posterior vestibular artery and the main cochlear artery. The latter enters the central canal of the modiolus, where it gen- erates the radiating arterioles, forming a plexus within the cochlea irrigating the spiral ganglion, the structures in the basilar membrane, and the stria vascularis. The posterior vestibular artery, a branch from the common cochlear artery, is the source of blood supply to the inferior part of the saccule and the ampulla of the posterior semicircular canal. The other primary branch of the labyrinthine artery, the anterior vestibu- lar branch, provides irrigation to the utricle and ampulla of the anterior and horizontal semicir- cular canals as well as some blood to a small portion of the saccule. Thus, different parts of the labyrinth can be selectively damaged by thrombotic or embolic events.
The anterior vestibular vein drains the utri- cle and the ampullae of the anterior and hori- zontal canals; the posterior vestibular vein drains the saccule, the ampulla of the posterior canal, and the basal end of the cochlea (Fig. 2–9B).51,52 The confluence of these veins and the vein of the round window becomes the vestibulocochlear vein. Blood from the cochlea is carried primarily by the common modiolar vein and, when joined by the vestibulocochlear vein, becomes the vein at the cochlear aque- duct. This large venous channel enters a bony canal near the cochlear aqueduct to empty into the inferior petrosal sinus. The semicircular canals are drained by veins that pass toward the utricle and form the vein of the vestibular aque- duct, which accompanies the endolymphatic duct and drains into the lateral venous sinus.
Blood flow (BF) from the arterioles to the venules is determined by the ratio of the driv- ing force (F) to the resistance (R) of the walls such that BF = F/R and the value of F is given by the blood pressure difference between the arterioles and the venules. The value of R includes the wall resistance and any outside pressure acting on the vessel walls. As in other organs, veins in the inner ear have lower R val- ues than those of arterioles with intraluminar pressure of 5–20 cm H2O and will collapse or
expand, depending on the value of F, with
venules operating as effective blood reservoirs. However, when the pressure outside the venules becomes greater than the intravenous pressure, R will increase and there will be a collapse of the walls, with impairment of the blood flow. Experimental and clinical data cor- roborate the possibility of inducing ischemia and damage to the sensory cells in the auditory and vestibular organs in combination or sepa- rately, either by occluding the vessels or increasing intralabyrinthine pressure.
The physiological and anatomical effects of permanent and temporary ischemia on the inner ear by occluding the internal auditory artery have been studied extensively in ani- mals.53–54 Cochlear function is affected within 15–30 sec but can recover even after 5–10 min of complete blood flow obstruction. If the dys- function is of a longer duration, the damage is irreversible and associated with pathological inner ear changes, including sensorineural degeneration and even new bone formation destroying the inner ear spaces. Shorter inter- vals of ischemia produce mixed functional and
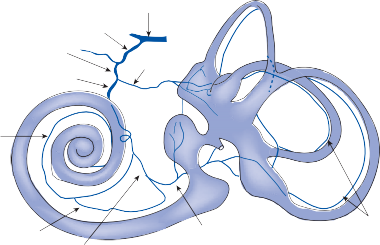
Anterior inferior cerebellar artery
Labyrinthine artery
Common cochlear artery
Basilar artery
Anterior vestibular artery
Main cochlear artery
Cochlear ramus
Vestibulo-cochlear artery
Posterior vestibular artery
Arteries of the canal
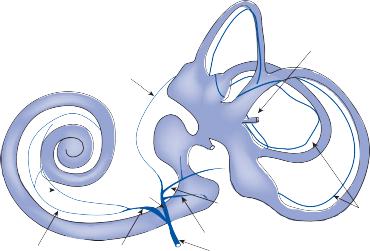
Anterior vestibular vein
Vein of the vestibular aqueduct
Anterior spiral vein
Posterior spiral vein
Vestibulo cochlear vein
Common modiolar vein
Posterior vestibular vein
Vein of the round window Vein at the
cochlear aqueduct
Veins of the canals
Figure 2–9. Arterial (a) and venous (b) labyrinthine circulation. (From Schuknecht HF. Pathology of the Ear. Harvard University Press, Cambridge, 1974, with permission.)
morphological changes. Interfering with endo- lymph circulation (experimental hydrops), and thus increasing inner ear pressure, can impair labyrinthine blood flow.55,56
Innervation
The medial end of the internal auditory canal opens into the cerebellopontine angle cistern;
the lateral end is closed by a thin bony plate, the lamina cribrosa.57 Through tiny perfora- tions in the lamina cribrosa, the afferent and efferent vestibular and cochlear nerve fiber endings pass into the labyrinthine cavity to contact the sensory organs. The lamina crib- rosa is divided into an upper and a lower section by the crista falciformis; each of these halves is in turn divided by vertical bony cristae into an anterior and a posterior section.
The auditory nerve, consisting of approxi- mately 30,000 fibers, occupies the anteroinfe- rior part of the internal auditory canal, and the vestibular nerve, containing approximately 15,000 fibers, occupies the posterior half (both superior and inferior parts).58 The facial nerve is located in the remaining anterosuperior quadrant.
The afferent bipolar ganglion cells of the vestibular nerve (Scarpa’s ganglion) are arranged in two cell masses in a vertical column within the internal auditory canal—the supe- rior group forming the superior division of the vestibular nerve and the inferior forming the inferior division (see Fig. 1–6 in Chapter 1).59,60 The superior division innervates the cristae of the anterior and horizontal canals, the macule of the utricle, and the anterosuperior part of the saccular macule. It leaves the internal audi- tory canal through the posterosuperior fossa of the lamina cribrosa. The inferior division inner- vates the crista of the posterior canal and the main portion of the macule of the saccule and leaves the internal auditory canal through the posteroinferior area of the lamina.
Embryonic Development
Embryonic development of the inner ear largely mirrors the phylogenic development discussed earlier.61–64 In the embryo, the mem- branous labyrinth begins as an ectodermal thickening, the otic placode, on each side of the rhombencephalon (Fig. 2–10). The primi- tive otocyst forms by invagination of the otic placode, which becomes the inner layer of the membranous labyrinth. Three components develop through infolding of the walls of the otocyst: (1) the endolymphatic duct and sac,
the utricle and semicircular canals, and
the saccule and cochlear duct. The walls of the membranous labyrinth consist of an inner layer of ectodermal origin and an outer layer of mesodermal origin separated by a basement membrane. These regions of the inner layer subsequently develop into specialized sensory organs.
The timing of the development of the differ- ent inner ear structures is important to know since developmental defects can occur at each stage of development. The inner ear begins to develop approximately 3 weeks after concep- tion with development of the otic placode.
The placode invaginates into the otic pit, which becomes pinched off to form the otocyst (Fig. 2–10a-c). Concurrent with the placode-otocyst development, the statoacousticofacial ganglion forms from the neural crest at the end of the third week. The geniculate ganglion then migrates away leaving the vestibulocochlear ganglion next to the otocyst.
The otocyst differentiates into the endolym- phatic, vestibular, and cochlear ducts (Fig. 2–10d). By the fifth week, the vestibular duct begins to differentiate into the three semicircular canals and the vestibule (Fig. 2–10e). Shortly after formation of the otocyst the medial part, the endolymphatic diverticulum, separates from the utriculosaccular chamber. This chamber then differentiates into an utric- ular chamber that gives rise to the utricle and semicircular canals and a saccular chamber that gives rise to the saccule and cochlea. The utricular chamber rapidly expands into three diverticula, the centers of which fuse, leaving the spaces around the perimeter to become the three semicircular ducts. The superior canal forms first at about 6 weeks followed rapidly by the posterior and then the horizontal canals. One end of each canal dilates to form the ampulla and both ends remain connected to the utricle.
The cochlear duct becomes separated from the saccule by a narrowing at its dorsal end to form the ductus reunions. The cochlear turns begin to form by the sixth to seventh week, with completion of two and one-half turns by the eighth week. By the end of the fifth month, the primitive organ of Corti has formed within the cochlear duct. The vestibulocochlear gan- glion divides into a superior portion that sends fibers to the utricle and ampullae of the ante- rior and horizontal semicircular canals and into an inferior portion that sends fibers to the sac- cule and the ampulla of the posterior semicir- cular canal. The remainder of the ganglion becomes the spiral ganglion of the cochlea.
The hair cells in the sensory epithelium do not develop until the afferent nerve endings arrive. By the end of the third week a large area of specialized neuroepithelium appears. The upper part of the neuroepithelium becomes the utricular macule and the cristae of the supe- rior and horizontal semicircular canals and the lower part becomes the saccular macule and the crista of the posterior semicircular canal. Vestibular hair cells showing typical synapses
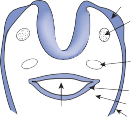
Pharynx
Otic placode
Acoustico facial ganglion
Dorsal aorta
Entoderm Mesoderm Ectoderm
(b)
Hindbrain
Otic pit

Early phase

of otocyst Hindbrain
Differentiation of otocyst (otic vesicle)
Dorsal aorta
Mesenchymal condensation for future ossicles

Endolymphatic duct
Semicircular canals
Vestibular duct
Cochlear duct
Vestibule
Cochlea
Developing eustachian tube
Figure 2–10. Embryological development of the ear: (a) otic placode stage, (b) otic pit stage, (c) otocyst-otic vesicle development, (d) and (e) labyrinthine development. (Adapted from Pearson AA. The development of the ear: A manual. American Academy of Ophthalmology and Otolaryngology, Rochester, MN, 1967.)
with nerve endings are present by 9 weeks. The sensory epithelium is mature in the macules by about 15 weeks, in the cristae by about 23 weeks, and in the organ of Corti by about 25 weeks.
The molecular mechanisms underlying the development of individual sensory organs in the inner ear have largely been worked out in chicks, zebrafish, and mice.65 Differences occur between species, but certain patterns are common. All of the sensory organs develop
from a prosensory region of the otocyst defined by the asymmetric expression of transcription factors. The Notch signaling pathway is key for specifying the prosensory region and for determining whether a cell differentiates into a hair cell or a supporting cell (through the process of lateral inhibition). The level of Notch activation determines whether a cell becomes a hair cell (low), sup- porting cell (high) or a prosensory progenitor cell (intermediate).
Morphologic Characteristics
In the vestibular organs of avians and mam- mals, there are two different types of hair cells—type I and type II (Fig. 2–11, also see Fig. 1–1 in Chapter 1). Type I hair cells are globular and are completely surrounded by a large calyx nerve terminal. The afferent fibers that give rise to these nerve calices are among the largest in the body, measuring more than 20 µm in diameter in some lower animals and 10 µm in humans. Efferent nerves synapse on the outside surface of the calices. Type II hair cells are cylindrical and receive numerous small
synaptic terminals from afferent and efferent neurons. The hair cells are surrounded by sup- porting cells whose top surface is covered with microvilli. The supporting cells extend the whole length of the sensory neuroepithelium from the basal membrane to the surface. Their nuclei line up in a row immediately above the basal membrane (Fig. 2–11A,B). By contrast, the nuclei of the hair cells are midway between the basal membrane and the luminar surface. This pattern of nuclear organization is similar throughout all vertebrates. Supporting cells can differentiate into new hair cells following destruction of the sensory epithelium. This was initially seen in the cochlea of quail and chicken after acoustic trauma66,67 and then in the cochlea
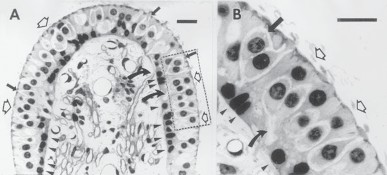
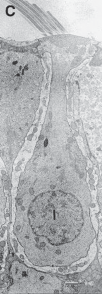
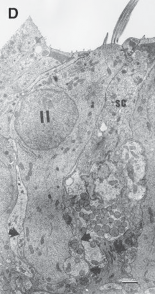
Figure 2–11. Mammalian hair cells. (A, B) Photomicrographs of chinchilla crista (cross section). Long arrows, type I hair cells; open arrows, type II hair cells; arrow heads, supporting cells, curved arrows, afferent nerve fibers. B is an enlargement of the box outlined in A. Bar, 10 µm. (C, D) Electron micrographs of type I and type II hair cells from the chinchilla. Type I hair cells are surrounded by the chalice ending of an afferent nerve fiber, whereas type II hair cells are contacted by afferent nerve boutons (arrows). SC, supporting cell. Bar = 1 µm.
and vestibular labyrinth of mammals after drug ototoxic exposure.68,69
TIP-LINKS
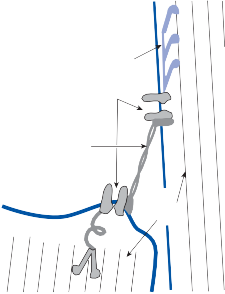
The tips of the cilia are connected by extracel- lular linkages called tip-links.70–72 These struc- tures are already seen in primitive aquatic animals. The basic elements of a tip link include an ion channel, an interconnecting tether, and a motor protein (Fig. 2–12). The motor protein moves along actin filaments and is critical for adaptation to prolonged stimuli.73 Mechano- sensory ion channels are already present in single-cell organisms where they function to prevent osmotic shock as the bacterium moved from salt to fresh water.74,75 These primitive mechanosensory channels have an iris-like opening that enlarges with tension acting in the plane of the cell membrane. The next major development is a tether that couples the chan- nel to intracellular or extracellular structures so that stretch on the tether opens the channel. The tethers are attached to stiff elements within the cell such as microtubules or actin filaments or outside the cell to protruding parts
Motor proteins Mechanosensory
channels
Tip link
Actin filaments
Motor proteins Mechanosensory
channels
Tip link
Actin filaments
Figure 2–12. Schematic model of hair cell transduction. Shearing with positive deflection increases tension in tip links, which pull open a transduction channel at each end. Myosin motors slip or climb on actin filaments to restore resting tension. An elastic gating spring likely exists between a channel and the actin cytoskeleton. (Adapted from Vollrath MA, Kwan KY, Corey DP. The microma- chinery of mechanotransduction in hair cells. Ann Rev Neurosci. 2007;30:339, with permission.)
of the same or other cells or the extracellular matrix.70,71 Many of the genes and proteins associated with mechanosensory transduction in the vestibular organs of invertebrates and vertebrates have been identified and some are highly conserved, particularly those coding for proteins in the tethers.70,75 Cadherins and pro- tocadherins form part of the tethers from the most primitive metazoan phyla to vertebrates, including primates. Mutations in the rare tip- link motor protein, myosin VIIa, cause vestibu- lar and auditory loss in humans and flies. Channel genes are less conserved, and so far the gene for the putative mechanosensory channel in vertebrates has not been identified. The transient receptor potential (TRP) super- family of mechanosensory channels is critical for hearing in the fruit fly and touch in the nematode but not for hearing or vestibular function in mammals.76
RIBBON SYNAPSE
The basal portion of the hair cells makes contact with afferent nerve terminals by way of ribbon synapses (see Fig. 1–1 in Chapter 1).77,78 These structures that are remarkably efficient in con- verting Ca2+ influx into neurotransmitter release are only seen in the inner ear and eye.79 There are approximately 10 to 20 synapses per hair cell, and each synapse contains 100 to 200 synaptic vesi- cles. Of these about 15 vesicles are docked beneath the ribbon ready to release their con- tents into the extracellular space. Within each receptor central hair cells of both types have more synaptic ribbons than do peripheral hair cells. 80
Sequence of Hair Cell Activation
Bending of the hair bundle toward the kinocil- ium opens the mechanically gated ion channels, causing an influx of potassium. The resting potential of the hair cells is between –50 and
–70 millivolts and as the potential rises above
–50 millivolts, voltage-gated calcium channels at the base open, allowing an influx of calcium ions. The calcium binds to a calcium sensor pro- tein in the ribbon synapse (otoferin in cochlear hair cells), activating the SNARE complex of proteins releasing packets of glutamate into the synaptic cleft.77,81 AMPA receptors in the affer- ent nerve terminals are activated, leading to an approximate linear relationship between Ca2+ influx and afferent nerve firing.82 A glutamate
transporter removes glutamate from the synap- tic cleft.83 About 13% of the transduction chan- nels are open at rest, resulting in a resting current flow through the hair cells and a resting firing rate of the afferent nerves (see Hair Cell Influence on Afferent Nerve Activity).84
Relationship between the Direction of Force and Hair Cell Activation
The adequate stimulus for hair cell activation is a force acting parallel to the top of the cell, resulting in bending of the hairs (a shearing force).85 A force applied perpendicular to the cell surface (a compressional force) is ineffec- tive in stimulating the hair cell.86,87 The stimulus is maximal when the force is directed along an axis that bisects the bundle of stereocilia and passes through the kinocilium (see Fig. 1–1 inset in Chapter 1). Deflection of the hairs toward the kinocilium decreases the resting membrane potential of the sensory cells (depolarization). Bending in the opposite direction produces the reverse effect (hyperpolarization).88 The effect is minimal when hair deflection is perpendicu- lar to the axis of maximal excitation.
Mechanism of Hair Cell Activation
The top surface of hair cells in mammals faces the endolymph, a fluid rich in K+ (like the intra-
cellular space), while the basolateral membrane is surrounded by perilymph rich in Na+ (like the extracellular space). In the cochlea, the peri- lymph is at zero voltage in relation to the rest of the extracellular space of the body, but the endolymphatic space has a positive potential (+80 mV). In the vestibular labyrinth, the posi- tive potential is smaller (+5 to 10 mV). The hair-bearing surface of the cell membrane is morphologically different from the rest, being thicker and more electron dense. This part of the cell membrane is depolarized because of the equal K+ concentration outside of the cell in the endolymph and inside of the cell. It acts as a passive resistor between the inside and outside of the hair cell whose value is modulated by the mechanical deformation associated with the dis- placement of the hairs (Fig. 2–13). This concept represents the essence of the Davis mechano- electric theory of hair cell function.89–93 Because of the electrical gradient across the luminal (top) part of the membrane, a current flow exists from the endolymph to the inside of the cell and out, through the basolateral membrane, which is known as the “current of silence,” a resting cur- rent. The basic concept of the Davis theory has been upheld by demonstration of transmembrane potential changes and associated impedance modulation during deflection of the stereocilia. Intracellular recordings from hair cells of amphibians and mammals have expanded our knowledge of the mechanoelectric transduction process.92,93 When hair cells are stimulated,
![]()
Hair cell displacement
![]()
Hair cell resistance
![]()

+
Endolymph voltage
–
![]()
Hair cell current
![]()
VIIIth nerve activity
Figure 2–13. Mechanism of hair cell activation. Sinusoidal displacement of the stereocilia produces a sinusoidal modula- tion or the vestibular nerve firing rate. See text for details.
there is a change in the electric current begin- ning at the tips and the lumen of the stereocilia to the inside of the cell. This “transduction” cur- rent causes a series of additional changes in the permeability of different ion channels in the basolateral membrane, leading to depolariza- tion of the hair cell membrane and release of neurotransmitters (see earlier discussion). There is a diversity of ion channels expressed in the hair cells.94–96 For example, type I hair cells express a K+ channel that results in an unusually low input resistance compared to that expressed by type II hair cells.97
Measurement, with intracellular electrodes, of the hair cell responses to cilia deflection shows that the curve relating the receptors’ potential to the stimulus has greater sensitivity and linearity for small signals.98,99 Larger stimuli exhibit saturation or nonlinearity that is greater for hyperpolarizing than for depolarizing stim- uli, leading to smaller responses, hence lower gains for deflection away from the kinocilium (Fig. 2–14). The voltage drop produced in the vicinity of the hair cells by the changing current is known as the microphonic potential, the so- called generator potential of these receptor organs.89 It is maximal at the tips of the hairs.91,99
Flexion
In contrast to nerve action potentials, the gen- erator potentials have no refractory period (fol- lowing the frequency of the stimulation above several thousand hertz), are highly resistant to anoxia, and may remain partially active after the animal’s death. The electric current associ- ated with the generator potentials acts upon the synaptic contacts between hair cells and nerve terminals by activating chemical trans- mitters to modulate the firing of action poten- tials by the afferent neurons (Fig. 2–13).
% Full responce
% Full responce
Hair cells are not passive elements; they actively participate in the mechanotransduction process.100 In particular, outer cochlear hair cells, which contain several contractile pro- teins,101 vary their length under direct electrical stimulation.102,103 This electromotility of the outer hair cells is dependent on a protein, prestin, isolated with comparative cDNA analysis of the inner and outer hair cells.104 Recombinant prestin introduced into cultured kidney cells provides them with contractile properties normally not present. Presumably, during acoustic stimulation, prestin experiences an electric charge realignment that results in morphological changes in the shape of the outer hair cells, elongating during hyperpolarization and contracting during depolarization. These conformational changes would influence the
10°
Responce (mV)
Responce (mV)
6
4
2 0
2
0 10°
100
0
displacement of the basilar membrane in a pos- itive feedback configuration facilitating the physiological stimulation of inner hair cells, in essence acting as the amplifier of the acoustic energy entering the ear.105 In vestibular hair cells, the stereocilia contain actin molecules and can carry out flagella-type movement.97,106,107 The cilia length varies among hair cells and location, but it is logical to expect that anatomic differences in stereocilia will result in differ- ences in the process of transducing head-motion
1.0 0 1.0
Displacement (m)
Figure 2–14. Intracellular voltage changes (mV) associ- ated with displacement of cilia of a hair cell from the frog saccule. Cilia bending toward the tallest stereocilia produce a positive depolarizing change whereas motion in the oppo- site direction results in a negative hyperpolarizing change. Note that the curve relating the receptor potential to the degree of deflection (µm) has the greatest sensitivity and linearity for small deflections and exhibits a saturation non- linearity for large displacements that is greater for hyper- polarizing than depolarizing stimuli. (From Hudspeth AJ, Corey DP. Sensitivity, polarity, and conductive change in response of vertebrate hair cells to controlled mechanical stimuli. Proc Natl Acad Sci USA. 1977; 74:2407–2411 with permission.)
information into neural signals. Although not proven, it is possible that hair cells at the periph- ery of the vestibular organs actively pull the cupula or otolithic membrane to influence the response of the more centrally placed hair cells, analogous to the effect of cochlear outer hair cells on inner hair cell walls.
Hair Cell Influence on Afferent Nerve Activity
One of the most significant findings concerning hair cell function was the discovery by Hoagland
in 1932 that the afferent nerves from lateral-line organs of fish generated continuous spontaneous activity.108 This observation has subsequently been confirmed in all other hair cell systems and represents a fundamental discovery in sensory physiology. As noted earlier, baseline current flow through the hair cells generates the sponta- neous activity and depolarization and hyperpo- larization of the hair cells’ membrane potential modulates the spontaneous activity. Bending of the hairs toward the kinocilium results in an increase of the spontaneous firing rate, and bending of the hairs away from the kinocilium results in a decrease of the firing rate.109 The spontaneous firing rate varies in different animal species and in different sensory receptors. It is thought to be greatest in the afferent neurons of the semicircular canals of mammals (up to 90 spikes/sec) and lowest in some of the acoustic nerve fibers innervating mammalian cochlear hair cells (1 to 2 spikes/sec).110,111 Given the non- linear behavior of the hair cell transduction mechanism, it is not surprising that the modula- tion of the spontaneous neuronal firing rate is likewise nonlinear. Responses to excitatory stim- uli are more than those to inhibitory stimuli. This asymmetry in response is of great physiological and clinical significance, as will be shown later.
Signal Processing at the Hair Cell/ Afferent Nerve Junction
The hair cell is a relatively simple force transducer mirroring the biomechanics of the forces acting on the surrounding tissues. Yet complex signals originate from the afferent nerves at the base. Signal processing must be interposed between the hair cell and the afferent nerve to account for the wide range of afferent nerve responses.112,113 This signal processing can be traced to at least four different processes: (1) neurotransmitters released by the hair cells, (2) neurotransmitters released by efferent terminals, (3) adaptation at the ribbon synapse, and (4) a diversity of receptors and ion channels in the afferent terminals. Glutamate is the main neurotransmitter at the hair cell–afferent nerve junction, but other trans- mitters, including gamma-aminobuteric acid (GABA), are also released. Hair cells in different receptors and in different locations within the receptors release different combinations of trans- mitters. Acetylcholine (Ach), released by the efferent system, modulates afferent nerve firing
through both presynaptic and postsynaptic mech- anisms. The number of vesicles (both total and docked) at the ribbon synapse can be up and down regulated based on a number of factors, including synaptic activity.78 The afferent nerve terminals express AMPA, NMDA, and GABA type B recep- tors along with a wide range of ion channels, all of which can be up and down regulated. For exam- ple, the number of AMPA receptors (GLuR 2-4) expressed depends on the amount of glutamate released and on NMDA receptor activity.114
THE INNER EAR VESTIBULAR RECEPTORS
The vestibular system monitors the forces asso- ciated with angular and linear accelerations of the head by means of five organs located within the labyrinthine cavities of the temporal bones on each side of the skull.115 The saccular and utricular macules sense linear acceleration, and the cristae of the three semicircular canals sense angular acceleration of the head.
Anatomy of the Semicircular Canals
The semicircular canals are three membranous tubes with a cross-sectional diameter of 0.4 mm; each one forms about two-thirds of a circle with a diameter of about 6.5 mm.116 They are aligned to form a coordinate system (see Fig. 1–4c in Chapter 1).117,118 The plane of the horizontal semicircular canal with two openings on the lateral wall of the utriculus makes a 30-degree angle with the horizontal plane. The other two canals are in vertical positions almost orthogo- nal to each other. The anterior canal is directed medially and laterally over the roof of the utric- ulus, and the posterior canal is directed down- ward and laterally behind the utriculus. The two vertical canals share a common opening on the posterior side of the utriculus. Precise mea- surement of the planes of the canals, however, indicates that they are not aligned perfectly orthogonal. All angular movements stimulate at least two canals and often all three.
CRISTA
At the anterior opening of the horizontal and anterior canal and the inferior opening of the
posterior canal, each tube enlarges to form the ampulla. A crest-like septum, the crista, crosses each ampulla in a perpendicular direction to the longitudinal axis of the canal (see Fig. 1–4a,b in Chapter 1). It rests on the bone of the canal and consists of sensory epithelium lying on a mound of connective tissue, where blood vessels and nerve fibers reach the sen- sory receptor area. In the human vestibular organ, there are approximately 23,000 hair cells (type I and type II) in the three cristae and about 52,000 in the two macules.119,120 The number of neurons innervating the three cris- tae is approximately 5700 and the two macules, approximately 8600, for a total of approximately 14,300 nerve fibers.121 In the chinchilla, for which more accurate measurements are avail- able, the number of hair cells (type I and type
II) in the crista of the horizontal semicircular canal is about the same as the number of sup- porting cells (about 5000 of each). In the mon- key crista, type I hair cells outnumber type II hair cells by almost 3:1 with the ratio being
>4:1 in the central zone and <2:1 in the periph- eral zone.122 In the chinchilla crista the ratio of type I to type II hair cells is near 1:1 through- out.122
CUPULA
The cupula is composed of a mixture of glycoproteins and proteoglycans secreted into the endolymph by specialized endothelial cells.123 It is composed of densely packed filaments 40–60 nm in diameter cross bridged by thinner filaments. Between the crista and the cupula a subcupular meshwork is composed of long branching filaments (50–70 nm in diameter) oriented parallel to the main axis of the stereocilia. The subcupular filaments are connected to the cupular filaments on one side and to the sensory epithelium on the other side. The cupula extends all the way to the roof of the ampulla. The tips of the stereocilia are tightly connected to the subcupular meshwork and sometimes the longest stereocilia or the kinocilia protrudes into the cupula and is connected to the cupular filaments. The subcupular meshwork may help transmit the shearing force of the cupula to the ciliary bundle and dampen unwanted vibrations.
Physiology of the Semicircular Canals
HISTORICAL BACKGROUND
The functional role of the semicircular canals was first linked to their gross anatomic features by Flourens in 1842.124 While studying the audi- tory labyrinth in pigeons, he noted that opening a semicircular canal resulted in characteristic head movements in the plane of that canal. Several subsequent investigators proposed that movement of endolymphatic fluid within the canal was responsible for excitation of the cris- tae.125–127 It was not until the studies of Ewald in 1892, however, that a clear relationship was established between the planes of the semicir- cular canals, the direction of endolymph flow, and the direction of induced eye and head movements.128 Exposing the membranous laby- rinth of the semicircular canals of pigeons, Ewald applied positive and negative pressure to each canal membrane to cause ampullopetal (toward the ampulla) and ampullofugal (away from the ampulla) endolymph flow. Three important observations, which became known as Ewald’s laws, were (1) the eye and head movements always occurred in the plane of the canal being stimulated and in the direction of endolymph flow, (2) ampullopetal endolymph flow in the horizontal canal caused a greater response (i.e., induced movements) than did ampullofugal endolymph flow, and (3) ampull- ofugal endolymph flow in the vertical canals caused a greater response than did ampullopetal endolymph flow.
Steinhausen129 and later Dohlman130 visual- ized the movement of the cupula during endo- lymph flow. By injecting India ink into the semicircular canals of fish, these investigators demonstrated that the cupula formed a seal with the ampullary wall and moved with the endo- lymph. Steinhausen, noticing the similarity between the cupular movement and that of a pendulum in a viscous medium, proposed a model for the description of cupular kinematics that became known as the pendulum model. Although the large movements observed by Steinhausen were later realized to be artifactual, the basic principle has been upheld by most experimental131 and theoretical studies.132,133
Physiologic verification of the model has been made by detailed study of the relationship
between angular head acceleration and the flow of action potentials in isolated ampullary nerve fibers. These studies were first conducted in elasmobranchs by Lowenstein and Sand,134 later in frogs,135,136 pigeons,137 and mam- mals,138–141 and first in primates by Goldberg and Fernández.111,142,143
cupula–endolymph system can be obtained if the values of these coefficients are known.
For natural to-and-fro head movements, the magnitude of the elastic and inertial forces is negligible and the following simplified equation describes the kinematics of the cupula system:
![]()
PENDULUM MODEL
C![]() (t)
(t)
[2]
The pendulum model is the most useful didac- tic model for describing the physiologic prop- erties of the semicircular canals and, as will be shown later, for describing the semicircular canal-induced reflexes, especially the vestib- ulo-oculomotor reflexes.132,144
The cupula acts as the coupler between the force due to angular acceleration of the head and the hair cells (the transducer of mechanical to biological energy), leading to the production of action potentials in the vestibular afferent fibers. Because of the configuration and dimen- sions of the canals, the endolymph can move in only one direction along the cylindrical canali- cular cavity. According to Newton’s third prin- ciple, when an angular acceleration [and hence
a force M¨ h(t)] is applied to the head, displace- ment of the cupula–endolymph system acting
as a solid mass is opposed by three restraining forces: (1) an elastic force [Kc (t)] due to the cupula’s spring-like properties (which is pro-
portional to the magnitude of its displacement),
the force due to the cupula–endolymph vis-
The force applied to the cupula–endolymph system during angular head acceleration is opposed mostly by the viscous drag of the cup- ula. Integrating Equation 2 we have
![]()
![]()
![]()
h
h
M ( [3]
Thus, the displacement of the cupula system during natural head movements is proportional to the velocity of head motion rather than head acceleration. The magnitude of the proportion- ality constant (M/C) relating angular deviation of the cupula in degrees to the velocity of the head in degrees per second has been estimated to be approximately 0.003 sec, based on the physical characteristics of the canals and endo- lymph.145 Most likely, during fast head move- ments with velocities as great as 800 degrees / sec, the deviation of the cupula does not exceed 1degree of deflection.120
Figure 2–15 illustrates the relationship between the time course of head acceleration, head velocity, and cupular displacement as
c
c
cosity [C˙ (t)] (whose magnitude is propor-
predicted by the pendulum model for three
tional to the velocity of its displacement), and
an inertial force [M¨ c(t)] due to the fluid’s mass (proportional to the acceleration of the
fluid–cupula complex). Cupular displacement can be described by the following equation, which is referred to as the equation of the pen- dulum model of semicircular canal function:
different types of angular rotation commonly used in clinical testing. The description of cup- ular displacement during constant angular acceleration (Fig. 2–15a) can easily be derived from Equation 1. At the beginning of head acceleration, endolymph movement lags behind the displacement of the head and thereby that of the walls of the semicircular
![]()
![]()
![]()
M t ![]() K (t) M (t) [1]
K (t) M (t) [1]
canals. After a few seconds, however, a balance is established between the applied and restrain-
where c is the angular displacement of the cupula–endolymph system with respect to the
ing forces, and the endolymph moves simulta- neously with the walls of the labyrinth. At this
c
c
wall of the canals, ˙ and ¨ are the first (veloc-
c
time the position of the ring of fluid within the
ity) and second (acceleration) time derivatives of the cupular displacement, and ¨ h is the angular acceleration of the head. M is the
moment of inertia; C, the moment of viscous friction; and K, the moment of elasticity. A complete description of the kinematics of the
canal and therefore the position of the cupula
c(t) differ from the initial conditions, having been displaced by a certain amount in the
direction of the force. The magnitude of the displacement can easily be calculated. Once the endolymph is stationary, the cupula
Stimulus
Stimulus
0
Constant acceleration stimulus
![]()
Acceleration
Velocity
Cupula displacement
Cupula displacement
100%
63
37
0
Stimulus
Stimulus
100%
T1 T1

![]()
Impulse stimulus
Acceleration
0
Velocity

Cupula displacement
Cupula displacement
100%
37
0
![]()
T1 Time

Stimulus
Stimulus
max
0
max
Sinusoidal stimulus Acceleration
Velocity

Cupula displacement
Cupula displacement
100%
0
100%
Figure 2–15. Relationship between cupular displacement and three types of angular acceleration of the head (a–c) as predicted by the pendulum model.
velocity ˙ (t) and its acceleration ¨ (t) in
The relationships embodied in Equations 3
c c
relation to the walls are zero and, consequently, the terms for viscous and inertial restrain- ing forces vanish in Equation 1, which now reduces to
![]()
K![]() (t)
(t)
or
and 4 are two of the fundamental concepts of cupular function. To restate them: the maxi- mum deviation of the cupula increases propor- tionally to the magnitude of head velocity during sinusoidal head rotations at the frequen- cies of natural head movements and to the magnitude of head acceleration during rotation with constant angular acceleration.
![]()
![]()
![]()
h
h
M (t [4]
That is, the final displacement of the cupula depends on a proportionality constant and on the magnitude of the constant angular acceleration.
Cupula displacement after a constant angu- lar acceleration stimulus follows an exponential time course (Fig. 2–15a) that can be deter- mined by a more detailed mathematical treat- ment of Equation 1. Sixty-three percent of the total cupular deviation, regardless of its final value, always takes place after a fixed delay
determined by what is known as the long time
constant (T1) of the system. The subsequent deviation of the cupula increases at the same
rate (63% of the remainder every T1 seconds), so that 95% of the final deviation will take place
after approximately 3T1 sec. The magnitude of the time constant depends on the viscous and
elastic coefficients: T1 = C/K. That is, the time the cupula takes to reach a maximum deviation
is directly proportional to the viscosity of the endolymph and inversely proportional to the elasticity of the cupula. T1 cannot be measured
directly, but it has been estimated to be
about 7 sec, based on the average response of primary afferent neurons in the squirrel monkey.111,142,143
According to the pendulum model, not only is the initial deviation of the cupula related to the constant acceleration stimulus, but after the stimulus is terminated the cupula returns to the resting position with the same exponential time course. It was precisely the observation by Steinhausen119 of the slow exponential-like return of the cupula to the resting position after it had been deviated that led to the formulation of the pendulum model.
The cupular displacement following a brief impulse of angular acceleration is given in Figure 2–15b. This type of angular accelera- tion, although the least natural, is of great value in clinical vestibular testing. An impulse of acceleration is generated by changing the
the cosine function at that instant in the cycle of motion. Since this value oscillates between
+ 1 and −1, the head velocity ranges between
+A and −A. These relationships are felt to apply for sinusoidal rotations between 0.1 and
4.0 Hz and therefore cover the range of most natural to-and-fro head movements.118
Anatomy of the Otolith Organs
MACULES
The two otolith organs consist of a sensory area (i.e., the macules) and a surface area (i.e., the otolithic membrane), which are located in the middle chamber of the inner ear (i.e., the vesti- bule). The vestibule is oval in shape, connecting to the membranous semicircular canals via five openings. The saccule lies on the medial wall of the vestibule in a spherical recess inferior to the utricle with which it is in contact but without direct connection.115 It communicates with the endolymphatic duct (and thus the utricular duct) by the saccular duct and with the cochlea by the ductus reuniens (see Fig. 2–8). The mac- ule of the saccule is a differentiated patch of membrane in the medial wall, is hood shaped, and lies predominantly in a vertical position (see Fig. 1-3c in Chapter 1). The macule of the utricle is located next to the anterior opening of the horizontal semicircular canal and lies mostly
velocity of the head (˙
acceleration possible.
h) with the maximum
maximum deviation
in a horizontal position in a recess on the anterior
part of the utricle. The utricle communicates by
The
of the cupula takes almost immediately
the utricular duct with the endolymphatic duct
place
and is proportional
magnitude of the
at the same level as, but by different openings
instantaneous
to the
head velocity
from, those of the saccular duct. Thus, the
c
c
h
h
(t) ˙ .
change in
Of particular note, the cupular
endolymph in the superior or utricular part of the labyrinth is separated from that of the sac-
deviation thereafter decays exponentially with
the same time constant to return to 63% of the maximum deviation.
The sinusoidal rotation in Figure 2–15c most closely resembles natural head movements because movement in one direction is followed by movement in the opposite direction. Most natural head movements can be broken down into a series of sine waves with different fre- quencies and amplitudes. According to Equation 3, the cupular displacement c(t) is
given by A cos t (the differential of head dis-
placement A sin t), where is the radian fre- quency (2 f) of head rotation and A is the angular head displacement. The head velocity at a given time t is proportional to the value of
cule and cochlea by these tiny ducts.
PRODUCTION AND MAINTENANCE OF OTOCONIA
Otoconia are complex calcium carbonate (CaCO3) biominerals that serve an important
role in both normal and abnormal vestibular
function (also see Chapter 10). Otoliths is another word for otoconia. Thousands of tiny calcium carbonate crystallites (0.1 to 25 micrometers) surround a glycoprotein/proteo- glycan core that is bound to the underlying amorphous gelatinous layer by fibrous proteins (Fig. 2–16, also see Fig. 1–3 in Chapter 1).147,148
![]()
Otogelin
-tectorin
-tectorin Otoancorin
Otopetrin 1
NOXA 1
NOX 3
tc hc sc
Gelatinous layer
Subcupular meshwork
Macula
Hair cells (hc) Support cells (sc) Transitional cells (tc)
Basement membrane
Figure 2–16. Schematic representation of the utricular macule. The macule is composed of sensory hair cells (hc) and sup- porting cells (sc); transitional cells (tc) border the edge of the macules. Directly above the macule, the otoconial membrane is composed of the subcupular meshwork, a fibrillar structure which rings the stereociliary projections of each hair cells, and the gelatinous membrane which is amorphous. Otoconia, CaCO3 biominerals precipitated around a proteinaceous core, are embedded in the gelatinous membrane and maintained in place by strands of noncollagenous extracellular matrix proteins that resemble beads on a string. Proteins that have been identified to influence the activity or structure of each of these layers are listed on the left. (Adapted from: Hughes I, Thalmann I, Thalmann R, Ornitz DM. Mixing model systems: Using zebrafish and mouse inner ear mutants and other organ systems to unravel the mystery of otoconial development. Brain Res. 2006;1091:58 with permission.)
Between the gelatinous layer and the sensory epithelium a dense reticular network of fibril- lar proteins ring the stereocillary projections of the hair cells. In the mouse, otoconia form over the sensory epithelium of the macules when core proteins (mostly Otoconin 90) coalesce into distinct structures at approximately embry- onic day 14.149 Calcification of the protein structures rapidly occurs over the next few embryonic days. Normal production of the otoconial membrane requires an ordered sequence of events, including localized pro- duction and export of otoconial matrix proteins, assembly of the protein core from free-floating matrix proteins, and locally increasing Ca2+ and CO3– concentrations. Inhibitors of calcification
in other areas prevent more generalized calcifi-
cation.
3
3
The rapid deposition of calcium carbonate on the protein matrix requires adequate Ca2+ and CO – ions. The plasma membrane calcium ATPase 2 (PMCA2) is the primary calcium pump for maintaining endolymph Ca2+ levels and carbonic anhydrase generates CO –.150,151
otoconia and may have an increased risk of developing benign positional vertigo.153
Much of the information regarding the genes and proteins critical for otoconia production and maintenance have been determined through the study of mutant mice and zebrafish.148 Many of these animals were first identified as having a possible vestibular disor- der after observing behavioral features that might suggest inner ear dysfunction (e.g., dancer, backstroke, head tilt, twirler). Knockout models have also provided insight into impor- tant proteins for otoconia production. Interestingly, despite the numerous animal models with absent or abnormal otoconia, so far no human counterparts have been found. Abnormal or reduced otoconia have been pro- duced in humans and experimental animals by a range of pharmacological agents, includ- ing aminoglycosides, phenytoin, carbonic anhydrase inhibitors, prostaglandins, and ethacrynic acid.154–159 It appears that mainte- nance of normal otoconia requires the mainte- nance of normal hair cells. Animal models that
3
Once formed there is normally a low rate of
lose hair cells with aging also lose otoconia.148
turnover of otoconial calcium in adult mice.152 Like bone, otoconia mineralization and turn- over may be sensitive to hypo and hypercalce- mia. Patients with osteoporosis have abnormal
Aminoglycosides damage hair cells and lead to otoconial degeneration. Alteration in endolym- phatic ion concentration may be the cause of both hair cell and otoconial degeneration.
Physiology of the Otolith Organs
HISTORICAL BACKGROUND
Over a century ago, Mach,125 Crum-Brown,126 and Breuer127 each concluded that linear and angular acceleration must be mediated by dif- ferent end organs, and Breuer in particular pos- tulated the mechanism by which the otoliths sense linear acceleration. As in the case of the semicircular canals, a gross anatomic feature of the macules—the dense, calcified otolithic membrane—suggested the mechanisms by which they sense the direction of gravitational force. The afferent neuronal activity from the macules associated with precise static and dynamic linear acceleration forces has only recently been investigated in primates.160–162 These studies confirm that the utricular and saccular macules are responsive to static tilt and dynamic linear acceleration, resolving an earlier controversy as to whether the saccular macule functions as an auditory or vestibular organ. However, we also know that the sacculus can be stimulated by loud sounds, which is the basis for the vestibular evoked myogenic potentials (VEMP) test (see Chapter 7). The pattern of afferent nerve response is complex, with vari- ous neurons exhibiting different resting activity, frequency response, and adaptation properties.
MECHANISM OF STIMULATION
During head displacement, the calcified otolithic membrane is affected by the combined forces of applied linear acceleration and gravity and tends to move over the macule, which is mounted in the wall of the membranous labyrinth (see Fig. 1–3b in Chapter 1). The otolithic mem- brane is restrained in its motion by elastic, vis- cous, and inertial forces analogous to the forces associated with cupular movement. De Vries146 measured the displacement of the large saccular otolithic membranes of several fish and obtained estimates of the forces restraining the mem- brane to the macules. He proposed a model, analogous to the pendulum model, that described the dynamics of otolithic membrane displace- ment as those of a low-pass filter. Displacements due to sinusoidal linear acceleration would be greatest at low frequencies, including static head tilts. At greater frequencies, the otolithic mem- brane displacement would decrease by one-half each time the frequency was doubled.
The nerve fibers of the macule are activated by linear accelerations and by changes in head positions in space.160 Each neuron has a charac- teristic functional vector that defines the axis of sensitivity for head acceleration. Individual neurons appear to be stimulated only by con- tiguous hair cells whose kinocilium is oriented with the same vector of excitability, creating a sensorineural unit (see Fig. 1–3c in Chapter 1). The combined functional units from the mac- ules cover all possible positions of the head in three-dimensional space. However, most of the units are oriented in the horizontal plane in the utricular macule and in the vertical plane in the saccular macule.160–162
In the utricular macule, tilting of the head ipsilaterally results in an increase in firing of units on the medial side of the striola and a decrease in firing of units on the lateral side of the striola. With the subject in the upright posi- tion, most of the units are at baseline because the vector orientation of the utricular macule is orthogonal to the gravitational vector. Because of the curvature of the macule, some afferent units are sensitive to forward and backward tilting of the head.
Since the saccular macule is oriented in the sagittal plane, the vectors of most of the func- tional units are parallel to gravity when the head is in the upright position. Because of the push–pull relationship of hair cells on either side of the striola, some functional units are excited, whereas others are inhibited. The stri- ola of the saccular macule has less curvature than that of the utricle and therefore most of the units have an orientation in the rostro- caudal direction. Baseline firing of neurons innervating the saccule and macule are about the same.160
Anatomy of Primary Neurons
The bipolar primary vestibular neurons of Scarpa’s ganglia are located within the internal auditory canal. The ganglia is shaped like an hourglass with a superior division, an inferior division, and an isthmus (see Fig. 1–6 in Chapter 1).163 The superior division innervates the horizontal and anterior canals, the utricular macule, and part of the saccular macule, while
the inferior division innervates the posterior canal and the rest of the saccular macule. During development the vestibular ganglia elongates with the growth of the nerves and temporal bone, leading to the fusiform appear- ance seen in adults. On average there are approx- imately 25,000 neurons in the human Scarpa’s ganglion.164,165 Neuronal diameters vary greatly in each division of the ganglia. The largest neu- rons innervate type I hair cells in the center of the cristae and macules, and the smallest neu- rons innervate type II hair cells in the periphery of the cristae and macules. Intermediate-sized neurons innervate combinations of type I and type II hair cells throughout the cristae and mac- ules (dimorphic units). Immunohistochemical studies show a differential expression of calcium- binding proteins in large and small neurons.166 Only large cells show immunoreactivity (IR) for calbindin and calretinen, while all cells show IR for parvalbumin. Large neurons also show greater IR for neurofilament proteins than do small neurons.167
Detailed study of the vestibular nerve in ani- mals and humans reveals a highly organized arrangement of the nerve fibers originating from the different inner ear receptors and from the two types of hair cells within each receptor.168–171 There is a similar unimodal distribution of pri- mary afferent neurons with regard to axon and
cell body diameter in several species studied (Fig. 2–17). As noted earlier, small fibers (<2.5 µm) project preferentially to the periph- ery of the sensory epithelium, whereas larger ones (>4.5 µm) project to the center.
INNERVATION OF THE CRISTAE
The highly organized innervation of the crista in the chinchilla is shown in Figure 2–18.172 Underneath the sensory organ, the main nerve divides into two smaller ones. Each of the two nerve branches innervates half of the crista and thus half of the hair cells. Within half a milli- meter from the sensory epithelium, each nerve divides into 8 to 10 bundles of fibers that align in two rows, one for each slope of the crista. Among these smaller bundles, those that inner- vate the center of the crista have a greater pro- portion of thick fibers, whereas the bundles that innervate the periphery have a preponder- ance of thin fibers (Fig. 2–18a, inset).
The bundles of fibers that innervate a dis- crete area of the crista travel together toward the nervous system and, in all probability, innervate groups of neighboring neurons in the vestibular nuclei. There appears to be a topo- graphical representation of the vestibular end organ in the central nervous system that is comparable to the topographical projection of
Diameter:
µm
< 2.5
2.5 – 4.5
> 4.5
Human Monkey Chinchilla

500 600 300
Number of fibers
Number of fibers
0
1.0 9.0
0
1.0
9.0
0
1.0 9.0
Binwidth = 0.5 µm
Figure 2–17. Distribution of primary afferent fibers of different diameters (including myelin) within the cristae of humans, monkeys, and chinchillas. The smallest fibers (< 2.5 µm) are concentrated in the periphery while the largest libers (> 4.5 µm) are more numerous at the center of the cristae. Intermediate-size fibers tend to be equally distributed throughout the cristae.
Figure 2–18. Cross sections of the superior semicircular canal nerve as it enters the crista in the chinchilla. (a) At 20 µm below the base of the crista the nerve fiber bundles are arranged in rows and each bundle consists of 30–50 nerve fibers. Fibers in the periphery have smaller diameters than those in the center. (b) At 400 µm below the surface of the crista. The nerve trunk is separated into two parts by a bony septum. (c) At 600 µm below the base of the crista. The nerve bundles of different sizes are surrounded by the fibrous perineurium to form a single nerve trunk.
different parts in the basilar membrane of the cochlea to the auditory nuclei. Each of the afferent bundles containing fibers of different diameters—thick, medium, and thin—is derived from the same restricted area of about
0.2 mm2 on the crista and carries information about localized cupula movement.
Classical morphologists identified three types of nerve endings in the receptors: large-diameter fibers had caliceal endings, small-diameter fibers had bouton endings, and intermediate-size fibers had both types of ending.173
With techniques for labeling individual neu- rons and fibers by intracellular injection of horseradish peroxidase, detailed information has been obtained in the chinchilla regarding the association of fiber diameter size with dif- ferent nerve endings in different parts of the receptors. In the crista, neurons with large axon diameters (2.8 ± 0.6 µm) innervate one or a few hair cells with caliceal endings (type I) in the center (Fig. 2–19).174,175 Neurons with interme- diate-size axon diameters (2.3 ± 0.6 µm) have both bouton and caliceal endings and are more
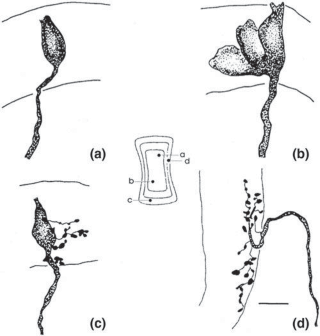
Figure 2–19. Different types of primary afferent nerve endings labeled by intracellular injection of horseradish peroxidase. Reconstructions of two calyx units with simple (a) and complex (b) endings, a dimorphic unit (c), and a bouton unit (d), all taken from a single horizontal canal crista, are shown. The points at which the parent axons of labeled afferents enter the sensory epithelium are indicated on a standard surface reconstruction (center). Bar = 10 µm. (From Fernandez C, et al. The vestibular nerve of the chinchilla. 1. Peripheral innervation patterns in the horizontal and superior semicircular canals. J Neurophysiol. 1988;60:167, with permission.)
or less evenly distributed throughout the crista. Neurons with small axon diameters (1.4 ± 0.4 µm) have only bouton endings and innervate multiple type II hair cells predomi- nantly in the periphery. Of a sample of 368 fibers, 40 (11.1%) were calyx units, 79 (21.5%) were bouton units, and 248 (67.4%) were dimorphic units. Approximately the same dis- tribution of fibers according to diameter size is seen in the crista of the squirrel monkey and in humans (Fig. 2–17).
INNERVATION OF THE MACULES
In the macules, as in the cristae, the diameter of the nerve fibers has a unimodal distribu- tion.176 Fibers of large diameter with only cal- iceal endings predominate near the striola, whereas the thinner fibers innervate the periph- ery. Fibers of intermediate diameter (dimor- phic) are distributed evenly throughout the macule. In the chinchilla macule, as in the crista, dimorphic units outnumber caliceal units by 3 to 1. Caliceal units typically innervate more hair cells (10–40) than dimorphic units (5–20).
NEUROTRANSMITTERS
Glutamate is the main excitatory neurotrans- mitter of primary afferent neurons.177 Immunohistochemical studies in a range of species, including primates, found glutamate immunoreactivity (IR) in all bipolar neurons in the vestibular ganglia. The majority of neu- rons also showed a graded IR to glycine and choline acetyltransferase (ChAT) in a highly overlapping neuronal population.178 Similar co-localization of glycine and ChAT was also seen in afferent terminals in the end organs. Glycine and acetylcholine presumably have a co-transmitter or modulatory function.
Physiology of Primary Neurons
SPONTANEOUS FIRING RATES
Just as there is a continuous spectrum in axon diameters, primary afferent neurons have a wide range of spontaneous firing rates and dynamic properties. It has proved useful to divide them on the basis of the regularity of
Table 2–1 Classification of primary afferent neurons based on their spontaneous firing rate
Firing regularity | Response dynamics | Size | Conduction velocity | Galvanic Stimulation | Epithelial zone | Afferent type |
Regular Irregular | Tonic Phasic | Small Large | Low High | Less sensitive More sensitive | Peripheral/ extrastriola Center/striolar | Bouton/ dimorphic Calyx/ dimorphic |
their spontaneous discharge rate (Table 2–1).179–181 Neurons with the most irregular baseline firing rate (given by the coefficient of variation [CV] of the mean interspike interval) are the most sensitive to galvanic stimulation, while neurons with the most regular baseline firing rate are least sensitive to galvanic stimulation. As a general rule, a primary afferent’s sensitivity to angular acceleration (in spikes/sec per degree/ sec2) is inversely related to the regularity of its baseline firing rate—that is, irregular units with high CV values have a higher sensitivity than regular units with low CV values.
Early in vivo experiments showed that regu- lar afferents have a different after-hyperpolariz- ing potential (AHP) following each spike than do irregular afferents.182 More recent in vitro studies of isolated primary afferent neurons confirmed these findings and suggest that dif- ferential expression of ion channels accounts
for these electrical properties (Fig. 2–20, Table 2–2).166 In isolated rat vestibular ganglia neu- rons, Kv1 potassium blockers converted phasic
firing cells to tonic firing cells, indicating
that Kv1 potassium channels control the firing patterns of irregularly discharging primary neu-
rons.183 Blocking of calcium-activated potassium channels decreased the number of spikes origi- nating from tonic primary afferent neurons. Ca2+ binding proteins and Ca2+ channels regu- late the activation of Ca2+-dependent potassium channels and thus regulate firing frequency.166
AFFERENTS FROM THE CRISTAE
As noted earlier, the primary vestibular afferent fibers maintain a constant baseline firing rate of action potentials. Recordings from the primary afferent fibers of the cristae in mammalian and nonmammalian species reveal that physiologic
Table 2–2 Pattern of expression of ion channels in large and small vestibular ganglion somata in rodents
![]()
Large neurons Small neurons
![]()
Firing regularity Irregular Regular
Spike shape Prominent AHP Brief AHP
Ca++ dependent K+ channels
Lower total density; proportionally more BK
More dense, more blocker resistant current
Voltage-gated K+ channels A A
KCNQ channels KCNQ4 ?
Voltage-gated Ca++ channels HVA, LVA (T) HVA
Voltage–gated Na+ channels Nav 1.5? Nav 1.8, 1.9?
HCN channels Yes Yes
Acid-sensing ion channels More Less
![]()
AHP – afterhyperpolarizing potential; BK – class of Ca++dependent K+ channels with large single-channel conductances; A – rapidly inactivating voltage gated K+ channels of heterogenous molecular composition; HVA – high voltage activated Ca++ channels; LVA – low voltage activated Ca++ channels (T current); HCN – hyperpolarization-activated cyclic- nucleotide-modulated channels. (Adapted from Eatock RA, Xue J, Kalluri R. Ion channels in mammalian vestibular afferents may set regularity of firing. Primary afferents J Exp Biol. 2008;211:1764, with permission.)
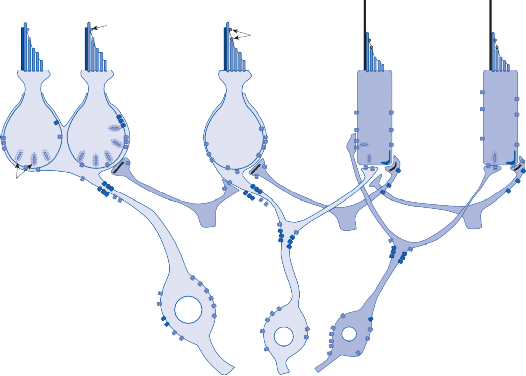
Tip links Mechanosensory ion channels
Nav 1.5
I I I
KCNQ4
Ribbon
Sunapses *Na
K
*
II II
A
Na
LVA
BK
IK
SK
C HCN
*
ASIC HCN
D B
*
Na HVA
HVA
A IK
BK
Figure 2–20. Schematic showing classification of vestibular afferent neurons by terminal morphology as pure-calyx (C), dimorphic (D) and pure-bouton (B). Trunkated fibres are efferents, arising from neurons in the brainstem. Pure-calyx afferents exclusively innervate the centre and striola and often form complex calyces around multiple type I hair cells, as illustrated. Pure-bouton afferents exclusively innervate the peripheral zone and extrastriola and can innervate tens of type II hair cells. Dimorphic afferents innervate both zones, but have more compact dendritic trees in the centre and striola than in the periphery and extrastriola (not shown). Pure-calyx afferents express calretinin, calbindin and parvalbumin; dimorphic afferents are thought to express calbindin and parvalbumin; and pure-bouton afferents, which are the thinnest, express only parvalbumin. Some differences in ion channel expression have been noted between large and small isolated ganglion somata and are indicated here on the pure-calyx and pure-bouton somata and summarized in Table 2–2. Whether dimorphic somata, which are likely to be mid-sized, have intermediate expression is not known. Asterisks indicate possible sites of spike initiation on each afferent. Abbreviations defined in Table 2–2. (Adapted from Eatock RA, Xue J, Kalluri R. Ion channels in mammalian vestibular afferents may set regularity of firing. Primary afferents J Exp Biol. 2008;211:1764, with permission.)
stimulation producing endolymph flow toward the ampulla (i.e., ampullopetal flow) in the horizontal semicircular canal increases the baseline firing rate. Conversely, endolymph flow away from the ampulla (i.e., ampullofugal flow) decreases the baseline firing rate. In the vertical canals the reverse occurs: ampullopetal endolymph flow decreases the baseline firing rate, and ampullofugal flow increases the firing rate. Considering these observations and the previous anatomic descriptions, it is apparent that endolymph displacement that deviates the hairs of the sensory cells toward the kinocilium results in increased firing of the afferent nerve, whereas displacement away from the kinocil- ium results in decreased firing of the afferent
nerve. The differences in the physiological characteristics of responses from the horizontal and vertical canals are the physiological basis to the formulation of Ewald’s second and third laws.
Detailed measurements of afferent nerve activity from the cristae of squirrel monkeys show that the firing rate associated with physio- logic rotatory stimulation follows qualitatively the prediction of the pendulum model;111 that is, the magnitude of change in frequency of action potentials is proportional to the theoretic devia- tion of the cupula. For example, during sinusoi- dal head rotation, the firing rate follows the time course of cupular displacement shown in Figure 2–15c. A sinusoidal change in firing frequency is
When the cristae are subjected to prolonged constant acceleration, a substantial proportion of nerve fibers undergo a slow decline in firing rate (adaptation) rather than maintaining a steady state as predicted in Figure 2–15a. Because of adaptation, the firing rate does not return to baseline after cessation of accelera- tion, but rather, drops to a lower level before slowly returning to the resting level.111,138 Similar overshooting of the baseline occurs after stimulation with an impulse of accelera- tion. Instead of the monotonic response pre- dicted by the pendulum model (Fig. 2–15b), the afferent nerve firing pattern exhibits a biphasic reaction with a prolonged secondary phase that slowly returns to baseline. It is not known whether the behavior is due to anatomic or synaptic processes. The process is more pro- nounced in “irregular” neurons. As will be shown later, the vestibulo-ocular reflex also reflects this deviation from the predicted pat- tern (see Fig. 7–19d).
How are these physiological properties related to the anatomical features of primary afferent neurons described earlier? The anatomical and physiological properties of a single primary afferent neuron can be studied by first record- ing the neurons’ dynamic response to angular acceleration with a micropipette and then injecting it with a tracer to study its anatomical connections. Initial studies in the bullfrog dem- onstrated that “irregular” neurons had thick, rapidly conducting fibers that preferentially innervated the central ridge of the crista, while “regular” neurons had thin, slowly conducting fibers that predominantly innervated the periphery.184 In mammals three patterns of
nerve terminals within the crista can be delin- eated: (1) caliceal endings, (2) bouton endings, and (3) combined caliceal and bouton endings (dimorphic units). All caliceal units are at the center of the crista and have “irregular” dynamic properties, whereas bouton units are in the periphery and have “regular” dynamic properties (Fig. 2–21). Dimorphic units can be either “irregular” or “regular,” with the former usually innervating the center of the crista and the latter, the periphery. Surprisingly, the cal- iceal units at the center of the crista have a lower rotational sensitivity than that of dimor- phic units with similar-size axons innervating the same region. Possibly the lower sensitivity of these caliceal units extends the dynamic range of vestibular reflexes—that is, they do not become saturated by the large velocity active head movements. Dimorphic units innervating different regions of the crista vary in their dynamic properties, even though they contacted similar numbers of type I and type II hair cells. Taken together, these findings indi- cate that the dynamic properties of a semicir- cular canal afferent neuron reflect the number and type of synaptic connections, and location within the crista. However, these factors alone cannot explain regularity of firing. For this one must look to the intrinsic membrane properties of the primary afferent neurons as described earlier.
AFFERENTS TO THE MACULES
As in the crista, the neurons of the macule can be classified according to the regularity of the spontaneous firing rate.162 Recordings from neurons whose nerve endings were visualized with intracellular labeling show that caliceal units (mostly near the striola) are irregular, whereas bouton units (in the periphery) are regular (Table 2–1). Dimorphic units that innervate the areas near the striola are more irregular than those innervating the periphery. In the chinchilla, regular units outnumber irregular units by a ratio of 3 to 1. As in the crista, irregular units are more sensitive and have a wider frequency range of response than that of regular units. They adapt promptly to stimuli of constant linear acceleration (e.g., head tilt). By contrast, regular units maintain a constant relation between the gravity vector and the firing rate during static tilts. As in the crista, large primary afferent neurons with an irregular

![]()
(a)
d c PI
c
c d
I I
d d
P P
d b

(b)
![]()
![]()
![]()
Figure 2–21. (a) Dynamic properties of afferent fibers originating in different parts of the chinchilla crista. This cross sec- tion of the crista is divided into peripheral (P), intermediate (I), and central (C) zones. Calyx (c) fibers innervate the central zone; bouton (b) fibers, the peripheral zone; and dimorphic (d) fibers innervate all three zones. (From Fernandez C et al. The vestibular nerve of the chinchilla. I. Peripheral innervation pattern of the horizontal and superior semicircular canals. J Neurophysiol. 1988;60:167–181, with permission.) (b) Locations of intraaxonally labeled fibers in the chinchilla cristae. (Left) Units are sorted by their normalized coefficients of variation into three categories: regular (open symbols), interme- diate (half-filled symbols), and irregular (filled symbols). (Right) Units are sorted into three categories according to their phases with regard to head velocity for 2 Hz sinusoidal head rotation: tonic (open symbols), intermediate (half-filled sym- bols), and phasic (filled symbols). (From Baird R et al. The vestibular nerve in the chinchilla. 11. Relation between afferent response properties and peripheral innervation patterns in the semicircular canals. J Neurophysiol. 1988;60:182–203, with permission.)
spontaneous firing rate have a different ion channel profile than do small afferent neurons with a regular spontaneous firing rate.156
In all vertebrates the inner ear efferent neu- rons are located in the hindbrain with cell bod- ies near brachial motor nuclei within the pons and medulla.185,186 Typically axons separate from the facial nerve and join the eighth nerve to innervate the different receptor organs. Even though they innervate sensory epithe- lium, efferent neurons are derived from the motor column and share a common embryo- logical origin with motor neurons. In fish and amphibians all efferent neurons are localized in a single nucleus, the octavolateralis nucleus, near the rostral end of the facial motor nucleus, whereas in mammals cochlear and vestibular efferent neurons are in separate nuclei. Cochlear efferent neurons are located near the
seventh nucleus, while vestibular efferent neurons are located near the sixth nucleus. Although there are only about 300 vestibular efferent neurons on each side of the brain stem, their axons branch extensively such that in the sensory epithelium efferent boutons out- number afferent boutons by a ratio of 3:1.187 Each labyrinth receives about an equal num- ber of efferent fibers from each side of the brain stem. Consistent with its role as a sensory feedback system, the efferent neurons receive input from the ipsilateral vestibular nerve and both vestibular nuclei.
Consistent with its embryological origin from motor neurons acetylcholine is the princi- pal neurotransmitter of the vestibular efferent system.188 Choline acetyltransferase (ChAT) immunoreactivity (IR) was found in nerve fibers and boutons adjacent to afferent nerve calyces and type II hair cells in the vestibular receptors of multiple animals including humans.189–193 GABA and the neuropeptide cal- citonin gene-related peptide (CGRP) IR was
found in a subpopulation of efferent neurons varying among species.194 Nitric oxide synthase (NOS) was also identified in a subpopulation of efferent neurons and peripheral efferent bou- tons, suggesting that nitric oxide might be another efferent transmitter.195
In mammals, including primates, electrical stimulation of brainstem efferent neurons results in a predominantly excitatory response, increased firing of afferent neurons.186,196 Efferent responses are larger and more rapid in irregularly discharging afferents and smaller and slower in regularly discharging efferent neurons. Only a few studies have used physio- logical stimulation of the vestibular efferent system, but overall the findings are similar to those obtained with electrical stimulation.186 Evoked responses are excitatory for rotations in both directions, are larger in more irregu- larly discharging afferents, and consist of a rapid and slow response component. These rotational responses were efferent mediated, since they were abolished when the vestibular nerve was cut between the recording electrode and the brain stem.
The functional role of the vestibular efferent
Duckworth EA, Silva FE, Chandler JP, Batjer HH, Zhao JC. Temporal bone dissection for neurosur- gery residents: identifying the essential concepts and fundamental techniques for success. Surg Neurol. 2008;69:93.
Leskinen K, Jero J. Acute complications of otitis media in adults. Clin Otolaryngol. 2005;30(6):511.
Hudde H, Weistenhofer C. Key features of the human middle ear. ORL J Otorhinolaryngol Relat Spec. 2006;68(6):324.
Wever E, Lawrence M. Physiological Acoustics. Princeton, NJ: Princeton University Press; 1954.
Rosson GD, Redett RJ. Facial palsy: anatomy, etiology, grading, and surgical treatment. J Reconstr Microsurg. 2008;24(6):379.
Dobe RA. Tests of facial nerve function. In: Cummings CW, Flint PW, Haughey BH, et al., eds. Otolaryngology—Head and Neck Surgery. 4th ed. St. Louis, MO: CJ Mosby; 2005.
Gray O. A brief survey of the phylogenesis of the laby- rinth. J Laryngol. 1955;69:151.
Budelmann BU. Morphological diversity of equi- librium receptor systems in aquatic invertebrates. In: Atema J, Fay RR, Popper AN, Tavolga WN, eds. Sensory Biology of Aquatic Animals. New York: Springer-Verlag; 1988.
Baird IL. Some aspects of the comparative anatomy and evolution of the inner ear in sub-mammalian verte- brates. In: Riss W, ed. Brain, Behavior and Evolution. Basel, Switzerland: S Karger; 1974.
Watson GM, Mire P, Hudson RR. Hair bundles of sea
system is still unclear.186 Although some have suggested that efferent signals might modulate afferent responses in anticipation of active head movement, there is little difference in afferent response to active and passive movements. Nor does the efferent system seem to be important for rebalancing labyrinthine tone after unilat- eral vestibular loss. Efferent responses are non- specific in that excitatory responses are evoked
anemones as a model for vertebrate hair bundles. Hear Res. 1997;107:53.
Watson GM, Mire P. A comparison of hair bundle mechanireceptors in sea anemones and vertebrate systems. In: Pedersen RA, Schatten GP, eds. Current Topics in Developmental Biology. Vol 43. New York: Academic Press; 1999.
Collewijn H. Oculomotor reactions in cuttlefish Sepia officinalis. J Exp Biol. 1970;52:369.
Williamson R, Chrachri A. Cephalopod neural net- works. Neurosignals. 2004;13:87.
Williamson R, Chrachri A. A model biological neural
by stimulation of any of the sensory organs on the same or opposite sides and by rotations that excite or inhibit the afferent nerve response. Furthermore, the responses are small com- pared to those produced by conventional affer- ent stimulation. The efferent system may play a role in the normal development of the periph- eral vestibular end organs.185
REFERENCES
Anson BJ, Donaldson JA. Surgical Anatomy of the Temporal Bone and Ear. 3rd ed. Philadelphia: WB Saunders; 1981.
Marsh M, Jenkins H. Temporal bone neoplasms and lateral cranial base surgery. In: Cummings CW, Flint PW, Haughey BH, et al., eds. Otolaryngology—Head and Neck Surgery. 4th ed. St. Louis, MO: CJ Mosby; 2005.
network: the cephalopod vestibular system. Philos Trans R Soc Lond B Biol Sci. 2007;362:473.
Budelmann BU, Sache M, Staudigl M. The angu- lar acceleration receptor system of the statocyst of Octopus vulgaris: morphometry, ultrastructure, and neuronal and synaptic organization. Phil Trans R Soc Lond B Biol Sci. 1987;315:305.
Baird IL. Some aspects of the comparative anatomy and evolution of the inner ear in sub-mammalian verte- brates. In: Riss W, ed. Brain, Behavior and Evolution. Basel, Switzerland: S Karger; 1974.
Dohlman GF. The mechanism of secretion and absorp- tion in the vestibular apparatus. Acta Otolaryngol (Stockh). 1965;59:275.
Sterker O, Ferrary E, Amiel C. Production of inner ear fluids. Physiol Rev. 1988;68:1083.
Salt AN. Dynamics of the inner ear fluids. In: Jahn AJ, Santos-Sacchi J, eds. Physiology of the Ear. 2nd ed. San Diego, CA: Singular/Thomson Learning Publ.; 2001.
Holden H, Schuknecht H. Distribution pattern of blood in the inner ear following spontaneous suba- rachnoid hemorrhage. J Laryngol. 1968; 82:321.
Perlman H, Lindsay J. Relation of the internal ear spaces to the meninges. Arch Otolaryngol. 1939;29:12.
Kimura RS, Schuknecht H, Ota C. Blockage of the cochlear aqueduct. Acta Otolaryngol (Stockh). 1974;77:1.
Suh KW, Cody DTR. Obliteration of vestibular and cochlear aqueducts in animals. Trans Am Acad Ophthalmol Otolaryngol. 1977;84:359.
Kimura RS. Distribution, structure and function of dark cells in the vestibular labyrinth. Ann Otol Rhinol Laryngol. 1969;78:542.
Salt AN. Regulation of endolymphatic fluid volume.
Ann N Y Acad Sci. 2001;942:306.
Steel KP. The benefits of recycling. Science. 1999;285:1363.
Hibino H, Kurachi Y. Molecular and physiological bases of the K+ circulation in the mammalian inner ear. Physiology (Bethesda). 2006;21:336.
Delpire E, Lu J, England R, Dull C, Thorne T. Deafness and imbalance with inactivation of the secretory Na-K- 2Cl co-transporter. Nat Genet. 1999;22:192.
Dixon MJ, Gazzard J, Chaudhry SS, et al. Mutation of the Na-K-Cl co-transporter gene Slc12a2 results in deafness in mice. Hum Mol Genet. 1999;8:1579.
Vetter DE, Mann JR, Wangemann P, et al. Inner ear defect induced by null mutation of the isk gene. Neuron. 1996;17:1251.
Beitz E, Zenner HP, Schultz JE. Aquaporin-mediated fluid regulation in the inner ear. Cell Mol Neurobiol. 2003;23(3):315.
Guild SR. The circulation of the endolymph. Am J Anat. 1927;39:57.
Lawrence M, Wolsk D, Litton WB. Circulation of the inner ear fluids. Ann Otol Rhinol Laryngol. 1961;70:753.
Lawrence M. The flow of endolymph: a unified con- cept. Otolaryngol Clin North Am. 1980;13:577.
Salt AN. Fluid homeostasis in the inner ear. In: Harris JP, ed. Meniere’s Disease. The Hague, Netherlands: Kugler Publications; 1999.
Takeuchi S, Takeda T, Saito H. Pressure relationship between perilymph and endolymph in guinea pigs. Acta Otolaryngol (Stockh). 1990;109:93.
Takeuchi S, Takeda T, Saito H. Pressure relationship between perilymph and endolymph associated with endolymphatic infusion. Ann Otol Rhinol Laryngol. 1991;100:244.
Kishimoto S, Naganara K, Fisch V, Dillier N. Inner ear pressure measurements: effects of obstruction of the cochlear aqueduct and endolymphatic duct on the perilymphatic pressure. Otolaryngol Clin North Am. 1983;16:21.
Andrews JC, Böhmer A, Hoffman LF. The measure- ment and manipulation of intralabyrinthine pressure in experimental endolymphatic hydrops. Laryngoscope. 1991;101:661.
Marchbanks RJ, Reid A. Cochlear and cerebrospinal fluid pressure: their inter relationship and control mechanisms. Br J Audiol. 1990;24:179.
Beentjes BIJ. The cochlear aqueduct and the pressure of cerebrospinal and endolabyrinthine fluids. Acta Otolaryngol (Stockh). 1972;73:112.
Kimura RS, Schuknecht H. Membranous hydrops in the inner ear of the guinea pig after obliteration
of the endolymphatic sac. Pract Otorhinolaryngol. 1965;27:343.
Smith CA, Lowry OH, Wu ML. The electrolytes of the labyrinthine fluids. Laryngoscope. 1954;64:141.
Silverstein H. Biochemical studies of the inner ear fluids in the cat. Ann Otol Rhinol Laryngol. 1966; 75:48.
Schuknecht H, El Seifi A. Experimental observations on the fluid physiology of the inner ear. Ann Otol Rhinol Laryngol. 1963;72:687.
Silverstein H, Schuknecht H. Biochemical stud- ies of inner ear fluid in man. Arch Otolaryngol. 1966;84:395.
Silverstein H. Inner ear fluid proteins in acoustic neu- roma. Meniere’s disease and otosclerosis. Ann Otol Rhinol Laryngol. 1971;80:27.
Mazzoni A. Internal auditory canal, arterial relations at the porus acusticus. Ann Otol Rhinol Laryngol. 1969;78:797.
Wende S, Nakayama N, Schwerdtfeger P. The internal auditory artery (embryology, anatomy, angiography, pathology). J Neurol. 1975; 210:21.
Mazzoni A. Internal auditory artery supply to the petrous bone. Ann Otol Rhinol Laryngol. 1972:81:13.
Perlman HB, Kimura RS, Fernández C. Experiments on temporary obstruction of the internal auditory artery. Laryngoscope. 1959;69:591.
Konishi T, Butler RA, Fernández C. Effect of anoxia on cochlear potentials. J Acoust Soc Am. 1961;33:349.
Yamamoto K, Kubo T, Matsunaga T. Autoregulation of inner ear blood flow in normal and hydropic guinea pigs. Acta Otolaryngol (Stockh). 1991:111:312.
Miller JM, Ren TY, Laurikainen E, et al. Hydrops- induced changes in cochlear blood flow. Ann Otol Rhinol Laryngol. 1995;104:476.
Anijad AH, Scheer AA, Rosenthal J. Human internal auditory canal. Arch Otolaryngol, 1969;89:709.
Rasmussen A. Studies of the VIIIth cranial nerve of man. Laryngoscope. 1940;50:67.
Lorente de Nó R. Anatomy of the eighth nerve. The central projection of the nerve endings of the internal ear. Laryngoscope. 1933;43:1.
Sando I, Black FO, Hemenway WG. Spatial distribu- tion of vestibular nerve in internal auditory canal. Ann Otol Rhinol Laryngol. 1972:81:305.
Pearson AA. The Development of the Ear: A Manual. Rochester, MN: American Academy of Ophthalmology and Otolaryngology; 1967.
Anson BJ. Developmental anatomy of the ear In: Paparella MM, Shumrick DA, eds. Otolaryngology. Vol 1. Philadelphia: WB Saunders; 1973.
Rinkwitz S, Bober E, Baker R. Development of the vertebrate inner ear. Ann N Y Acad Sci. 2001;942:1.
Tsai AC-H, Stool SE, Post JC. Phylogenetic aspects and embryology. In: Bluestone CD, ed. Pediatric Otolarygology. 4th ed. Philadelphia: WB Saunders; 2003.
Bryant J, Goodyear RJ, Richardson GP. Sensory organ development in the inner ear: molecular and cellular mechanisms. Br Med Bull. 2002;63:39.
Corwin TJ, Cotanche DA. Regeneration of sensory hair cells after acoustic trauma. Science. 1998;240:1722.
Ryals BM, Rubel EW. Hair cell regeneration after acoustic trauma in adult coturnix quail. Science. 1988;240:1774.
Forge A, Li L, Corwin JT, et al. Ultrastructural evi- dence for hair cell regeneration in the mammalian inner ear. Science. 1993;259:1616.
Tanyeri H, Lopez I, Honrubia V. Histological evidence for hair cell regeneration after ototoxic cell destruction with local application of gentamicin in the chinchilla crista ampullaris. Hear Res. 1995;89:194.
Vollrath MA, Kwan KY, Corey DP. The mechanoma- chinery of mechanotransduction in hair cells. Ann Rev Neurosci. 2007;30:339.
Tsuprun V, Goodyear RJ, Richardson GP. The struc- ture of tip links and kinocilial links in avion sensory hair bundles. Biophys J. 2004;87:4106.
Pickles JO, Comis SD, Osborne MP. Cross-links between the stereocilia of the guinea pig organ of Corti and their possible relation to sensory transduc- tion. Hearing Res. 1984;15:103.
Gillespie PG, Cyr JL. Myosin-1c, the hair cell’s adap- tion motor. Annu Rev Physiol. 2004;66:521.
Martinac B, Kloda A. Evolutionary origins of mecha- nosensitive ion channels. Prog Biophys Mol Biol. 2003;82:11.
Fritzsch B, Beisel KW, Pauley S, Soukup G. Molecular evolution of the vertebrate mechanosensory cell and ear. Int J Dev Biol. 2007;51:663.
Lin SY, Corey DP. TRP channels in mechanosensa- tion. Curr Opin Neurobiol. 2005;15:350.
Moser T, Neef A, Khimich D. Mechanisms underlying the temporal precision of sound coding at the inner hair cell ribbon synapse. J Physiol. 2006;576(pt 1):55.
Neef A, Khimich D, Pirih P, Riedel D, Wolf F, Moser T. Probing the mechanism of exocytosis at the hair cell ribbon synapse. J Neurosci. 2007;27(47): 12933.
Keen EC, Hudspeth AJ. Transfer characteristics of the hair cell’s afferent synapse. Proc Natl Acad Sci USA. 2006;103(14):5537.
Lisakowski A, Goldberg JM. A regional ultrastruc- tural analysis of the cellular and synaptic architecture in the cinchilla cristae ampullares. J Comp Neurol. 1997;389:419.
Beurg M, Safieddine S, Roux I, Bouleau Y, Petit C, Dulon D. Calcium- and otoferlin-dependent exo- cytosis by immature outer hair cells. J Neurosci. 2008;28(8):1798.
Goutman JD, Glowatzki E. Time course and calcium dependence of transmitter release at a single ribbon synapse. Proc Natl Acad Sci USA. 2007;104(41): 16341.
Obholzer N, Wolfson S, Trapani JG, et al. Vesicular glutamate transporter 3 is required for synap- tic transmission in zebrafish hair cells. J Neurosci. 2008;28(9):2110.
Hudspeth AJ. Mechanoelectrical transduction by hair cells of the bullfrog’s sacculus. Prog Brain Res. 1989;80:129.
Von Békésy G. Experimental models of cochlea with and without nerve supply. In: Rasmussen GL, Windle WF, eds. Neural Mechanisms of the Auditory and Vestibular System. Springfield, IL: Charles C Thomas; 1960.
Bauknight RS, Strelioff D, Honrubia V. Effective stimulus for the Xenopuslaevis lateral-line hair-cell system. Laryngoscope. 1976;86:1836.
Fernandez C, Goldberg JM. Physiology of peripheral neurons innervating of otolith organs of the squirrel monkey. II. Directional selectivity and force-response relations. J Neurophysiol. 1976;39:985.
Flock A, Jorgensen M, Russell I. The physiology of individual hair cells and their synapses. In: Miller A, ed. Basic Mechanisms in Hearing. New York: Academic Press; 1973.
Davis H. A model for transducer action in the cochlea.
Cold Spring Harbor Symp Quant Biol. 1965;30:181.
Honrubia V, Strellioff D, Sitko ST. Physiological basis of cochlear transduction and sensitivity. Ann Otol Rhinol Laryngol. 1976;85:697.
Hudspeth AJ. Mechanoelectrical transduction by hair cells in the acousticolateralis sensor system. Annu Rev Neurosci. 1983;6:187.
Dallos P. Membrane potential and response changes in mammalian cochlear hair cells during intracellular recording. J Neurosci. 1985;5:1609.
Hudspeth AJ. The cellular basis of hearing: the biophysics of hair cells. Science. 1985;230:745.
Rennie KJ, Correia MJ. Potassium currents in mam- malian and avian isolated type I semicircular canal hair cells. J Neurophysiol. 1994;71:1.
Eatock RA, Rüsch A, Lysakowski A, Salki M. Hair cells in mammalian utricles. Otolaryngol Head Neck Surg. 1998;119:172.
Housley GD, Marcotti W, Navaratnam D, Yamoah EN. Hair cells–beyond the transducer. J Membr Biol. 2006;209(2-3):89.
Guth PS, Perin P, Norris CH, Valli P. The vestibular hair cells: post-transductional signal processing. Prog Neurobiol. 1998; 54(2):193.
Hudspeth AJ, Corey DP. Sensitivity, polarity, and conductance change in the response of vertebrate hair cells to controlled mechanical stimuli. Proc Natl Acad Sci USA. 1977;74:2407.
Hudspeth AJ. Extracellular current flow and the site of transduction by vertebrate hair cells. J Neurosci. 1982;2:1.
Orman S, Flock A. Active control of sensory hair mechanics implied by susceptibility to media that induce contraction in muscle. Hear Res. 1983; 11:261.
Flock A, Flock B, Ulfendahl M. Mechanisms of move- ment in outer hair cells and a possible structural basis. Arch Otorhinolaryngol. 1986;243:83.
Brownell WE. Microscopic observation of cochlear hair cell motility. Scand Electron Microsc. 1984;3:1401.
Kachar B, Brownell WE, Altschuler R, Fex J. Electrokinetic shape changes of cochler outer hair cells. Nature. 1986;322:365.
Zheng J, Shen W, He DZ, Lnog KB, Madison LD, Dallos P. Prestin is the motor protein of cochlear outer hair cells. Nature. 2000;405:130.
Mountain DC. Electromechanical properties of hair cells. In: Altschuler RA, Hoffman DW, Bobbin RP, eds. Neurobiology of Hearing: The Cochlea. New York: Raven Press; 1986.
Flock A, Cheung H. Actin filaments in sensory hairs of the inner ear receptor cells. J Cell Biol. 1977;75:339.
Sans A, Griguer C, Lehouelleur J. The vestibular type I hair cells: a self-regulated system? Acta Otolaryngol Suppl (Stockh). 1994;513:11.
Hoagland H. Impulses from sensory nerves of catfish.
Proc Natl Acad Sci USA. 1932;18:701.
Lowenstein O, Wersall J. A functional interpreta- tion of the electron microscopic structure of the sensory hairs in the cristae of the elasmobranch Raja clavata in terms of directional sensitivity. Nature. 1959;184:1807.
Kiang NYS, Watanabe T, Thomas EC, Clark LF. Discharge Patterns of Single Fibers in the Cat’s Auditory Nerve. Research Monograph 35. Cambridge, MA: MIT Press; 1965.
Goldberg JM, Fernandez C. Physiology of periph- eral neurons innervating semicircular canals of the squirrel monkey. 1. Resting discharge and response to constant angular accelerations. J Neurophysiol. 1971;34:635.
Holstein GR, Rabbitt RD, Martinelli GP, Friedrich VL, Jr., Boyle RD, Highstein SM. Convergence of excitatory and inhibitory hair cell transmitters shapes vestibular afferent responses. Proc Natl Acad Sci USA. 2004;101(44):15766.
Highstein SM, Rabbitt RD, Holstein GR, Boyle RD. Determinants of spatial and temporal cod- ing by semicircular canal afferents. J Neurophysiol. 2005;93(5):2359.
Chen Z, Kujawa SG, Sewell WF. Auditory sensitiv- ity regulation via rapid changes in expression of sur- face AMPA receptors. Nat Neurosci. 2007;10(10): 1238.
Highstein SM, Fay R, Popper AN. eds. The Vestibular System Springer Handbook of Auditory Research. Vol. 19. New York: Springer; 2004.
Ramprashad F, Landolt JP, Money KE, Laufer J. Dimensional analysis and dynamic response charac- terization of mammalian peripheral vestibular struc- tures. Am J Anat. 1984;169:295.
Blanks RHI, Curthoys IS, Markham CH. Planar relationships of semicircular canals in the cat. Am J Physiol. 1972;223:55.
Blanks RHI, Curthoys IS, Markham CH. Planar relationships of the semicircular canals in man. Acta Otolaryngol (Stockh). 1975;80:185.
Rosenhall U. Mapping of the cristae ampullares in man. Ann Otol. 1972;81:882.
Rosenhall U. Vestibular macular mapping in man.
Ann Otol. 1972;81:339.
Lee W-S, Suárez C, Honrubia V, Gómez J. Morphological aspects of the human vestibular nerve. Laryngoscope. 1990;100:756.
Fernández C, Lysakowski A, Goldberg JM. Hair-cell counts and afferent innervation patterns in the crista ampullares of the squirrel monkey with a comparison to the chinchilla. J Neurophysiol. 1995;73:1253.
Takumida M. Functional morphology of the crista ampullaris: with special interests in sensory hairs and cupula: a review. Biol Sci Space. 2001;15(4):356.
Flourens P. Recherches Expérimentales sur les Propriétés et les Functions due Système Nerveux dans les Animaux Vertébrés. Paris: Crevot; 1842.
Mach E. Grundlinien der Lehre von den Bewegungsempfindungen. {in German] Amsterdam, Netherlands: Bonset; 1967, Reprint of 1875-edition.
Crum-Brown A. On the sense of rotation and the anatomy and physiology of the semicircular canals of the internal ear. J Anat Physiol. 1874;8:327.
Breuer J. Über die Funktion der Bogengänge des Ohrlabyrinthes. Wien Med Jahrb. 1874;4:72.
Ewald R. Physiologische Untersuchungen über das Endorgan des Nervous Octavus. Wiesbaden, Germany: Bergmann; 1892.
Steinhausen W. Über Sichtbarmachung and Funktionsprüfung der Cupula terminalis in den Bogengangs-ampullen der Labyrinths. Arch Ges Physiol. 1927;217:747.
Dohlman GF. Some practical and theoretical points of labyrinthology. Proc R Soc Med. 1935;28:1371.
McLaren JW, Hillman DE. Displacement of the semicircular canal cupula during sinusoidal rotation. Neuroscience. 1979;4:2001.
Wilson VJ, Melvill Jones G. Mammalian Vestibular Physiology. New York: Plenum Press; 1979.
Rabbit RD, Boyle R, Highstein SM. Sensory trans- duction of head velocity and acceleration in the toad- fish horizontal semicircular canal. J Neurophysiol. 1994;72:1041.
Lowenstein O, Sand A. The individual and integrated activity of the semicircular canals of the elasmobranch labyrinth. J Physiol. 1940;99:89.
Ledoux A. Les canaux semi-circulaires Etude électro- physiologique. Contribution à l’effort d’uniformisation des épreuves vestibulaires. Essai d’interprétation de la sémiologie vestibulaire. Acta Otorhinolaryngol Belg. 1958;12:109.
Precht W, Llinás R, Clarke M. Physiological responses of frog vestibular fibers to horizontal angular rotation. Exp Brain Res. 1971;13:378.
Lipschitz WS. Responses from the first order neurons of the horizontal semicircular canal in the pigeon. Brain Res. 1973;63:43.
Blanks RHI, Estes MS, Markham CH. Physiologic characteristics of vestibular first-order canal neurons in the cat. II. Response to constant angular accelera- tion. J Neurophysiol. 1975;38:1250.
Curthoys IS. The response of primary horizontal semicircular canal neurons in the rat and guinea pig to angular acceleration. Exp Brain Res. 1982;47:286.
Schneider LW, Anderson DJ. Transfer characteristics of first and second order lateral and vestibular neu- rons in gerbil. Brain Res. 1976;112:61.
Tomko DL, Peterka RJ, Schor RH, O’Leary DP. Response dynamics of horizontal canal afferents in barbiturate-anesthetized cats. J Neurophysiol. 1981;45:376.
Fernández C, Goldberg JM. Physiology of periph- eral neurons innervating semi-circular canals of the squirrel monkey. II. Response to sinusoidal stimula- tion and dynamics of peripheral vestibular system. J Neurophysiol. 1971;34:661.
Goldberg JM, Fernández C. Physiology of peripheral neurons innervating semicircular canals of the squir- rel monkey. III. Variations among units in their dis- charge properties. J Neurophysiol. 1971;34:676.
Van Egmond AAJ, Groen JJ, Jongkees LBW The mechanics of the semicircular canal. J Physiol. 1949;110:1.
McLaren JW, Hillman DE. Displacement of the semicircular canal cupula during sinusoidal rotation. Neuroscience. 1979;4:2001.
De Vries H. The mechanics of the labyrinth otoliths.
Acta Otolaryngol (Stockh). 1950;38:262.
Lundberg YW, Zhao X, Yamoah EN. Assembly of the otoconia complex to the macular sensory epithelium of the vestibule. Brain Res. 2006;1091(1):47.
Hughes I, Thalmann I, Thalmann R, Ornitz DM. Mixing model systems: using zebrafish and mouse inner ear mutants and other organ systems to unravel the mystery of otoconial development. Brain Res. 2006;1091(1):58.
Ballarino J, Howland HC. Otoconial morphology of the developing chick. Anat Rec. 1982;204:83.
Shull GE, Okunade G, Liu LH, et al. Physiological functions of plasma membrane and intracellular Ca2+ pumps revealed by analysis of null mutants. Ann NY Acad Sci. 2003;986:453.
Shiao JC, Lin LY, Horng JL, Hwang PP, Kaneko T. How can teleostean inner ear hair cells maintain the proper association with the accreting otolith? J Comp Neurol. 2005;488:331.
Erway LC, Purichia NA, Netzler ER, D’Amore MA, Esses D, Levine M. Genes, manganese, and zinc in formation of otoconia: labeling, recovery, and maternal effects. Scan Electron Microsc. 1986;pt 4: 1681.
Vibert D, Kompis M, Hausler R. Benign paroxysmal positional vertigo in older women may be related to osteoporosis and osteopenia. Ann Otol Rhinol Laryngol. 2003;112:885.
Harada Y, Kasuga S, Mori N. The process of otoco- nia formation in guinea pig utricular supporting cells. Acta Oto-Laryngol. 1998;118:74.
Johnsson LG, Wright CG, Preston RE, Henry PJ. Streptomycin-induced defects of the otoconial mem- brane. Acta Oto-Laryngol. 1980;89:401.
Lim DJ. Otoconia in health and disease. A review.
Ann Otol Rhinol Laryngol. 1984;suppl 112:17.
Minck DR, Erway LC, Vorhees CV. Preliminary find- ings of a reduction of otoconia in the inner ear of adult rats prenatally exposed to phenytoin. Neurotoxicol Teratol. 1989;11:307.
Takumida M, Zhang DM, Yajin K, Harada Y. Effect of streptomycin on the otoconial layer of the guinea pig. ORL J Otorhinolaryngol Relat Spec. 1997;59: 263.
Wright CG, Hubbard DG, Graham JW. Absence of otoconia in a human infant. Ann Otol Rhinol Laryngol. 1979;88:779.
Fernández C, Goldberg JM. Physiology of peripheral neurons innervating otolith organs of the squirrel monkey. I. Response to static tilts and to long-dura- tion centrifugal force. J Neurophysiol. 1976;39:970.
Fernández C, Goldberg JM. Physiology of peripheral neurons innervating otolith organs of the squirrel monkey. II. Directional selectivity and force-response relations. J Neurophysiol. 1976;39:985.
Fernández C, Goldberg JM. Physiology of periph- eral neurons innervating otolith organs of the squir- rel monkey. III. Response dynamics. J Neurophysiol. 1976;39:996.
Sato H, Sando I, Takahashi H. Three-dimensional anatomy of human Scarpa’s ganglion. Laryngoscope. 1992;102:1056.
Ishiyama A, Lopez I, Ishiyama G, Tang Y. Unbiased quantification of the microdissected human Scarpa’s ganglion neurons. Laryngoscope. 2004;114(8): 1496.
Ishiyama G, Finn M, Lopez I, Tang Y, Baloh RW, Ishiyama A. Unbiased quantification of Scarpa’s gan- glion neurons in aminoglycoside ototoxicity. J Vestib Res. 2005;15(4):197.
Eatock RA, Xue J, Kalluri R. Ion channels in mam- malian vestibular afferents may set regularity of fir- ing. Primary afferents. J Exp Biol. 2008;211(pt 11): 1764.
Ylikoski J, Pirvola U, Häppölä O. Characterization of the vestibular and spiral ganglion cell somata of the rat by distribution of neurofilament proteins. Acta Otolaryngol Suppl (Stockh). 1993;503:121.
Gacek RR. The innervation of the vestibular laby- rinth. Ann Otol Rhinol Laryngol. 1968;77:676.
Honrubia V, Hoffman LF, Sitko S, Schwartz IR. Anatomic and physiological correlates in bullfrog ves- tibular nerve. J Neurophysiol. 1989;61:688.
Honrubia V, Kuruvilla A, Mamekunian D, Eichel JE. Morphological aspects of the vestibular nerve of the squirrel monkey. Laryngoscope. 1987;97:228.
Lopez I, Ishiyama G, Tang Y, Frank M, Baloh RW, Ishiyama A. Estimation of the number of nerve fibers in the human vestibular endorgans using unbiased stereology and immunohistochemistry. J Neurosci Methods. 2005;145(1-2):37.
Naito E, Honrubia V, Naito Y, Beykirch K, Toga AW, Hoffman L. Arrangement of vestibular nerve fibers in the semicircular canal crista of the chinchilla. Audiol Neurootol. 1997;2:213.
Lorente de Nó R. Anatomy of the eighth nerve. The central projection of the nerve endings of the internal ear. Laryngoscope. 1933;43:1.
Fernández C, Baird RA, Goldberg JM. The vestibu- lar nerve of the chinchilla. I. Peripheral innervation patterns in the horizontal and superior semicircular canals. J Neurophysiol. 1988;60:167.
Baird RA, Desmadryl G, Fernández C, Goldberg JM. The vestibular nerve in the chinchilla. II. Relation between afferent response properties and periph- eral innervation patterns in the semicircular canals. J Neurophysiol. 1988;60:182.
Goldberg JM, Desmadryl G, Baird RA, Fernández
C. The vestibular nerve of the chinchilla. V. Relation between afferent discharge and peripheral innerva- tion patterns in the utricular macula. J Neurophysiol. 1990;63:791.
Straka H, Reichenberger I, Dieringer N. Synaptic transmission by vestibular nerve afferent fibers. In: Beitz AJ, Anderson JH, eds. Neurochemistry of the Vestibular System. New York: CRC Press; 2000: 47.
Bäurle J, Brüning G, Schemann M, Nishiike S, Guldin WO Co-localization of glutamate, choline acetyl- transferase and glycine in the mammalian vestibular ganglion and periphery. Neuroreport. 1999;10(17): 3517.
#. Goldberg JM, Highstein SM, Moschovakis A, Fernández C. Inputs from regularly and irregularly discharging vestibular-nerve afférents to second- ary neurons in the vestibular nuclei of the squir- rel monkey. I. An electrophysiological analysis. J Neurophysiol. 1987;58:700.
Goldberg JM, Smith CE Fernández C. Relation between discharge regularity and responses to externally applied galvanic currents in vestibular nerve afferents of the squirrel monkey. J Neurophysiol. 1984;51:1236.
Highstein SM, Goldberg JM, Moschovakis AK, Fernández C. Inputs from regularly and irregularly discharging vestibular-nerve afferents to secondary neurons in the vestibular nuclei of the squirrel mon- key. II. Correlation with output pathways of second- ary neurons. J Neurophysiol. 1987;58:719.
Highstein SM, Politoff AL. Relation of interspike baseline activity to the spontaneous discharges of primary afferents from the labyrinth of the toadfish, Opsanus tau. Brain Res. 1978;150(1):182.
Iwasaki S, Chihara Y, Komuta Y, Ito K, Sahara Y. Low-voltage-activated potassium channels underlie the regulation of intrinsic firing properties of rat ves- tibular ganglion cells. J Neurophysiol. 2008;100(4): 2192.
Honrubia V, Sitko S, Kimm J, et al. Physiological and anatomical characteristics of primary vestibular afferent neurons in the bullfrog. Int J Neurosci. 1981;15:197.
Simmons DD. Development of the inner ear effer- ent system across vertebrate species. J Neurobiol. 2002;53(2):228.
Sadeghi SG, Goldberg JM, Minor LB, Cullen KE. Efferent-mediated responses in vestibular nerve afferents of the alert macaque. J Neurophysiol. 2009;101(2):988.
Goldberg JM, Fernández C. Efferent vestibular system in the squirrel monkey: anatomical location and influence on afferent activity. J Neurophysiol. 1980;43:986.
Guth PS, Perin P, Norris CH, Valli P. The vestibular hair cells: post-transductional signal processing. Prog Neurobiol. 1998;54(2):193.
Lopez I, Meza G. Neurochemical evidence for afferent GABAergic and efferent cholinergic neu- rotransmission in the frog vestibule. Neuroscience. 1988;25:13.
Bernard C, Cochran SL, Precht W. Presynaptic actions of cholinergic agents upon the hair cell-afferent fiber synapse in the vestibular labyrinth of the frog. Brain Res. 1985;338:225.
Gacek RR, Nomura Y, Balogh K. Acetylcholinesterase activity in the efferent fibers of the stato-acoustic nerve. Acta Otolaryngol (Stockh). 1965;59:541.
Caston J, Rousell H. Curare and the efferent vestibu- lar system. Acta Otolaryngol (Stockh). 1984;97:19.
Schrott-Fischer A, Kammen-Jolly K, Scholtz A, Rask- Andersen H, Glueckert R, Eybalin M. Efferent neu- rotransmitters in the human cochlea and vestibule. Acta Otolaryngol (Stockh). 2007;127(1):13.
Matsubara A, Usami S, Fujita S, Shinkawa H. Expression of substance P, CGRP, and GABA in the vestibular periphery, with special reference to spe- cies differences. Acta Otolaryngol Suppl (Stockh). 1995;519:248.
Lysakowski A, Singer M. Nitric oxide synthase local- ized in a subpopulation of vestibular efferents with NADPH diaphorase histochemistry and nitric oxide synthase immunohistochemistry. J Comp Neurol. 2000;427(4):508.
Marlinski V, Plotnik M, Goldberg JM. Efferent actions in the chinchilla vestibular labyrinth. J Assoc Res Otolaryngol. 2004;5:126.
![]()
VESTIBULAR NUCLEI
Phylogeny Anatomy Neurotransmitters Physiology
VESTIBULO-OCULAR REFLEXES
Overview
Rotational Vestibulo-Ocular Reflexes Translational Vestibulo-Ocular Reflex Ocular Counterrolling
Semicircular Canal–Otolith Interaction
CERVICO-OCULAR REFLEXES
Anatomic and Physiologic Basis Characteristics of Neck-Induced Eye
Movements
VISUAL–VESTIBULAR INTERACTION
Visual Tracking Eye Movements Organization of Visually Guided Tracking
Eye Movements Comparison of Vestibular- and
Visual-Induced Eye Movements Visuo-Vestibulo-Ocular Connections Model of Visual–Vestibular Interaction
Adaptive Modification of the
Vestibulo-Ocular Reflext with Vision Cellular Basis for Visual Vestibular
Interaction
VESTIBULOSPINAL REFLEXES
Comparison of Ocular and Spinal Vestibular Reflexes
Vestibulospinal Connections Cerebellar–Vestibular Interaction Vestibulo-Collic Reflexes Cellular Mechanisms
SUBJECTIVE VESTIBULAR SENSATION
Vestibulothalamocortical Connections Response Properties of Thalamic Relay
Neurons
Response Properties of Vestibular Cortex Neurons
Functional Brain Imaging in Normal Human Subjects
Lesions of the Vestibulocortical Pathways in Patients
Psychophysical Studies
The vestibular system must integrate multiple internal representations of head and body movement obtained from several different sen- sory systems into a single internal coding of space that provides a frame of reference for encoding motor commands. Secondary vestibu- lar neurons are at the center of this sensorimo- tor transformation. They receive a multiplicity of signals originating in the vestibular end organs. Thousands of axons of primary vestibu- lar neurons enter each side of the brain stem to
innervate second-order neurons in four ana- tomically distinctive groups, located on the floor of the fourth ventricle.1–5 Afferents from the semicircular canals stay separate from each other, but they converge with afferents from the macules in a specific spatial pattern.6,7 Secondary vestibular neurons form a column of different lengths depending on the species, being the longest in humans (13.5 mm) and smaller in animals with evolutionarily smaller brains—frogs (2.2 mm), chinchillas (4.0 mm), and squirrel monkeys (5.0 mm). However, not all of the neurons in these nuclei receive
63
The main vestibular nuclei are the superior (angular or Bechterew’s), the lateral (Deiters), the medial (triangular nucleus of Schwalbe), and the descending (inferior or spinal) vestibu- lar nuclei. In addition, the vestibular nuclear complex includes several small groups of cells that are closely associated topographically with the main nuclei but have distinct morphologic characteristics and anatomic connections (e.g., the interstitial nucleus).8–10
There are a number of questions about the organization and function of the vestibular nuclei that are pertinent to clinical neurophysi- ology of the vestibular system. The amount of information arriving at the vestibular nuclei from different sources reaches staggering levels. In primates, since afferent vestibular neurons are characterized by their high level of sponta- neous activity, the nuclei receive about 100 action potentials/sec/nerve fiber (see Chapter 2). In humans, since each vestibular nerve has approximately 15,000 fibers, more than 1.5 million action potentials are received every second from the vestibular organs alone. How are the signals that originate in different receptor organs dis- tributed within the vestibular nuclei? Studies
using intracellular labeling techniques can trace the trajectory of afferent neurons to answer this important question. Each fiber usually inner- vates a restricted number of secondary neurons in all four of the vestibular nuclei. There are clear separations of afferent fibers such that specific areas in each nucleus preferentially receive afferents from specific receptors. At the same time, secondary vestibular neurons receive a converging input from different sensory organs from each ear—that is, where interactions between the organs of the two ears take place. The emerging picture is a complex one of both separation (channeling) and convergence of afferent signals at the level of the vestibular nuclei.
The biology of individual neurons in the ves- tibular nuclei is an important aspect of vestibu- lar physiology that is just beginning to be addressed. What transmitters are released and what ion channels and receptors are expressed in the different nuclei and within different neurons in the same nucleus? How do the characteristics of these secondary neurons influence signals arriving from the primary afferents and the organization of reflexes involved in the maintenance of gaze, equilib- rium, and orientation? Finally, how do neurons in this major sensorimotor integration center
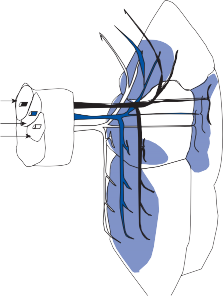
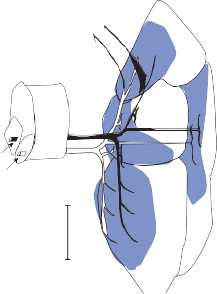
(b)
Cerebellum Cerebellum
S
L M
AC
HC UT
PC
SA D
1 mm
Figure 3–1. Distribution of primary vestibular afferent fibers (shaded blue areas) within the vestibular nucleus of the chin- chilla. (a) superior vestibular nerve, (b) inferior vestibular nerve. AC, anterior canal; D, descending nucleus; HC, horizontal canal; L, lateral nucleus; M, medial nucleus; PC, posterior canal; S, superior nucleus; SA, saccule; UT utricle.

GAD+ GAD– Dorsal
GAD+ GAD–
DVN
Lateral Medial Ventral
MVN
GAD+ GAD– GAD+ GAD–
Figure 3–2. Immunohistochemical staining of the vestibular nuclei (VN) in the chinchilla showing distribution of neurons that are glutamic acid decarboxylase (GAD) positive and GAD negative. Shaded blue areas receive primary vestibular afferents.
adapt to conflicting sensory perceptions that occur in daily life or after damage to the inner ear or motor apparatus?
Phylogeny
The vestibular nuclei are one of the first supraspinal cell groups that differentiate them- selves from the reticular formation.11,12 Lampreys have two discernible vestibular nuclear groups, the dorsal and ventral, com- posed of granular and spindle-shaped cells.
Modern fish (teleosts) have four discernible vestibular nuclei, although the nuclei contain relatively few cells. This basic organization of four vestibular nuclear groups is maintained throughout the higher vertebrates, although the relative size of each nuclear group varies from species to species (see earlier discussion). In invertebrates and early vertebrates, sec- ondary connections of the vestibular nuclei are primarily vestibulospinal, in keeping with their major role in maintaining body orientation.13 Vestibulocerebellar connections become pro- gressively more prominent in higher vertebrates.
The development of these “modern” vestibular pathways accompanies the development of increasingly complex somatic and ocular motor skills. In primates, vestibulocerebellar and vestibulo–ocular connections form a large part of the central vestibular pathways, and vestibu- lospinal connections are less prominent.13 The lateral vestibular nucleus (Deiters’ nucleus), a major source of vestibulospinal fibers, is the most prominent nuclear group in lower mam- mals, whereas in human beings it is small and almost confined to the vestibular root entry zone. By comparison, the superior vestibular nucleus is barely detectable in lower vertebrates but is prominent in humans, where it is the major source of vestibulo-ocular fibers. It extends ros- trally from the root entry zone (at the medullo- pontine junction) to the midpontine region.11
Anatomy
PRIMARY AFFERENT ENDINGS
The thousands of primary afferent vestibular nerve fibers arrive in the vestibular root in a specific orientation depending on their organ of origin (Fig. 3–1).1–5 After entering the brain stem, they divide into secondary ascending and descending branches that form a clearly defined vestibular tract in all animals studied. Branches from fibers in the ascending tract end in the rostral part of the vestibular nuclei or in the cerebellum, while branches from the descend- ing tract course in a ventrolateral direction in relation to the fourth ventricle, ending in the caudal vestibular nuclei.1–5
Individual primary afferent neurons provide multiple branches; in the bullfrog and cat, there is an average of 200 branches per affer- ent fiber.14–17 As illustrated in the chinchilla (Fig. 3–1), not all areas of the vestibular nuclei are innervated by the labyrinthine afferents. In addition, in the areas that receive primary afferents, signals from the vestibular organs interact with afferent fibers from other systems (visual and proprioceptive) and centers (especially the cerebellum).8,10
SUPERIOR VESTIBULAR NUCLEUS
The superior vestibular nucleus in humans extends from the caudal pole of the trigeminal motor nucleus approximately to the level of the
abducens nucleus.18–20 It is the smaller of the main nuclei with a length of 2.7 mm, contain- ing approximately 20,000 neurons.20,21 Medium- size neurons of about 15–30 µm in diameter predominate, with some large multipolar cells at the center. Most of the primary afferent pro- jections to the superior vestibular nucleus come from the cristae of the semicircular canals, arriving at the nucleus in the form of fascicles with a variety of fiber diameters.4 Large fibers terminate preferentially on the larger neurons in the center of the nucleus.2,10,22 Fibers from the superior semicircular canal are found medially, those from the horizontal and posterior canals more laterally (Fig. 3–1). Fibers from the utricle and saccule innervate only the periphery of the nucleus on the lateral side.
Another major group of afferent fibers origi- nates in the cerebellum. Those from the floc- culus end in the central region and those from the fastigial nucleus, nodule, and uvula end in the peripheral region.10,23 A group of fibers from the contralateral medial and descending nucleus connects the two sides.
Axons from the neurons in the superior ves- tibular nucleus run in the ipsilateral and con- tralateral medial longitudinal fasciculus (MLF) to innervate the motor nuclei of the extrinsic eye muscles; others project to the cerebellum and dorsal pontine reticular formation.24,25 Dendrites of neurons in the periphery of the nucleus extend into the adjacent reticular for- mation and into the principal trigeminal nucleus. Because of the pattern of afferent and efferent connections, the superior vestibular nucleus is a major relay center for ocular reflexes mediated by the semicircular canals.
LATERAL VESTIBULAR NUCLEUS (DEITERS’ NUCLEUS)
Beginning at the caudal end of the superior nucleus and ending below the level of the abducens nucleus, the lateral nucleus is trans- versed by the initial segments of the vestibular tract fibers corresponding to the root entry zone. In humans, the length of the lateral nucleus is 5.6 mm. It contains approximately 25,000 neurons.20,21 It is distinguished by the presence of giant cells (30 to 60 µm) that are relatively more numerous in the dorsocaudal than in the central ventral part.8,10 No sharp anatomic distinction divides these two parts of
the nucleus; in cats and chinchillas, only the rostroventral part receives primary vestibular afferents (the majority originating from the utricular macule). The dorsocaudal part receives afferent fibers from the vermis and fastigial nucleus of the cerebellum (see Fig. 3–26). Afferent components from other sources (spinal and commissural fibers) are few in comparison with those from the cerebellum and vestibular nerve. The lateral nucleus sends most of its efferent fibers to the spinal cord as the ipsilateral vestibulospinal tract (see Fig. 3–25). This projection is somatotopically organized in that fibers to the cervicothoracic cord originate from the rostroventral part of the nucleus, while fibers to the lumbosacral cord originate from the dorsocaudal part.26,27 The lateral nucleus also sends efferent fibers bilaterally to the MLF, which connect with the various oculomotor nuclei. Based on its fiber connections, the lateral vestibular nucleus is an important station for control of vestibulospi- nal reflexes, particularly those involving the forelimbs.28
MEDIAL VESTIBULAR NUCLEUS
The medial vestibular nucleus is located beneath the floor of the fourth ventricle caudal to the superior nucleus and medial to the descending (inferior) nucleus (see Fig.1–7). In humans, it is the largest nucleus (about 10 mm in length and a total volume of about 30 mm3) with by far the greatest number of neurons (about 125,000).20,21 It consists of cells of many different sizes (12 to 33 µm in diameter) and shapes that lie rela- tively close together, embedded in a fine mesh- work of very thin fibers that course in almost all directions.8,10,18 It differs from the other nuclei in that it does not receive large-diameter fibers.29 Anatomic separation from the superior nucleus is not well defined. Neurons in the upper part of the nucleus receive afferent fibers from the cristae of the semicircular canals as well as from the fastigial nucleus and flocculus of the cerebellum. Saccular and utricular affer- ents project to the medial lateral section of the nucleus.2,5,7 The caudal part receives its main afferents from the cerebellum (the ipsilateral and contralateral fastigial nucleus and the ipsi- lateral nodule). Other afferent contributions include a large projection from the contralat- eral medial vestibular nucleus and a small projection from the reticular formation.
Efferent connections from the medial nuclei run in the descending MLF to the cervical and thoracic spinal levels by way of the medial ves- tibulospinal tract (see Fig. 3–25). From the rostral area (receiving afferent input from the cristae), efferent fibers pass to the ascending MLF bilaterally to reach the nuclei of the ocul- omotor nerves.30 Other efferents are distrib- uted to the vestibular cerebellum, the reticular formation, and the contralateral vestibular nuclei.18 Because of its projections in the MLF to extraocular muscles and the cervical cord, the medial vestibular nucleus appears to be an important center for coordinating eye, head, and neck movements.31 The prominent com- missural connections are probably important for the compensatory processes following peripheral vestibular lesions.
DESCENDING (INFERIOR) VESTIBULAR NUCLEUS
The descending, or inferior, vestibular nucleus is difficult to differentiate anatomically from the adjacent medial vestibular nucleus. In humans, it has a length of about 8 mm with approximately 55,000 neurons.20,21 It consists of small and medium-sized cells with occasional giant cells.4,10,18 Projections from the labyrinth are restricted to the lateral side, with those from cristae extending more to the center and those from the macules to the periphery (utric- ular being more ventral, saccular–dorsal). Cerebellar afferents from the flocculus, nod- ule, and uvula are scattered throughout the nucleus, intermingling with the vestibular afferents. Projections from other sources, including spinal afferents, are minimal. Most of the efferent fibers from the descending nucleus pass to the cerebellum and to the reticular formation.28 Numerous commissural fibers supply the contralateral superior, descending, medial, and lateral nuclei.8,32 The descending nucleus apparently integrates ves- tibular signals from the two sides with signals from the cerebellum and reticular formation.
INTERSTITIAL NUCLEUS OF THE VESTIBULAR NERVE
Of the small groups of cells associated with the vestibular nuclei (such as groups x, f, p, m, and others), the interstitial nucleus is most clearly defined.4,10 It consists of small strands of
Neurotransmitters
Signal processing in the secondary vestibular neurons depends not only on anatomical con- nectivity but also on the type of synaptic recep- tors expressed and neurotransmitters released. At least eight neurotransmitters and four neu- ropeptides are involved.34–38 Among the major neurotransmitters, -aminobutyric acid (GABA), glutamate (glu), and glycine (gly) are believed to be the most important in both the synaptic input and output.37
All primary vestibular fibers release gluta- mate, an excitatory neurotransmitter, at their synapses in the vestibular nuclei.39 Glycine acts as a cotransmitter in large-diameter fibers. Most other neural inputs to the nuclei utilize terminals that express GABA immunoreactiv- ity, which suggests that they have an inhibitory influence upon vestibular nuclei neurons.39–41 The afferent excitatory action of glutamate on secondary vestibular neurons is mediated by both -amino-3-hydroxy-5-methyl-4- isoxasoleproprionic acid (AMPA) and N-methyl-d-aspartate (NMDA) glutamate receptors.42–44 In situ hybridization techniques found that all secondary vestibular neurons express the AMPA-selective receptor subunit GluR2 with the highest levels of expression in the giant Deiters’ cells.44 GABA receptors were found ubiquitously in the vestibular nuclei neurons by Lopez et al.41
Among the second-order vestibular neurons, most use glutamate as their excitatory37 and GABA as the inhibitory neuro- transmitter.36,39,40,45,46 In the chinchilla, the vestibular nuclei contain approximately 40,000 neurons—about 15,000 each in the medial and descending nuclei and the rest in the remain- ing nuclei. A large percentage of these neurons (about 60%) express glutamic acid
decarboxylase (GAD)—the enzyme used for synthesizing GABA—and consequently may act as inhibitory neurons.47 The distribution of neurons capable of releasing GABA (GAD+ neurons) among the four major vestibular nuclei is shown in Figure 3–2. Ninety percent of neurons < 7 µm in diameter are GAD- positive neurons, whereas 80% of neurons with a diameter >15 µm are GAD negative. Overall, it appears that most large neurons are excitatory and most small neurons are inhibitory.
Physiology
Vestibular signals originating in the two laby- rinths first interact with signals from other sen- sory systems at the neurons of the vestibular nuclei.32 Only a fraction of the neurons receive direct vestibular connections and, with the exception of the interstitial nucleus of the ves- tibular nerve, the neurons that receive primary vestibular afferent fibers also may receive affer- ents from the cervical area, the cerebellum, the reticular formation, the spinal cord, and the contralateral vestibular nuclei.31,48 Consequently, efferent signals from the ves- tibular nuclei reflect the interaction of these various afferent systems.49 For example, visual signals relayed through the cerebellar flocculus to neurons in the superior and medial vestibu- lar nucleus modulate the activity of the VOR.50–54 Inputs from neck proprioceptors modulate the vestibulocollic reflexes.55 The cerebellum influ- ences the vestibulospinal reflexes by means of connections between the vermis and the lateral and descending vestibular nuclei.56 Through connections with the reticular substance, ves- tibular neuron outflow interacts with descend- ing corticobulboreticular and reticulospinal signals.57
TYPES OF SECONDARY VESTIBULAR NEURONS
Following stimulation of the vestibular nerve with a single brief electric pulse, two different groups of secondary vestibular neurons have been identified on the basis of field potentials produced in areas of the brain stem receiving vestibular inputs (Fig. 3–3).58–60 This field potential consists of three components: an ini- tial positive–negative deflection from action

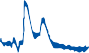
N1
N2
1.2
0.36
1.35
Figure 3–3. a Field potential recorded in the medial vestibular nucleus after electric stimulation of the ipsilateral ves- tibular nerve. N1 is generated by monosynaptic activated secondary vestibular neurons and N2 by multisynaptic activated neurons. b Response of a monosynaptic activated neuron N1 field potential is not seen because of superposition of spikes. c Response of a multisynaptic activated neuron demonstrating spikes timed with N2 field potential. Each recording is com- posed of about 20 superimposed traces. (Adapted from Precht W, Shimazu H. Functional connections of tonic and kinetic vestibular neurons with primary vestibular afferents. J Neurophysiol.1965;28: 1014.)
currents in the primary vestibular fibers, a neg- ative deflection wave (N1) with a short latency of
<1.0 msec (generated by monosynaptically
activated secondary vestibular neurons and fibers), and a delayed negative deflection (N2)
with a latency of about 2.5 msec (generated by
multisynaptically activated neurons and fibers) (Fig. 3–3a). By carefully placing microelec- trodes in the vicinity of or inside secondary ves- tibular neurons and tailoring the electric stim- uli, it has been demonstrated that some neurons produce action potentials at the time of the extracellular N1 wave with latencies between
0.5 and 1.0 msec (Fig. 3–3b), suggesting
that they receive monosynaptic input. Other neurons produce delayed action potentials (Fig. 3–3c), which suggests that they might be activated through multisynaptic connections. Only about 75% of neurons are activated by nerve stimulation and approximately half of these are monosynaptically activated.58,60 All monosynaptic connections are ipsilateral and excitatory. Among the monosynaptically acti- vated neurons, about 37% respond to small electrical stimuli with very short latencies that activate only the thickest, most sensitive irreg- ular primary afferents.61 The rest of the neu- rons respond to larger electrical currents. This suggests that they receive a predominant input from thinner, regular afferents. This differen- tial input is at least in part reflected in the output of the secondary neurons with vestibulospinal neurons receiving more input from irregular afferents and vestibulo-ocular neurons receiv- ing more input from regular firing afferents. However, it would be wrong to view secondary vestibular neurons as narrowly tuned channels, with each receiving only a single kind of pri- mary afferent input.61 Most vestibular nuclei neurons, even those predominantly related to regular and irregular afferents, receive a broad range of afferent inputs.
The second-order vestibular neurons can be divided into two groups: type I neurons, which are excited, and type II neurons, which are inhibited by ipsilateral head rotations.48,54,62,63 Type I neurons are also excited by electrical stimulation of the ipsilateral vestibular nerve. The physiological response of type I neurons is similar to that of primary vestibular afferents. Type II neurons respond to electrical stimula- tion of the contralateral vestibular nerve with greater latency than do type I neurons to ipsilat- eral stimulation. The physiological response of type II neurons is opposite that of type I neu- rons—that is, type II neuronal response is qualitatively similar to the response of primary afferent neurons innervating the contralateral ear. Among the type I neurons there are two groups (Fig. 3–4). One group activates the agonist motor neurons—for example, motor neurons innervating the lateral rectus of the opposite side and the medial rectus of the ipsi- lateral side for the horizontal semicircular canal reflex. These neurons are excitatory, that is, GAD-negative neurons, as illustrated in Figure 3–2. The second group of type I neurons inhib- its the antagonist muscles—for example, the ipsilateral lateral rectus and the contralateral medial rectus. These neurons are GAD posi- tive. Type II neurons are activated by type I neurons from the contralateral side.62 After destruction of the labyrinth, excitation of type II neurons by stimulation of the contralateral nerve inhibits neighboring type I neurons. Type II neurons are GAD positive, which is con-
sistent with their inhibitory action (Fig. 3–4).
In summary, during physiological rotatory stimulation of the horizontal semicircular canals, the central vestibular neurons on one side receive ampullopetal signals (increased firing), while the neurons on the opposite side receive ampullofugal signals (decreased firing). However, since excitatory type I neurons are
Middle Line
Type II GAD+
Left
Horizontal Semicircular Canal
Scarpa’s Ganglion
Type I GAD–
Type I GAD+
Vestibular Nucleus
Rt MR Lt LR
Lt MR Rt LR
Rt MR Lt LR
Lt MR Rt LR
Oculomotor Neurons
Figure 3–4. Interrelation of type I and type II secondary vestibular neurons. Blue neurons are inhibitory and white neurons are excitatory. LR, lateral rectus; MR, medial rectus.
connected to inhibitory type II neurons of the opposite side, the excitatory type I neurons receive a combination of stimulation from the two ears, corresponding to the sum of the ampullopetal (excitatory signals) from the ipsi- lateral side and the inverted ampullofugal (dis- inhibitory) signal mediated by the type II (inhibitory) neurons (Fig. 3–4). Most of the cells in the vestibular nuclei also respond to visual stimulation.64,65 The main visual input to the vestibular neurons comes from the cerebel- lum, particularly the flocculonodular lobe.52,53,66,67
INTRINSIC MEMBRANE PROPERTIES OF SECONDARY NEURONS
In most physiological studies, secondary ves- tibular neurons are identified either by their location within the know boundaries of the ves- tibular nuclei or by their activation by stimula- tion of the eighth nerve or some of its branches. Secondary neurons are most clearly identified in whole-brain preparations where the eighth nerve or nerves from individual canals or macules can be selectively stimulated. These preparations have been particularly useful for studying vestibulo-motor signal processing in central networks. By contrast, slice prepara- tions that are disconnected from most inputs and outputs have been particularly useful for studying membrane properties in individual neurons with the disadvantage that neurons
can only be identified as secondary vestibular neurons based on their location within the boundaries of the known vestibular nuclei.
Just as with primary vestibular neurons sec- ondary neurons can be classified as regular (tonic) and irregular (kinetic) based on their spontaneous firing pattern.58 Spontaneous activity is probably due to a combination of intrinsic membrane properties of the second- ary vestibular neurons and excitatory and inhibitory input from primary afferents.68–71 Based on an average spontaneous firing rate for secondary vestibular neurons of about 30 spikes/sec one can estimate that about half of the spontaneous rate is generated by intrinsic pacemaker activity and half by spontaneous primary afferent input since the spontaneous rate is reduced to 16 spikes/sec in alert animals that undergo bilateral labyrinthectomy.32,72
In vitro slice preparations from the medial vestibular nucleus of rat and guinea pig have allowed a classification of secondary vestibular neurons based on action potential shapes and membrane properties (Table 3–1).68,73–75 Type A secondary neurons show a single deep after- phyperpolarization (AHP) after each action potential and a rectifying IA-like K+ current when
released from hyperpolarization (Fig. 3–5a). By
contrast, type B secondary neurons have an ini- tial fast AHP followed by a delayed slow AHP
and no IA-like rectification (Fig. 3–5b). Also, type A neurons have wider action potentials than
type B neurons. Type A secondary neurons
Table 3–1 Functional classes of different secondary vestibular neurons identified in the rodent medial vestibular nucleus
![]()
Type A Neurons Type B Neurons
![]()
After-hyperpolarization Monophasic, large amplitude Biphasic, small amplitude A-like rectification Strong Weak
Action Potential Broad Thin
Discharge regularity Regular Irregular
Dynamic response Mostly tonic Phasic-tonic
Major afferent inputs Thin afferents; no input from
flocculus
Thin and thick afferents; some receive floccular input
![]()
predominantly receive input from thin primary afferents, have a regular spontaneous firing rate, and have a wide linear range, whereas type B neurons receive input from thick and thin affer- ents, have an irregular spontaneous firing rate, and have a relatively smaller linear range.32 It appears that type A neurons correspond to the tonic secondary neurons and type B neurons correspond to kinetic neurons in the earlier classification of Shimazu and Precht.58
Potassium channels are critical for determin- ing resting potential, shaping action potentials, and controlling discharge regularity; prelimi- nary studies suggest that K+ channels have differential expression in type A and type B secondary vestibular neurons.32,74,75 As with primary vestibular neurons Ca2+-dependent K+ channels play a key role in shaping the AHP.76 Initial studies attempting to correlate physio- logical properties with neurochemistry indicate
that the majority of type A secondary neurons are GABAergic neurons, while type B neurons can be either GABAergic or glutamatergic.77
COMPENSATION AFTER LABYRINTHECTOMY
Knowledge of the different types of secondary vestibular neurons and their interconnecting pathways is important for understanding the sequence of recovery following a unilateral loss of labyrinthine function.78–80 On the basis of connections depicted in Figure 3–4 it can be anticipated that immediately after a labyrinth- ectomy, the ipsilateral type I neurons lose their spontaneous activity and become unresponsive to ipsilateral angular rotation due to excessive inhibitory activity arriving via commissural pathways. At the same time, contralateral healthy type I neurons lose their inhibitory
a b c
Type A MVN neuron
20 mV
50 ms
Type B MVN neuron
Spike threshold
Contralesional side Control
Ipsilesional side
5 mV
2 ms
Figure 3–5. Changes in spike profiles of guinea pig medial vestibular nucleus (MVN) neurons after unilateral labyrinth-
ectomy. A: typical spikes of a type A MVN neuron a with a monophasic after-hyperpolarization (AHP; arrow) and a type B MVN neuron b with a biphasic AHP (double arrow) recorded on the contralesional side of slices taken one month post-lesion; superposition at spike threshold of averaged spikes of type B MVN neurons at the resting membrane potential c recorded in control slices and from the ipsi- and contralesional sides of slices taken one month after the lesion. All neurons were identified as type B MVN neurons by the presence of the biphasic AHP at hyperpolarized membrane potentials. Note the inverse changes in the amplitude of the AHP of spikes recorded on the two sides. (From Straka H, Vibert N, Vidal PP, Moore LE, Dutia MB. Intrinsic membrane properties of vertebrate vestibular neurons: function, development and plasticity. Prog Neurobiol. 2005;76:349, with permission.)
contralateral input, and their spontaneous activity increases in comparison to normal levels.81 Contralateral type II neurons lose their inputs from deafferented excitatory type I neu- rons and cannot be identified electrophysiolog- ically. Specifically, an imbalance in the muscle tone takes place, resulting in the signs of laby- rinthectomy—spontaneous nystagmus, yaw and roll tilt of the head, asymmetric tone in extensor muscles, and falling toward the side of the lesion.
A few days after a labyrinthectomy, the pre- viously silent type I neurons on the damaged side recover their spontaneous activity and begin to respond to physiologic stimulation of the contralateral labyrinth,82–84 as a result of their connections with ipsilateral type II neu- rons. These reactivated type I units are inhib- ited when the type I neurons on the healthy side are excited and are disinhibited when the contralateral type I neurons are inhibited. The recovery of sensitivity in the ipsilateral type I neurons after a labyrinthectomy parallels the time course of or improvement in clinical symptoms and signs. Experimental studies in rodents confirm that immediately after a uni- lateral labyrinthectomy there is a marked increase in GABA release in the ipsilesional vestibular nucleus that is not prevented by bilateral flocculectomy, indicating that it is due to hyperactivity of the inhibitory commissural pathway.85 With vestibular compensation and recovery of vestibular symptoms and signs, the elevated GABA levels on the ipsilesional side return toward normal levels.86 At the same time there is a downregulation of GABA receptor efficacy, decreasing the response to commis- sural inhibitory drive.
The return toward normal spontaneous fir- ing in secondary neurons after labyrinthectomy is associated with changes in both active and passive membrane properties.87–90 Overall the resting membrane properties of secondary neurons on the ipsilesional side shift toward type A-like properties and those on the contral- esional side shift toward type B-like properties (Fig. 3–5c). The AHP and discharge regularity of type B neurons on the ipsilesional side are selectively augmented, while the reverse occurs on the contralateral side.91,92 These changes result from changes in expression of a variety of genes and proteins that determine each cell type.32 On average, secondary neurons on the ipsilesional side develop a more regular
spontaneous firing rate with tonic dynamics, whereas those on the contralesional side have a more irregular spontaneous rate with phasic dynamics. The result is a switch from the balanced “push-pull” mode in the normal con- dition to a state where the ipsilesional side provides more tonic activity in vestibular cir- cuits while the contralesional side provides more of the dynamic responses.32
After labyrinthectomy, in parallel with the changes in membrane properties, there is a reorganization of synaptic inputs onto the deaf- ferented secondary neurons, changes in synap- tic sensitivity to neurotransmitters, and changes in vestibular neuronal networks.80,89,91,92 Similar lesion-induced plasticity is seen in other sen- sory systems and appears to be a general reac- tion pattern to injury, the so-called distributed process described by Llinas and Walton.32,93 Triggers for the plastic changes after unilateral labyrinthectomy are not entirely clear. Intracellular messenger systems are activated to initiate the gene/protein changes required for the compensation process.94 The transcrip- tion factor c-Fos is activated in secondary neu- rons on both sides and could be a trigger for changes in expression of channel proteins and transmitter receptor subtypes.95–97 There is an increased expression of the transmembrane phosphoprotein GAP-43, known to be involved in growth, regeneration, and remodeling of neuronal pathways.98 The neurotrophic factor BDNF is transiently expressed in some sec- ondary vestibular neurons after labyrinthec- tomy and mutant mice deficient in BDNF have impaired vestibular compensation.99 Increases in intracellular Ca2+ concentration seen in sec- ondary vestibular neurons after labyrinthec- tomy might be a link between changes in genetic regulation and changes in membrane and discharge properties.32
Overview
All sensorimotor reflexes have a reference frame for coding spatial information.100 The vestibular receptors are fixed in the head so they have a head-centered reference frame. They provide no information about how the head moves relative to the body or how the
The inner ear does provide information in a world-reference frame generated by the gravi- ty-sensing otolith organs. As will be seen later, the velocity storage system uses this informa- tion to align the low-frequency rotational VOR with gravity. Angular velocity from the semicir- cular canals is combined with gravitational information from the otoliths into two compo- nents, one parallel to gravity and the other perpendicular to gravity.102,103
As noted earlier, the otolith organs detect net linear acceleration but do not distinguish between translational movements and gravity, the so-called tilt-translation ambiguity. One solution to the tilt-translation ambiguity is to frequency filter the otolith signals such that low-frequency otolith components are inter- preted as gravitational acceleration and high- frequency components are interpreted as inertial accelerations.104 Another solution is convergence of canal and otolith signals begin- ning at the vestibular nuclei.105,106 Unlike pri- mary afferent neurons that fire the same regardless of the type of linear acceleration, many secondary neurons fire selectively for translational motion and are relatively silent during changes in head position relative to gravity (Fig. 3–6). These translational sensitive neurons are not only found in the vestibular nuclei but also in the cerebellum, particularly in areas that receive primary vestibular affer- ents. The translation-only secondary neurons disappear after canal plugging, proving that they result from convergence of canal and otolith signals. The tilt-translation ambiguity is not always resolved at the perceptual level, however, because the canals do not provide a
reliable estimate of angular velocity at very low frequencies (<0.1 Hz).107,108 It was the illusion of tilt experienced when riding a curve in the railroad line that lead Mach to postulate a “sixth sense” for the perception of acceleration in the head. Somatogravic and oculogravic illusions commonly occur during airplane landings and takeoffs due to the tilt-translation ambiguity. Extravestibular signals are required to avoid these illusions.
The brain must also differentiate between self-generated and externally applied move- ments. Primary afferents fire the same regard- less of whether the movement is active or pas- sive. A common strategy for solving this problem is the principle of reafference—a copy of the expected sensory result of a motor com- mand is subtracted from the sensory signal.109 This principle has been well studied in mechanosensory systems of primitive animals where self-generated behaviors are selectively suppressed in central neurons.
Rotational Vestibulo-Ocular Reflexes
The basic organization of the rotational VOR in the horizontal plane is shown in Figure 3–4. Type I secondary neurons make direct contact with oculomotor neurons and provide axon col- laterals to other secondary neurons and to the cerebellum (not shown).110 Commissural con- nections connect the two sides. These and other feedback pathways form the velocity stor- age system, which allows signals from different vestibular receptors and other sensory systems to interact with vestibular signals while sustain- ing the activity in the vestibular nuclei beyond the time of arrival of the primary afferent signal.111
The effect of velocity storage is graphically illustrated in Figure 3–7 (see also Fig. 1–5). After an impulse of head acceleration, the time constant (TCOR) of the oculomotor response is
prolonged beyond that of the primary afferent
response (T1) because of feedback onto the secondary vestibular neurons. Quantitatively, the
positive feedback loops perform the equivalent of a partial mathematical integration on the primary afferent signal.112 The interneurons in these feedback pathways can be viewed as valves con- trolling the spontaneous activity and dynamic properties of the secondary vestibular neurons.
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Translation only net accel: 0.2 g
Roll tilt only net accel: 0.2 g
Roll tilt-translation net accel: 0 g
Roll tilt + translation net accel: 0.4 g
![]()
![]()
![]()
![]()

![]()

![]()
![]()
![]()
Otolith afferent
IFR ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Htrans ![]()
100 sp s–1
20 cm
![]()
Hroll
![]() 12º
12º
Vestibular nucleus neuron
IFR ![]()
![]()
![]()
![]()

![]()
![]()
Htrans
100 sp s–1
![]()
![]()
20 cm
![]()
![]()
Hroll
![]() 12º
12º
![]()
Figure 3–6. Instantaneous firing rate (IFR) from a primary otolith afferent (top) that encodes net linear acceleration, and a central vestibular nucleus neuron (bottom) that encodes translational motion during four movement protocols: Translation only, Tilt only, Tilt – Translation and Tilt + Translation (0.5 Hz). The stimulus (bottom) traces show sled position (Htran) and roll tilt position (Hroll). (From Angelaki DE, Cullen KE. Vestibular system: the many facets of a multimodal sense. Annu Rev Neurosci. 2008;31:125, with permission.)
Velocity storage
Canal
signal
T1
TCOR
to EOM’s
Figure 3–7. Prolongation of the dominant time constant of the canal ocular reflex (TCOR) by velocity storage within the feedback pathways shown in Figure 3–4. A step change in head angular velocity occurred at time 0 (vertical arrows). T1 represents the long time constant of the cupula measured from the average response of primary afferent neurons. EOM, extra ocular muscle.
From the above-described experimental data and model simulations, the rotational VOR is thought to be mediated by two components: a rapid, or “direct,” and a slow “indirect,” or “velocity storage,” pathway.111 The three- neuron chain that conveys neural signals from the labyrinth to the motor neurons of the extraocular muscles constitutes the direct path- way. It has been modeled as a direct pathway because it does not substantially alter the dynamics of the semicircular canal responses transferred to the eye muscles. The indirect VOR pathway also receives semicircular canal input, but it has slow charge and discharge time constants. It holds, or “stores,” activity from the vestibular periphery and discharges it over a more prolonged time span, thus the term “velocity storage” and the designation “indi- rect” in models of the VOR. Commissural path- ways between the two vestibular nuclei are involved in both direct and indirect pathways, but the fibers appear to cross at different lev- els.113 In monkeys, an experimental midline section of rostral medullary commissural fibers abolishes oculomotor and vestibular functions attributable to velocity storage, whereas the direct VOR pathway remains intact.113 Utrastructural studies after such lesions indi- cate that the commissural neurons related to velocity storage are located in the lateral cres- cents of the rostral medial vestibular nucleus. Immunohistochemical staining of the neurons in this portion of the medial vestibular nucleus in monkeys identified at least two types of GABAergic neurons: neurons related to the velocity storage pathway and vestibular interneurons.114 A variety of GABAergic bouton endings was also identified in this region of the medial vestibular nucleus: type A boutons cor- responding to Purkinje cell afferents, type B
terminals from axons of GABAergic medial ves- tibular neurons, and type C boutons originating from the vestibular commissural pathway of velocity storage.115
SEMICIRCULAR CANAL–OCULAR CONNECTIONS
Detailed information about the connections that link vestibular receptors and different eye muscles was initially obtained by recording the eye muscle response following either physiologic or electric stimulation of each receptor.116–118 By measuring the muscle tonus, the excitatory or inhibitory nature of each connection was established. Table 3–2 summarizes the primary excitatory and inhibitory connections of each semicircular canal with the muscles of both eyes.117 Note that each semicircular canal is connected to the eye muscles in such a way that stimulation of the canal nerve results in eye movement approximately in the plane of that canal. For example, stimulation of the left posterior canal nerve causes excitation of the ipsilateral superior oblique and the contralat- eral inferior rectus muscles while inhibiting the ipsilateral inferior oblique and the contralateral superior rectus. An oblique downward move- ment in the plane of the left posterior canal is the end result. As suggested in Chapters 1 and 2, the spontaneous activity of neurons in each canal leads to a resting level of contraction at each eye muscle from all the labyrinthine organs and provides an important background upon which these more specific reflexes act.116 By systematically recording responses in different vestibular and oculomotor nuclei after selective stimulation of each semicircular canal, it has been possible to trace the main disynaptic excitatory and inhibitory pathways
Table 3–2 Connections of the Semicircular Canals with Muscles of the Eyes
Semicircular Canal | Excitation | Inhibition |
Horizontal | I–MR C–LR | C–MR I–LR |
Posterior | I–SO C–IR | I–IO C–SR |
Anterior | I–SR C–IO | I–IR C–SO |
C, contralateral; I, ipsilateral; IO, inferior oblique; IR, inferior rectus; LR, lateral rectus; MR, medial rectus; SO, superior oblique; SR, superior rectus.
connecting the semicircular canals with the extraocular muscles, as shown in Figure 3–8 and Table 3–2.117,119,120 As a general rule, excitatory connections run in the contralateral MLF and inhibitory connections run in the ipsilateral MLF.121
Each muscle receives an excitatory input originating from the ampullopetally activated canal and a disinhibiting input from the ampull- ofugally activated canal pair on the opposite side operating in reciprocity. For example, the right medial rectus receives an excitatory input from the right horizontal semicircular canal via the ipsilateral type I excitatory (GAD-negative) neurons and a disinhibition input mediated through the contralateral type I (GAD-positive) neurons (see Fig. 3–4). Consequently, during clockwise rotation, the muscle will experience an increase in the excitatory input from the ampullopetally activated canal (right horizontal semicircular canal [Rt HSC]) and a decrease in the inhibitory signal from the ampullofugally
activated canal (Lt HSC). This push–pull organization provides more stability to the reflexes and improves the dynamic linear range of the system. The muscles are in a constant state of tension and respond to minimal modulation of activity of the peripheral organs.
The connections illustrated in Table 3–2 are only part of the picture, however. Since the planes of the semicircular canals are not exactly aligned with the planes of the three pairs of eye muscles, a spatial transformation from canal to muscle coordinates must occur if eye move- ments are to compensate for head movements. In other words, it is not adequate to simply connect afferents from a single canal to a set of eye muscles as shown in Figure 3–8; other con- nections must also exist. Studies of labeled sec- ondary vestibular neurons, identified as part of the canal ocular reflex, indicate that the spatial transformations occur through both a conver- gence of signals at the level of the vestibular
a b c
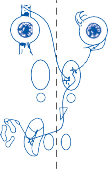
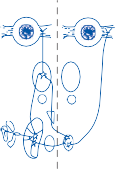
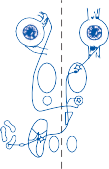
SR MR LR SO
III IO
IV
AC
VI
M
VN
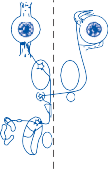
![]()
d
ATD
HC
D
e
IR
MLF
S
L
PC
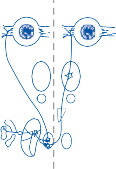
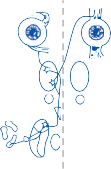
![]()
f
SO
IR III
SR
LR MR
IO
IV
AC
HC
VI
VN PC
Figure 3–8. Excitatory (a, b, c) and inhibitory (d, e, f) pathways between the individual semicircular canals and eye muscles in the cat.96,97 AC, anterior canal; ATD, ascending tract of Dieters; HC, horizontal canal; III, oculomotor complex; IO, inferior oblique; IR, inferior rectus; IV, cochlear nucleus; L, lateral nucleus; M, medial nucleus; D, descending nucleus; LR, lateral rectus; MLF, medial longitudinal fasciculus; MR, medial rectus; PC, posterior canal; S, superior nucleus; SO, superior oblique; SR, superior rectus; VI, abducens nucleus; VN, vestibular nuclei.
nuclei and a divergence of signals at the level of the oculomotor nuclei.122,123
COMPENSATORY EYE MOVEMENTS
The semicircular canal–ocular reflexes produce eye movements that compensate for head rota- tions. Angular and sinusoidal head rotation of small amplitude within the frequency range of natural head movements (0.1 to 4.0 Hz) results in compensatory sinusoidal eye movement ampli- tudes that are 180 degrees out of phase with the head (i.e., eye movements in the opposite direc- tion as the head movement) (as illustrated in Fig. 1–12a, b). The various transformations involved in this process are illustrated in Figure 3–9. The natural stimulus for the semicir- cular canals is head angular acceleration, as shown in Figure 3–9b. During sinusoidal rota- tion, at the frequencies of natural head move- ments, the viscoelastic properties of the canal– cupula complex (as defined by the pendulum model in Chapter 2) produce the equivalent of one step of mathematical integration (a 90-degree phase shift) so that the vestibular nerve firing rate (Fig. 3–9e) is in phase with head
velocity (Fig. 3–9c) rather than head acceleration (Fig. 3–9b). But the normal reflex response pro- duces a compensatory eye movement amplitude that is equal and opposite to the amplitude of the head movement (compare a and g in Fig. 3–9). This eye movement results from activation of, among others, the abducens nerve to the left lat- eral rectus muscle (Fig. 3–9f) during ampullo- petal stimulation of the right cupula–vestibular nerve (Fig. 3–9d, e). However, the timing of the recorded activity in the abducens nerve lags behind the activity in the vestibular nerve by an additional 90-degree delay. This raises a key question first asked by Skavinski and Robinson: what produces the phase shift (i.e., increase from 90 degrees to 180 degrees) between the firing rates of the vestibular nerve and abducens nerve (between Fig. 3–9e and Fig. 3–9f)?124 To answer this question, they introduced the concept of an oculomotor integrator, a hypothetical neural net- work that integrates, in a mathematical sense, velocity-coded signals, such as those originating in the vestibular end-organ, to position-coded signals required by the oculomotor neurons. Although the concept of neural integration is now generally accepted, the specifics are
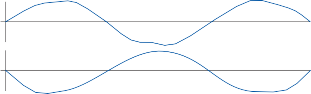
![]()
R a
L
+ b
0
–
+
c
0
–
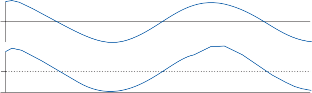
Ap
center d
Af
e
spikes/sec
spikes/sec
baseline
0
![]()
f
baseline
0
![]()
R g
L
Head position
Head acceleration
Head velocity
Right cupula displacement
Right vestibular nerve firing
Left abducens nerve firing rate
Left eye position
Figure 3–9. Mechanism by which sinusoidal change in head position (a) is converted to an equal and opposite-eye position (g). R, right; L, left; Af, ampullofugal (i.e., away from the ampulla); Ap, ampullopetal (i.e., toward the ampulla). See text for details.
still debated. Some investigators feel it is “local- ized” in a region of the brain stem125,126 or cere- bellum,127 whereas others consider it a “distrib- uted property” of the feedback pathways shown in Figure 3–4. Mathematical models show how these feedback pathways, particularly those via the commissural connections, can produce the necessary integration.128,129
Although the VOR operates as an effective integrating angular accelerometer for frequen- cies >0.1 Hz, at lower frequencies the VOR is progressively less effective, which is manifest by a phase lead of eye velocity relative to head velocity, reaching a maximum of 90 degrees at about 0.001 Hz. Velocity storage within the central VOR feedback pathways improves the low-frequency gain and phase deficit of incom- ing primary afferent signals but does not cor- rect it completely. As will be shown later, this low-frequency gain and phase deficit of the VOR is of little functional significance, since natural head movements combine visual and vestibular stimulation and the combination of visuovestibulo-ocular responses results in essentially perfect compensation, even at the lowest frequencies. However, the low- frequency phase shift does have important implications for clinical testing since an increase in the low-frequency phase two-standard devi- ations above the mean normal value is a non- specific sign of damage to the canal-ocular reflexes (see Chapter 7).
In summary, the rotational VOR involves the activity of many nuclei and a countless number of neurons whose group behavior may differ from that of the isolated units. One must keep this complexity in mind when attempting to evaluate the effects of lesions on VOR activity. It is often impossible to interpret the results of vestibular tests in terms of deficits in a single neural pathway.
NEURONAL MECHANISMS
Based on recordings from the vestibular nuclei in animals three groups of secondary vestibular neurons likely provide the main premotor drive for the rotational VOR: (1) position-ves- tibular pause (PVP) neurons, (2) eye-head (EH) neurons, and (3) burst tonic (BT) neurons.130–132 PVP neurons modulate their firing rate during visual suppression of the VOR and during smooth pursuit and stop firing during saccades. Their response to angular
head velocity and eye velocity is in the opposite direction so that the signals are complimentary during visualization of an earth-fixed target. EH neurons also modulate their firing rate during both smooth pursuit and vestibular sup- pression but in the opposite direction of PVP neurons such that the two signals oppose each other during visualization of an earth-fixed tar- get. BT neurons do not modulate their activity during vestibular suppression, but their firing rate is strongly correlated with all types of eye movements. Based on these features it appears that BT neurons carry a fully transformed motor signal, whereas PVP and EH neurons carry signals with intermediate features in the transformation of sensory input from the semi- circular canals to motor signals required by the oculomotor neurons.
The relationship between the firing rate of oculomotor neurons and the movements of the eyes during each phase of nystagmus has been studied extensively. During the production of an agonist slow component, the membrane potential is slowly depolarized by excitatory postsynaptic potentials arriving via the vestibulo- ocular pathways discussed in the previous sections.133,134 Toward the end of the slow com- ponent, the membrane potential rapidly becomes hyperpolarized and the motoneuron abruptly terminates its discharge. This hyper- polarization is produced by inhibitory activity of a group of neurons different from those producing the slow component.
Figure 3–10 illustrates the firing rate of a single right abducens nerve fiber during sinu- soidal angular rotation at three different mag- nitudes.135 The concurrent nystagmus of the left eye is shown above each firing record. With slow components to the right, the right abducens nerve innervates an agonist muscle, and a steady increase in nerve firing occurs that is roughly proportional to the eye displace- ment. Just before initiation of the fast compo- nent in the opposite direction (to the left), the firing of the right abducens nerve suddenly decreases and, in many instances, stops com- pletely. During the subsequent slow compo- nent, the nerve fiber remains silent until the eye reaches a position in the orbit that is above threshold for this particular abducens neuron. With slow components to the left, an abrupt increase in firing rate occurs just before the onset of the fast component, followed by a slow decrease during the slow components.
Measurement of the relationship between motoneuron firing rates and eye movement induced by vestibular or visual stimuli has shown that the motoneurons behave the same regardless of the nature of the stimulus.136 Almost all oculomotor neurons exhibit a thresh- old above which they increase their firing rate roughly in proportion to the change in eye posi- tion in the orbit. A small percentage of the change in firing rate (approximately 20%) is proportional to the velocity of the eye move- ment. It is as though the firing rate of the ocul- omotor neurons were designed to overcome the elastic and viscous forces (roughly in a ratio of 5 to 1) restraining the eye in the orbit. This relationship can best be appreciated by exam- ining the rate of firing of an oculomotor neuron associated with a visually induced refixation saccade (Fig. 3–10d), where the goal is to move the eyes as rapidly as possible from one posi- tion in the orbit to another and to maintain the new position once it is reached. During the high-velocity saccade, the oculomotor neu- ron increases its firing rate to a high level to compensate for the viscous drag of the eye
ligaments.137 Once the new position is reached, a much lower rate of discharge produces com- pensation for the elastic restraining force and maintains the new position. Although the reflex pathways for vestibular and visually induced eye movements involve different neuronal cir- cuits, the motoneurons governing the extrinsic eye muscles fire in the same manner regardless of the original sensory input.
FAST COMPONENT GENERATION
As noted in Chapter 1, neurons in the parame- dian pontine reticular formation (PPRF) fire in short bursts just before the onset of horizontal fast components and voluntary saccades.138–140 Fast eye movements, whether voluntary or involuntary, apparently are generated by a common neuronal mechanism.138,141 The PPRF is not a discrete anatomic structure but rather a region that has been designated as such because ofitsapparentfunctionalspecificity. Stimulation in the PPRF produces ipsilateral slow and rapid eye movements, depending on the stimulus variables.139,140 The latency of induced eye movements suggests that one or two synapses lie between the pontine neurons and the oculomotor neurons. Anatomic pathways between this area of the reticular formation and the eye muscle motor nuclei were
![]()
a
![]()
![]()
![]()
100 ![]()
0
![]()
![]()
b
![]()
100
0
![]()
c
![]()
![]()
100
0
![]()
![]()
d
![]()
100
0
![]()
![]()
0 10 20 30
Seconds
Figure 3–10. Right abducens motoneuron activity during induced nystagmus in the cat. In each pair of traces, the top trace represents the electrooculographic recording of eye movement and the bottom trace, the motoneuron firing fre- quency, (a–c) The animal was rotated at the frequency of 0.1 Hz at peak velocities of 30°, 60°, and 120°/sec, respectively,
Spontaneous eye movements (saccades).
first reported by Lorente de Nó110 and subse- quently confirmed by other investigators.140 Numerous documented anatomic pathways also interconnect the vestibular nuclei with the PPRF.140,142
PATTERN OF EYE MOTION
Intuitively one might assume that the slow phases of nystagmus deviate the eyes toward the periphery of the orbit and the fast compo- nents reset them back to the center. Indeed, this pattern occurs in the rabbit (see Fig. 1–12 in Chapter 1). In animals with more developed visual oculomotor function, however, the fast components act as anticipatory movements taking the eyes toward the periphery.143 The fast components of the initial beats of nystag- mus are larger than the preceding slow compo- nents, and the eyes deviate in the direction of the fast component (see Fig. 7–19a, b, and c in Chapter 7). In humans, the exact threshold position for the generation of a saccade varies with the velocity of the slow component of nystagmus, but it is usually near the mid- position.144 The apparent advantage of this strategy is that the eyes are ready to focus on newly arriving targets in the field of rotation, and fixation can be maintained during the subsequent slow component.
EFFECT OF EXPERIMENTAL LESIONS
Spontaneous vestibular nystagmus is produced by lesions of the labyrinth, the vestibular nerve, and the vestibular nuclei.118,145 A key ingredient for the production of spontaneous nystagmus is an imbalance of tonic activity within the vestibulo-ocular pathways. If a pro- cess simultaneously removes both labyrinths, spontaneous nystagmus does not result, dem- onstrating that, for production of nystagmus, the relative balance of input is more important than the absolute magnitude of input. Spontaneous nystagmus produced by section- ing of the vestibular nerve duplicates that resulting from labyrinthectomy. The slow com- ponent is directed toward the side of the lesion. The direction of spontaneous nystagmus asso- ciated with lesions of the vestibular nuclei, however, is less predictable and depends on the location and extent of the lesion. Typically, the slow phase of nystagmus is contralateral to lesions in the superior and rostral medial
nuclei and ipsilateral to lesions in the lateral and caudal medial nuclei. The imbalance between inhibitory and excitatory secondary vestibular neurons undoubtedly determines the direction of spontaneous nystagmus.
Lesions involving the vestibulo-ocular path- ways in animals may affect either the slow or the fast component and occasionally both phases of induced nystagmus. Interruption of the connec- tions linking the semicircular canals to the oculo- motor neurons decreases the velocity of the slow components of induced nystagmus. Lesions involving the peripheral vestibular structures (end organ and nerve) affect the nystagmus in both eyes equally, since the central pathways are symmetrically connected. A single remaining labyrinth senses angular rotation in both direc- tions and produces conjugate nystagmus in both directions. The maximum slow component veloc- ity of induced nystagmus may be asymmetric, however, because of the asymmetry in afferent nerve firing rate produced by ampullopetal and ampullofugal endolymph flow. Central lesions lying anywhere from the vestibular nuclei to the oculomotor neurons often produce disconjugate nystagmus, since the pathways to the eye muscles diverge beginning at the vestibular nuclei.146 A lesion of the MLF, for example, impairs slow and fast components made by the ipsilateral medial rectus muscle but leaves normal slow and fast components at the contralateral lateral rectus (see Fig. 7–29d in Chapter 7).
The proposed role of the PPRF in the pro- duction of rapid horizontal eye movements is supported by the results of experimental lesions in several species of animals. Animals with uni- lateral lesions of the PPRF lose all types of rapid ipsilateral eye movement, and the eyes move into the contralateral hemifield.147,148 Ipsilateral voluntary saccades and quick phases of vestibular and optokinetic nystagmus are affected equally. Stimuli that normally would produce nystagmus with ipsilateral fast compo- nents simply cause a strong tonic contralateral deviation of the eyes (see Fig. 7–29c in Chapter 7). By contrast, vestibular stimuli that produce contralateral fast components result in normal nystagmus, that is, ipsilateral slow phases that are normal. Lesions in the pretectal region have a similar effect on vertical rapid eye movements without affecting horizontal eye movements,149,150 an effect consistent with the separate neural organization of horizontal and vertical saccades.
LEVEL OF AROUSAL AND HABITUATION
Since the turn of the twentieth century, numer- ous investigators have noted a relationship between the magnitude of induced nystagmus and the state of arousal of the animals or human subjects receiving vestibular stimulation.151 In animal studies, amphetamines are routinely used to maintain alertness. During rotational and caloric testing in human subjects the veloc- ity of the slow components of induced nystag- mus depends on the type of mental activity.152 If subjects are instructed to relax and daydream, the velocity is less than when they are instructed to perform continuous mental arithmetic (suc- cessive division). Although other techniques of mental alerting, such as having the subject report on the turning sensation or estimate the time of auditory stimuli, are also effective, men- tal arithmetic tasks are most effective in main- taining mental alertness. The mental task has to achieve a certain degree of complexity since simple forward-counting may not be effective in maintaining the nystagmus response.151
If a normal subject is continuously rotated at low sinusoidal frequencies in the dark, there is a gradual decrease in gain and an increase in the phase lead of eye velocity relative to head velocity (so-called habituation).153,154 The effect peaks in about an hour and can persist for days. Presumably, with habituation, there is a gradual decrease in velocity storage within the multi- neural pathways, illustrated in Figure 3–7, shifting the low-frequency response of the canal-ocular reflex toward that of the primary afferent signal (i.e., the VOR time constant decreases toward that of the cupula). Since a caloric stimulus is equivalent to a very low- frequency rotational stimulus (approx- imately 0.002–0.004 Hz), habituation with repeated caloric testing is explained on a similar basis. Alerting techniques (including stimulant drugs) may work by activating the multineural feedback pathways and thereby improving the low-frequency response of the canal–ocular reflex.
SUMMARY
Several basic principles underlie the connec- tions between the semicircular canals and eye muscles. First, a receptor organ is connected to a group of motoneurons whose activity
produces an eye muscle contraction that com- pensates for a specific head movement with the objective of maintaining gaze stability. Second, null spots do not exist in the receptive field of the semicircular canals because the organs in each ear form a complementary set of accel- eration sensors capable of reacting to the indi- vidual components of angular acceleration associated with head movement in any direc- tion in three-dimensional space. Third, each receptor organ simultaneously activates an excitatory and an inhibitory pathway to agonist and antagonist muscles, resulting in a push– pull system of control. Fourth, most natural head movements activate several receptors simultaneously and inputs from multiple recep- tors converge on secondary neurons. Fifth, alternate pathways complement the elemen- tary disynaptic connections. These pathways consist of chains of interneurons that form reverberating circuits by means of which dif- ferent reflexes interact and “fine-tune” the more specific end-organ reflexes. Finally, it is becoming evident that, because of multisen- sory interactions and neurochemical weights, the strength and even the specificity of the reactions from these pathways can be physiologically modulated.
Translational Vestibulo-Ocular Reflex
OVERVIEW
Sensory cells of each macule are oriented along either side of an equatorial boundary created by the striola, as shown earlier. The orientation of the cells in multiple directions in each mac- ule is different from that in the cristae. All the sensory cells of a semicircular canal are aligned in the same direction and are either excited or inhibited by a stimulus acting in the plane of the canal. The study of the otolith-ocular reflexes is, therefore, more complex than that of canal–ocular reflexes and also technically more difficult.155
To simplify the discussion of otolith-ocular reflexes, it has been traditional to consider the otolith organs (utriculus and sacculus combined) as a unitary sensor capable of resolv- ing all of the linear forces acting on the head into a single resultant vector force. This “uni- tary” three-dimensional otolith receptor is
positioned at the center of the head with the x- and y-axes orthogonal to and the z-axis paral- lel to the earth’s vertical axis. The receptor computes the angle () between the resultant vector force and the earth’s vertical axis and sends this information to the central nervous system (CNS), where a compensatory eye devi- ation is generated with the goal of maintaining the eyes at normal orientation to the earth’s vertical axis. The perfect macular reflex would be one that rotates the eyes at an angle equal and opposite to . In the case of head tilt in the sagittal plane, as illustrated in Figure 3–11, the efficiency or gain of the reflex can be represented by the relation of the angle of eye deviation to the angle of head tilt (gain equals /).
Following this simple model of otolith func- tion, one would predict that linear acceleration along the occipitonasal axis (y-axis) would result in vertical eye movements and linear acceleration along the interaural axis (x-axis), in torsional eye movements (just as lateral tilt produces ocular counterrolling). However, lin- ear acceleration in the occipitonasal axis pro- duces minimal vertical eye movements, and acceleration along the interaural axis induces predominantly horizontal eye movements.155,156 This is reasonable from a functional point of view, since the logical function of the reflex is to augment visual pursuit during linear dis- placement of the head (analogous to the role of the canal-ocular reflex during angular displace- ment of the head).157 Lateral head movements require horizontal, not torsional, eye move- ments to maintain fixation on an earth-fixed
target. Similarly, with fore–aft movements, vertical eye movements would impair rather than improve fixation on an earth-fixed target in the axis of movement.
Furthermore, the translational VOR can only stabilize images on one area of the retina. Consider the circumstances one experiences when walking straight ahead. Objects in the periphery of the visual field appear to move faster than those on the fovea and objects move in different directions based on their location within the visual field. The amplitude and direction of the translational VOR is designed to minimize image slip on the fovea.
As noted earlier, most natural head move- ments are composed of a combination of linear and angular displacements so that the canal- and otolith-ocular reflexes must work together to ensure steady fixation. There is an important difference, however, between the geometry of target displacement during angular and linear accelerations. With the latter, the angle of the required compensatory eye movement increases as the target moves closer to the subject. A simple model of canal-otolith-ocular reflex interaction during horizontal head movements assumes the gain of the canal- ocular reflex is fixed while the otolith-ocular reflex gain increases with decreasing target distance.158 This simple model ignores interoc- ular spacing and the separation of the vestibu- lar organs from the eyes (i.e., it assumes a central cyclopean eye), but this is not a major problem as long as the target distance is greater than a meter. With this model, if the head rotates with angular velocity A and translates
(b) (c)

a
q
a Fg
Fg
Ft
q
F´g
Figure 3–11. Compensatory eye movement induced by static head tilt (b) and by linear acceleration tangential (Ft) to the “unitary” otolith receptor (c); = the angle of eye rotation and = the angle between the resultant force of gravity (Fg) and a line orthogonal to the receptor.
with linear velocity T, then the eye angular velocity = −A − KT, where K inversely depends on target distance. Studies in primates includ- ing humans found that the magnitude of induced eye movements measured during combined linear and angular accelerations (by varying the radius of head rotation) was depen- dent on target location.159,160 Furthermore, the adjustments for target distance occurred too fast (within 10 msec) to be visually guided.
OTOLITH–OCULAR CONNECTIONS
The pathways from the macules to the extraoc- ular muscles are less clearly defined than those from the semicircular canals. The latency of eye muscle activation after stimulation of the utricular and saccular nerves is similar to that recorded after semicircular canal nerve stimu- lation; disynaptic pathways also exist from the macules to the extraocular muscles.161–163
However, electricallyinducedmuscleresponses are not as useful for determining the specific connectivity as responses to semicircular canal nerve stimulation. Because of the varied orien- tation of hair cells within the macules, simulta- neous electrical stimulation of all the nerve fibers coming from a macule produces the equivalent of a heterogeneous activation of dif- ferently oriented hair cells.164 The induced eye movements fail to represent those seen with naturally occurring hair cell stimulation. Likewise, direct mechanical displacement of the otolithic membrane has similar limitations.165–167 As one would expect, stimula- tion on each side of the striola produces oppo- site-directed eye movements. In the utricle, on the basis of the type of eye movements observed experimentally, the macule can be divided into three sections: anterior, posterior, and central (Fig. 3–12).166 Each section is divided by the striola into two unequal parts—peripheral and
Anterior
(a)
Lateral
Central
Medial
Posterior
Superior
Posterior
Anterior
Inferior
Figure 3–12. Separation of the utricular (a) and saccular (b) macules into different sections based on the eye muscles activated by electrical stimulation of each section (as shown in Table 3–2). Arrows indicate polarity of hair cells on each side of the striola (dark blue area).
medial. The eye muscles activated by stimula- tion of each section are those required to produce compensatory eye movements in the plane of the hair cells activated—that is, those required to compensate for linear head move- ments or head tilts in that plane (Table 3–3). The saccular macule is much simpler in its organization. It can be divided into a superior and an inferior area (Fig. 3–12).167 As with the utricular macule, the eye movements produced by discrete electrical stimulation of the saccu- lar macule occur in the direction expected to compensate for the physiological stimulus (Table 3–3).
Unlike the horizontal semicircular canal that makes strong disynaptic excitatory connections with the contralateral abducens motoneurons and disynaptic inhibitory connections with the ipsilateral abducens neurons, abducens motoneurons and internuclear neurons receive only weak disynaptic excitation after stimula- tion of the ipsilateral utricular nerve and only trisynaptic inhibition after stimulation of the contralateral utricular nerve.168–173 Only trisyn- aptic connections have been found between the utricle and the medial rectus motoneurons. Also unlike the canal activated secondary ves- tibular neurons, only about half of utricular activated secondary neurons receive commis- sural inhibition and almost none of the saccular activated secondary neurons receive commis- sural inhibition. However, due to the opposite
Table 3–3 Connections of the Utricle and Saccule with Muscles of the Eyes
![]()
UTRICLE
![]()
Lateral Medial
![]()
Anterior I–SR I–IR
C–IO C–SO
Central C–MR I–MR
I–LR C–IR
Posterior I–IO I–SO
C–SR C–IR
SACCULE
![]()
Superior Inferior
![]()
I–SR I–SO
C–IO C–IR
![]()
C, contralateral; I, ipsilateral; IO, inferior oblique, IR, inferior rectus, LR, lateral rectus; MR, medial rectus; SO, superior oblique; SR, superior rectus.
polarity of hair cells on each side of the striola, convergence of input from both sides of the striola on secondary neurons could take the place of commissural inhibition.172,173 Cross- striolar inhibition was observed in two-thirds of saccular activated and one-third of utricular activated secondary vestibular neurons.174,175 This cross-striolar convergence on secondary neurons might make it possible for one laby- rinth alone to generate the translational VOR.
There is an important difference between the signals coded by the canal and otolith affer- ents. Within the range of natural head movements, otolith afferents code linear accel- eration while canal afferents code angular velocity. An additional integration or low-pass filter of the otolith signals as compared to canal signals is needed in the VOR pathways. Several models have been developed to account for the needed extra low-pass filter.155,176,177 Otolith sig- nals could undergo a separate neural integra- tion prior to convergence on the common neural integrator shared with canal ocular sig- nals or the otolith signals could enter the shared but distributed velocity-to-position integrator network at different sites such that signals from the canals are only integrated once while those from the otolth organs are integrated twice. One way to achieve the latter is to assume that both the angular and translational VOR share the same neural integrator pathway but have completely different direct pathways (a reason- able assumption based on the above). The direct pathway of the rotational VOR is strong while that of the translational VOR is weak. The strong rotational VOR pathway generates a lead that can compensate for the low-pass dynamics of the eye plant while the weak trans- lational VOR pathway does not generate a lead. The low-pass filter dynamics of the eye plant, which remains uncompensated, can provide the second integration for the translational VOR but only at high frequencies above the dominant time constant of the eye plant (a prediction consistent with experimental findings).
CHARACTERISTICS OF EYE MOVEMENTS INDUCED BY TRANSLATION
Brief high-acceleration steps of the whole body along the interaural axis have been used to assess the function of the otolith-ocular reflex
in primates and in normal human subjects and patients. Paige and Tomko178,179 studied the translational VOR in squirrel monkeys and found a short latency response that was depen- dent on viewing distance. Lempert et al.180 studied humans with step accelerations of
g using a cart on wheels and recorded eye movements with electrooculography. Normal subjects have symmetrical otolith-ocular reflexes with short latencies (<130 msec). Abnormalities in gain, symmetry, and latencies were seen in patients with bilateral loss of caloric responses, but none of these findings seemed specific for loss of the otolith-ocular reflex. Crane et al.181 produced brief high- acceleration steps (0.4 g) using a pneumatically driven chair moving along the interaural axis and recorded eye movement with the precise scleral search coil technique. Normal subjects showed stereotyped short-latency (about
30 msec) otolith-ocular responses that depended on the distance of a fixation target that was extinguished just before the onset of the head acceleration (Fig. 3–13). Patients with bilateral loss of caloric response showed pro- longed latencies and decreased gain of the otolith-ocular reflex and these responses were minimally affected by the distance of a remem- bered visual target. In all of these studies, the gain of the otolith-ocular reflex was much less
than ideal (i.e., that required for compensatory eye stabilization), implying that during most natural head movements, the angular VOR plays the major role in gaze stabilization.
Ocular Counterrolling
Compensatory eye movements produced by static head tilt have been easier to measure in animals that have limited spontaneous eye movement. They usually are either rotational or torsional, depending on the direction of tilt and the position of the orbits in the skull. For example, in rabbits and fish, lateral tilt causes a vertically directed rotational movement, and forward–backward tilt causes a torsional eye movement. These results are consistent with the compensatory movement of the eye in rela- tion to the earth reference coordinates. In humans, compensatory torsional movements are produced by lateral tilt (ocular counter- rolling), whereas vertical rotation results from forward–backward tilt (see Fig. 3–11). Eye movements associated with static tilt have been studied most extensively in the rabbit. Head tilt in the dark within a range of ± 45 degrees of the normal position causes a compensatory eye deviation with a gain of approximately 0.6182; that is, the angle of eye
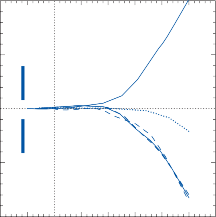
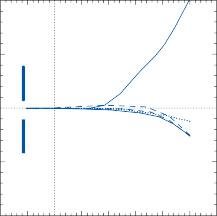
Normal Subject Bilateral Vestibulopathy 10 10
Head Position (cm)
Head Position (cm)
Head Head
5 5
Eye Position (deg)
Eye Position (deg)
0
–5
–10
3 cm
3º
Eye
200 cm
25 cm
0
–5
–10
3 cm
3º
Eye
200 cm
25 cm
–100 –50 0 50 100 150 200 250 300 –100 –50 0 50 100 150 200 250 300
Time (msec)
Figure 3–13. Short-latency otolith-ocular responses to brief high-acceleration (> 0.5 g), linear displacements along the intraaural axis in a normal subject and a patient with bilateral vestibulopathy (absent caloric responses). Subjects fixated on a target at 25 cm and 200 cm that was extinguished just before the onset of the acceleration in total darkness. Eye move- ments were recorded with scleral search coils. (Courtesy Crane B, Wiest G, Demer J, Departments of Ophthalmology and Neurology, UCLA.)
rotation () is approximately 60% of the angle of tilt (). In human subjects, the ocular response to tilt is much less efficient. The maximum ocular torsion for a lateral tilt of 50 degrees is only 5 to 6 degrees (a gain of approximately 0.1).183,184 After an acute unilat- eral lesion, there is a static ocular torsion (about 10 degrees) toward the side of the lesion185 and decreased ocular counterrolling with tilt toward the side of the lesion,186 but these asymmetries disappear with compensation. Ocular counterrolling responses may be transiently decreased after prolonged space flight.187
Semicircular Canal–Otolith Interaction
Two techniques have been routinely used to study the interaction of semicircular canal and otolith induced eye movements: off-center axis rotation (OCAR) and off-vertical axis rotation (OVAR). With both techniques the otolith component of the induced eye movement can be studied in isolation once a constant velocity of angular rotation is achieved.
OCAR, which induces combined centripetal and angular acceleration, has been used exten- sively to study the interaction of the rotational and translational VOR in animals.188–192 If a monkey undergoes OCAR by rapidly accelerat- ing to a constant angular velocity, the induced nystagmus is similar to that seen after a compa- rable rotational stimulus about the z axis but with several important differences (Fig. 3–14).193 The axis of induced eye move- ments shifts toward the gravito-innertial accel- eration (GIA) so that the amplitude of the horizontal component decreases and torsional and vertical components develop. A DC offset develops in the roll plane (counterrolling) and the time constant of nystagmus decay shortens. When the canals are plugged isolating the con- tribution of the otolith organs, nystagmus dis- appears and only the counterrolling remains (Fig. 3–14b). Both the shift in the axis of eye velocity and the change in time constant are quantitatively predicted by models of the spa- tial orientation properties of velocity storage.193 By contrast if monkeys undergo OCAR at high frequencies (1–4 Hz) where there is minimal tilt of the GIA, only horizontal eye movements are induced and the eye movements represent
the sum of the angular and translational VOR.192 This finding is consistent with the concept that the translational VOR functions mainly in the frequency range of natural head movements.
If a monkey is rotated at a constant velocity about a tilted vertical axis (OVAR), the slow- phase velocity of induced nystagmus does not decay to zero (as when the monkey is vertical), but rather persists at a steady-state level.191,194 If the animal is suddenly stopped, the post- rotatory nystagmus after OVAR shows a shift in axis toward the GIA and is shorter than when the animal is stopped in the upright position. Blocking the semicircular canals does not alter the steady-state response during OVAR, indi- cating that the otoliths generate the signals necessary for the continuous nystagmus. It has been postulated that sequential excitation and inhibition of the otolith hair cells by the rotat- ing gravity vector produces a traveling wave whose velocity is estimated centrally and then passed on to the velocity storage integrator, which produces the continuous horizontal nystagmus.195 The velocity storage system can be activated by many types of stimuli (canal, otolith, vision) and through a three-dimensional gravity-dependent structure; the system is capable of storing information to produce eye movements in all planes.196–197
Ocular stability during most natural head movements results from a coordinated interac- tion of signals originating in vestibular, visual, and neck receptors. The compensatory nature of neck-induced eye movements has been well documented in animals. In 1924 De Kleyn198 showed that if one holds an animal’s head sta- tionary and displaces the body, a compensatory eye deviation occurs that tends to preserve the relationship between gaze and the body axis. Nonfoveated animals, such as the rabbit, exhibit clear compensatory eye deviations.199 Cervico- ocular and vestibulo-ocular reflex interaction is more difficult to study in primates because of the dominance of voluntary and visually con- trolled eye movements and because the gain of the cervico-ocular reflex is so low compared to that of the vestibulo-ocular reflex. Very few investigators have quantitatively assessed eye,
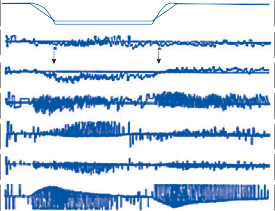
ANG VEL
–400º/S
GIA
PRE-OP M9308 ANG VEL
GIA
52º
60º
X
Y
![]()
Z
b
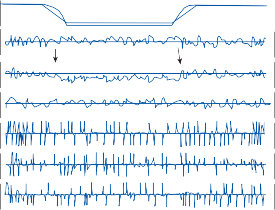
![]()
ANG VEL
POST-OP M9308 ANG VEL
![]()
20 Sec
30º
60º
500º/S
500º/S
500º/S
GIA
Facing Motion GIA Ag

º
![]()
Ag
–400º/S
X
Y
Z
GIA
52º
60º
30º
60º
100º/S
100º/S
100º/S
![]()
![]()
![]()
20 Sec
Figure 3–14. Response to centrifugation after plugging the 6 semicircular canals. a: preoperatively, the animal had normal horizontal eye velocity and normal counterrolling during the period of gravito-inertial acceleration (GIA) tilt (between the arrows). b: after plugging the canals, normal counterrolling was still present during the period of GIA tilt (between the arrows) indicating normal otolith function. No horizontal or vertical eye velocity was present. (From Wearne S, Raphan T, Cohen B. Effects of tilt of the gravito-inertial acceleration vector on the angular vestibuloocular reflex during centrifugation. J Neurophysiol. 1999 May;81:2175, with permission.)
head, and neck movement coordination in humans, and the clinical significance of lesions involving the cervico-ocular reflex pathways is controversial.200
Anatomic and Physiologic Basis
Animal studies have shown that the cervico- ocular reflex originates from nerve endings in the ligaments and capsules of the upper
cervical articulations.55,201 The reflex can be induced by electrically stimulating the capsules of the upper cervical joints, the C1 to C3 dorsal
roots, and the high cervical spinal cord. The
reflex is not induced by stimulating the superfi- cial muscles or skin of the neck. Bilateral sec- tioning of the high cervicodorsal roots or the application of local anesthetic around the cer- vical articulations abolishes the cervico-ocular reflexes. Unilateral interruption of the neck- ocular reflex pathways produces nystagmus in
rabbits, cats, and monkeys when fixation is inhibited, although no consistent relationship exists between the side of dorsal root involve- ment and the direction of nystagmus.202,203 As with the VOR, the eye muscles are either excited or inhibited by neck stimulation, depending on whether the muscle is agonistic or antagonistic for the required compensatory movement.
Electrophysiologic experiments suggest that the cervico-ocular reflexes are mediated via the vestibular nuclei (primarily in the medial and descending nuclei).55,204 The precise projec- tions of the neck afferents to each vestibular nucleus are only partially known, but it can be anticipated that since the neck-induced eye movements compensate for displacement in the precise plane of body motion, the vestibu- lar nuclei must contain a discrete topographic representation of cervical afferents that is simi- lar to that of the vestibular afferents.
Electric stimulation of the high cervicodor- sal roots in the cat produces evoked potentials in the contralateral vestibular nuclei55 followed by excitation of the abducens nucleus ipsilat- eral to the neck stimulation and inhibition of the contralateral abducens nucleus. In addi- tion, stimulation of the cervicodorsal roots enhances the amplitude of action potentials in the ipsilateral abducens nerve induced by con- tralateral vestibular nerve stimulation, and it inhibits action potentials in the contralateral abducens nerve induced by ipsilateral vestibu- lar nerve stimulation. Vestibulo-ocular and cervico-ocular reflex interactions, therefore, result from a convergence of neck and semicir- cular canal afferents on secondary vestibular neurons.
The firing properties of VOR-related sec- ondary vestibular neurons and floccular Purkinje cells were studied during neck rota- tions in monkey.205–208 In squirrel monkey, pas- sively moving the neck produced changes in firing rate related to neck position and/or neck velocity in PVP and EH neurons.205,206 Floccular Purkinje neurons were relatively insensitive to neck-induced eye movements, but when eye movements were visually suppressed the firing rate of these neurons was modulated by neck movements. Thus, it appears that the flocculus plays a role in suppressing the cervico-ocular reflex when it would cause inappropriate move- ment of a visual target on the retina (similar to its role in modulating the VOR). On the other
hand, in rhesus monkey there was no modula- tion in EH secondary vestibular neurons with passive neck rotations.208 These different find- ings are consistent with the fact that squirrel monkeys have a relatively robust cervico-ocular reflex compared to the rhesus monkey.208
Characteristics of Neck-Induced Eye Movements
Figure 3–15 illustrates the synergistic interac- tion of neck and vestibulo-ocular reflexes. When the rabbit’s head is turned to the right (clockwise about the cephalocaudal axis), the eyes turn counterclockwise in the orbit because the movement stimulates the horizontal semi- circular canals and neck reflexes (Fig. 3–15b). The direction of the eye movement is the same as if the whole animal had been rotated, stimu- lating only the semicircular canals (Fig. 3–15c). The characteristics of the neck-ocular reflex alone are evaluated by rotating the body while the head is stationary (Fig. 3–15a). The same relationship between head and torso is pro- duced as in Figure 3–15b and the eyes deviate in the same direction. In both instances, the normal relationship between eyes and torso is maintained.
Since the time of Bárány, rotating the body with the head stationary and measuring the eye movements has been considered a potential functional test of the human neck-ocular reflex pathways.209–213 Several methodologic problems have been encountered, however. It is difficult to induce body motion and concurrently main- tain the head completely stationary so as to avoid vestibular stimulation. As with vestibular- induced eye movements, care must be taken to inhibit fixation while monitoring the neck- induced eye movement. Even if these prob- lems are overcome, the gain of the cervico- ocular reflex in humans is very low (<0.1 for the frequency range between 0.1 and 1.0 Hz).210,213 The gain of the cervico-ocular reflex does appear to increase with age, but this increase is only seen at low frequencies (<0.1 Hz) and peak velocities (<15 degrees/sec) so it is unlikely that it could help compensate for VOR loss in the frequency and velocity range of natural head movements. Studies in monkeys and humans after unilateral vestibular loss found no increase in cervico-ocular reflex gain, but similar studies after bilateral vestibular loss
![]()
![]()
![]()
Angle of:
![]()
![]()
![]()
Head motion
qh qh
Eye motion
Torso motion
qe qe qe
q
q
qt
t
Figure 3–15. Synergistic interaction of cervico-ocular and vestibulo-ocular reflexes. See text for details.
found an increase in cervico-ocular reflex gain. Again, it is unclear whether this increase would be beneficial at the frequencies of natural head movements.
Three visually controlled ocular stabilizing sys- tems produce versional eye movements: the saccadic, the smooth pursuit, and optokinetic systems.214 In everyday life, these three systems interact with the vestibular system for the maintenance of gaze as subjects attempt to identify moving objects in space and maintain stability in gaze. The optokinetic is the phylo- genetically older system; the other two are related to the anatomical development of the fovea. The saccade system responds to an error in the direction of gaze with respect to the posi- tion of an object of interest by initiating a rapid eye movement (a saccade) to correct the “reti- nal position error,” bringing the object to the fovea in the shortest possible time. The brain- stem centers for generating saccades were briefly discussed earlier (for review, see Shinoda et al.215). The smooth pursuit system is responsible for maintaining gaze on a moving target, that is, it keeps the target within the foveal visual field. It compares the eye velocity with that of the target velocity and produces a continuous match of the eye and target
position. The optokinetic system is generally considered to be a primitive form of smooth pursuit involving the whole retina instead of the fovea alone. The eye tracking motion is periodically interrupted by corrective saccades in the opposite direction to relocate the gaze on new targets coming into the visual field.
Visual Tracking Eye Movements
OPTOKINETIC
Afoveate animals, such as rabbits, have been useful models for studying the organization of the optokinetic reflex response. In the rabbit, monocular stimulation results in a prominent asymmetry in response; temporonasal target motion induces much greater slow-phase veloc- ity than nasotemporal motion. 216,217 The peak optokinetic nystagmus (OKN) slow-phase velocity rarely exceeds 30 degrees/sec, and there is a gradual buildup in slow-phase velocity after the step onset of an optokinetic stimulus (over 20 to 30 sec). In foveate animals, including humans, OKN slow-phase velocity is symmetrical and may exceed 100 degrees/sec.218
Although OKN can be elicited in primates with parafoveal stimulation, the strongest responses are induced when the fovea is included in the field of stimulation.219,220 A tem- poronasal preponderance occurs during mon- ocular stimulation in the first few months of life
while the fovea is immature.221,222 Also, mon- ocular asymmetry has been observed in patients with maldeveloped foveas223 and with congeni- tal or early acquired amblyopia.224
In rhesus monkeys the OKN response to a step change in stimulus velocity has two dynamic components (Fig. 3–16). There is a rapid initial jump in slow-phase velocity (Fig 3–16B,a) followed by a gradual rise to a steady- state level (Fig. 3–16B,b).225 If the lights are turned off after the steady state is achieved, the eye velocity immediately drops to a value near that achieved after the initial rapid rise (Fig. 3–16B,c) from which it slowly decays as optoki- netic after-nystagmus (OKAN) (Fig. 3–16B,d). The eye velocity reached after the initial fast jump varies from 40% to 80% of the stimulus velocity; the steady-state values are almost equal to stimulus velocities up to 60 degrees/ sec but progressively decrease for higher-stim- ulus velocities.225 Also, OKN is more irregular at high-stimulus velocities, so that average gain values are less than peak values. The maximum initial velocity of OKAN varies with stimulus velocity but can exceed 70 degrees/sec.225 With sinusoidal optokinetic stimulation in monkeys, the gain of OKN is near 1 at low frequencies
(<0.1 Hz) but progressively decreases at higher frequencies (e.g., 0.3–0.5 at 2–4 Hz).226,227
A slow buildup in OKN slow-phase velocity is not observed in normal humans,220 but it has been seen in patients with lesions of the retina,223,224 parietal lobe,228 and cerebellum.229 If normal subjects are instructed to follow the stripes on an optokinetic drum, the subjects produce a large-amplitude, low-frequency nys- tagmus (“look” OKN); if they are instructed to stare straight ahead at the drum surface, they produce a small-amplitude, high-frequency nystagmus (“stare” OKN). In normal human subjects the slow-phase velocity of “look” OKN can exceed 100 degrees/sec, whereas that of “stare” OKN rarely exceeds 60 degrees/sec.230 The OKAN responses in humans are much less consistent than those in monkeys; the maxi- mum initial slow-phase velocity does not exceed 20 degrees/sec.231,232
SMOOTH PURSUIT
In foveate animals, the smooth pursuit system functions to stabilize a moving target on the fovea.233 The system operates optimally for low velocities and low frequencies of
A
Smooth pursuit velocity
OKN
OKAN
![]()
30º/sec

Horizontal EOG
![]()
30º
![]()
Drum velocity
160º/sec
15 sec
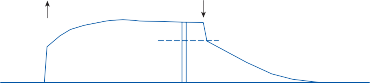
B
Smooth pursuit velocity
OKN
b c
OKAN
a d
Figure 3–16. A Optokinetic nystagmus (OKN) and optokinetic after-nystagmus (OKAN) in response to a velocity step of the optokinetic drum. First trace, velocity of horizontal eye movements, fast phases clipped; second trace, horizontal eye position; third trace, velocity profile of the optokinetic drum. Upward arrow, lights on; downward arrow, lights out. B Velocity envelope (a–d) of the slow phases of OKN and OKAN redrawn schematically from the first trace in A. (From Waespe W, Henn V. Gaze stabilization in the primate. The interaction of the vestibulo-ocular reflex, optokinetic nystagmus and smooth pursuit. Rev Physiol Biochem Pharmacol. 1987;106: 37, with permission.)
target motion.234,235 As the velocity and fre- quency increase (e.g., above 60 degrees/sec or
1 Hz in humans), the eyes continually fall behind and frequent corrective saccades are required to bring the target to the fovea. The gain of smooth pursuit is not only a function of target velocity but also of target acceleration.236 Subjects better pursue a target with continu- ously changing velocity if the pattern of move- ment is predictable (as with a sinusoidal pattern).234 The stimulus for smooth pursuit may be perceived target motion rather than actual target motion since subjects can pursue apparent target motion in the absence of a tar- get moving across the retina.237 Also, subjects can pursue a stabilized retinal image eccentric to the fovea.238,239 Probably retinal velocity error is the main driving force for smooth pursuit eye movements, with a lesser contribution coming from retinal position error and perceived target motion.240
Organization of Visually Guided Tracking Eye Movements
A simple scheme of visually guided tracking eye movements based on the concepts of Cohen et al.241 is shown in Figure 3–17. Visual motion information reaches the oculomotor neurons via two pathways: a direct pathway with fast dynamics and an indirect pathway with slower dynamics. A key feature of the indirect pathway is the velocity storage element shared with the VOR. Optokinetic stimulation activates both pathways, whereas pursuit (according to Robinson112) activates only the
direct pathway. The velocity storage element accounts for the slow buildup in optokinetic nystagmus and OKAN. The direct pathway accounts for the initial rapid rise in OKN and the rapid drop after turning off the lights.
In 1936 Ter Braak242 performed a series of experiments in which he confirmed the pres- ence of cortical and subcortical optokinetic pathways in several animal species. Cortical OKN was elicited by movement of a series of relatively small objects that attracted the ani- mal’s attention (so-called active nystagmus), while subcortical OKN was produced by movement of the whole optical environment (passive nystagmus). Presumably, the cortical pathway corresponds to the direct (pursuit) pathway and the subcortical pathway to the indirect (velocity storage) pathway. In animals without a fovea, such as the rabbit, only passive OKN can be induced, and bilateral occipital lobectomy produces a minimal effect on induced OKN.243 In cats and dogs, passive and active OKN can be induced, but only the latter is abolished by bilateral occipital lobectomy.242 In monkeys bilateral occipital lobectomy abol- ishes smooth pursuit and the initial rapid rise in OKN (leaving the slow buildup and OKAN intact), but after a few months the animals regain some smooth pursuit and part of the rapid phase of OKN.244
Since human subjects have poor OKAN and do not exhibit a buildup in OKN slowphase velocity, the subcortical (indirect) pathway must be much less prominent in humans than in other animals. However, patients with occip- ital infarction often have the ability to perceive visual motion and avoid objects when walking,
Retinal signal
![]()
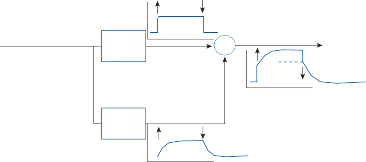
Direct
Velocity storage
to EOM’s
Figure 3–17. Schematic drawing of the direct and indirect (velocity storage) visuomotor pathways. A constant velocity optokinetic stimulus begins at the first arrow and the lights are turned off at the second arrow. The signals from the two pathways add together to produce the characteristic slow-phase velocity envelope of OKN and OKAN (far right). EOM- extra ocular muscle.
even though they are not consciously aware of “seeing.”245 One such patient exhibited a slow buildup in OKN.246 As noted earlier, patients with lesions of the parietal lobe and midline cerebellum also exhibit a slow buildup in OKN; the indirect pathway is uncovered after loss of the direct pathway.228,229
Comparison of Vestibular- and Visual-Induced Eye Movements
The schematic diagrams of the visuo-ocular (pursuit and optokinetic) and the vestibulo- ocular reflexes in Figure 3–18 illustrate impor- tant similarities and differences between the two types of reflexes. In both instances the
e
e
objective of the reflex eye movement (˙ ) is to
match that of the stimulus velocity if the system is functioning perfectly. In the case of the visuomotor system, the stimulus is the target
contrast, the gain of the VOR is about 0.6 when a normal human subject is sinusoidally rotated in the dark at 0.1 Hz and a maximum velocity of 30 degrees/sec. Unlike the visuomotor sys- tem, however, the VOR responds with a gain near 1 for frequencies from 1 to 4 Hz and velocities >100 degrees/sec.234,241 The reader can test the increased efficiency of the vestibu- lar system over the visuomotor system at high input velocities and frequencies by a simple maneuver. Rapidly move your hand back and forth with increasing velocity with your head stationary until your hand appears blurred. Then hold your hand stationary and rapidly move your head back and forth at the same high speed. Despite the rapid head move- ment, the smallest detail of the palm remains clear.248
Visuo-Vestibulo-Ocular
t
t
velocity ˙ (or optokinetic drum velocity), while
for the vestibular system it is the head velocity
h
h
˙ . The eye movement takes the form of either
smooth pursuit or nystagmus. In the latter case,
Connections
The existence of neurons in the vestibular nuclei whose responses reflected visual inputs
e
e
the ˙ response is that of the slow component
represented a new concept in the organization
of the nystagmus. The target velocity and head velocity must have opposite signs to produce
e
e
(˙ ) with the same sign. The visuomotor system
functions as a closed-loop system with negative feedback to compare eye and target velocity, whereas the VOR is an open-loop system.
For both systems, the gain of induced eye movements is dependent on the velocity and frequency of the stimulus. The visuomotor sys- tem is most efficient at low target velocities and frequencies. Normal human subjects can track a target moving sinusoidally at 0.1 Hz, peak velocity 30 degrees/sec, with a gain near 1.236,247 The gain rapidly falls off for target velocities
>100 degrees/sec and frequencies >1 Hz. By
of the vestibular reflexes. Shortly after it was demonstrated by Dichgans et al.249 that neu- rons in the vestibular nuclei of goldfish responded to visual inputs, similar observations were made by other investigators in a variety of animals under a variety of experimental conditions.8,250 The need for a site for interac- tion of vestibular and visual inputs had been recognized, but the realization that the interac- tion took place within the vestibular nuclei rep- resented a departure from the rules of sensory specificity for the vestibular system.
In afoveate animals, the subcortical, accessory optic system is the predominant pathway for visual–vestibular interaction.59,217,251 This system
Negative Feedback Loop

· † Target Retina velocity
Visuoocular reflex
Eye velocity
· e
![]()
h
h
Head
velocity
Semicircular canals
Vestibuloocular reflex
· e
Figure 3–18. Schematic diagrams of the visuo-ocular and the vestibulo-ocular reflexes. The former is a closed-loop negative feedback system and the latter is an open-loop system.
includes a group of nuclei at the mesodienceph- alic border that, like the lateral geniculate nucleus, receives direct retinal projections but, unlike the lateral geniculate, projects directly to the brain stem and cerebellum. The most prom- inent cell group of the accessory optic system, the nucleus of the basal optic root, is identifi- able in all classes of vertebrates. Lázár252 found that optokinetic responses are abolished in frogs after destruction of the basal optic root nuclei, whereas ablation of the lateral geniculate nuclei and superior colliculi did not affect optokinetic responses. As in the rabbit, only subcortical pas- sive OKN can be elicited in the frog.
Electrophysiological studies in rabbits have demonstrated projections from the retina to the flocculonodular lobe of the cerebellum via the accessory optic system.253–255 Microelectrode recordings in the accessory optic nucleus of the rabbit and the cat reveal units that show a strong response to a slow full- held retinal stimulation.217,256 Temporonasal movements of large patterns rich in texture evoke the strongest response. Neuroanatomical studies using horseradish peroxidase to map the connections between the accessory optic system and the flocculus show two separate pathways—one direct and the other an indirect pathway synapsing in the inferior olive.257,258
The cerebellovestibular pathways involved in visual–vestibular interaction were first described by Ito on the basis of his studies in rabbits
(Fig. 3–19).117 Retinal sensory information reaches the inferior olives by way of the acces- sory optic tract and the central tegmental tract and then the cerebellum, where they activate Purkinje cells in the flocculus, nodule, and other adjacent areas. These parts of the cere- bellum also receive primary vestibular afferent fibers and secondary vestibular fibers originat- ing mostly in the medial and descending ves- tibular nuclei. Outflow from the cerebellar Purkinje cells terminates at secondary vestibu- lar neurons and neurons in the adjacent reticu- lar substance.
With the development of the fovea, cortical pathways become progressively more impor- tant in visual–vestibular interaction. Anatomical and physiological studies in primates indicate that visual signals reach the brain stem for interaction with vestibular signals via a complex cascade of interconnecting pathways (Fig. 3–20). In contrast to the rabbit and cat, neurons in the pretectal complex of the mon- key receive predominant input from the visual cortex and respond equally well to small spots or large random dot patterns moving through their receptive field.222,259 Furthermore, they respond in the same manner to monocular or binocular stimulation—that is, they do not exhibit the temporal-nasal preponderance seen in afoveate animals. Electrical stimulation of the nucleus of the optic tract (NOT) in alert monkeys evokes horizontal nystagmus with
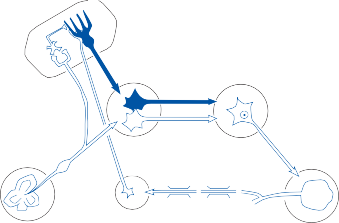
PU
FL
GR
OM
VN
MF
VO CF
CTT AOT
Eye
IO
Figure 3–19. Anatomic pathways of visual–vestibular interaction in the rabbit. AOT, accessory optic tract; CTT, central tegmental tract; Fl, flocculus; GR, granule cell; IO, inferior olive; MF, mossy fiber; OM, ocular motoneuron; PU, Purkinje cell; VO, vestibular end organ; VN, vestibular nucleus. Inhibitory neurons are dark blue. (From Ito M. The vestibulo- cerebellar relationships: vestibulo-ocular reflex arc and flocculus. In: Naunton RF (ed). The Vestibular System. Academic Press, New York, 1975, with permission.)
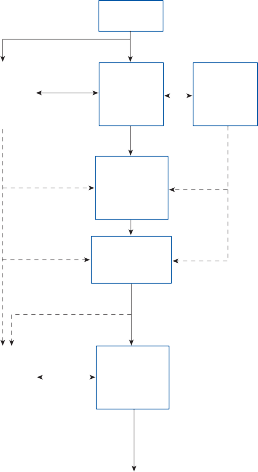
Pretectal nuclei AOS, NOT
Pretectal nuclei AOS, NOT
Primary
visual cortex
Frontal eye field cortex
Visual association cortex
MT, MST, PP
Pontine nuclei
DLPN, RTPN
Other Brain stem nuclei VN, PH, IO |
|
Other Brain stem nuclei VN, PH, IO |
|
Cerebellum FI, Ve
![]()
Oculomotor neurons
Oculomotor neurons
Figure 3–20. Schematic drawing of visual–vestibular pathways in the primate (see text for details). AOS, accessory optic system; DLPN, dorsal lateral pontine nuclei; DMPN, dorsal medial pontine nuclei; Fl, flocculus; IO, inferior olive; MST, medial superior temporal area; MT, middle temporal area; NOT, nucleus of the optic tract; PH, prepositus hypo- glossi nucleus; PP, posterior parietal cortex (area 7a); Ve, vermis; VN vestibular nucleus. Dashed lines indicate probable pathways.
a slow buildup in slow-phase velocity followed by after-nystagmus in the same direction.260 The rising time course in slow-phase velocity is similar to the slow buildup in OKN and the falling time course of the after-nystagmus parallels that of OKAN. The striate cortex,261 the superior temporal sulcus (particularly the middle temporal [MT] and medial superior temporal [MST] areas)262–266 and the posterior parietal cortex267,268 are the key cortical areas in the monkey for processing retinal motion information. The frontal cortex contains key pursuit-related regions: the caudal part of the
frontal eye fields (FEFs) and the supplemen- tary eye fields (SEFs).269,270 Both regions receive visual and vestibular inputs and both are criti- cal for integrating extraretinal pursuit informa- tion. The FEF plays an important role in pre- dictive pursuit while the SEF is specialized for different task conditions. These cortical cen- ters project to the dorsolateral pontine nucleus (DLPN) and the neighboring rostral reticularis tegmenti pontes nucleus (RTPN), which are a primary source of afferents to the flocculus and vermal areas VI and VII, two cerebellar areas involved in the regulation of eye
movements.271–274 Neurons in the DLPN exhibit a directionally selective response to movement of discrete spots, and large backgrounds and micro- stimulation in the region of the DLPN cause a short-latency modification of the velocity of an ongoing pursuit eye movement.272
target and the head move, the driving stimulus to the visuomotor system is the angular velocity of the target relative to the head— that is, the difference between the target
t
t
velocity relative to space (˙ ) and the head angular velocity relative to space (˙ ). In
In the monkey, lesions of the parietotemporal
the absence of head movement (˙
h
h=0),
the
region,275 DLPN,272 and the flocculus276 result in an impairment of (1) smooth pursuit, (2) the ini- tial rapid rise in OKN slow-phase velocity, and
(3) visual vestibular interaction, requiring the “direct” visuomotor pathway, for example, fixa- tion suppression of vestibular nystagmus with a foveal target. By contrast, lesions of the pretec- tal nuclei (nucleus of the optic tract) impair OKN but not pursuit.277 Taken together, these data suggest that the cortical and subcortical pathways illustrated in Figure 3–20 roughly cor- respond to the direct and indirect visuomotor pathways of the model shown in Figure 3–17.
Model of Visual–Vestibular Interaction
Figure 3–21 gives a simple linear interaction model for the visual and vestibular oculomotor systems.195 The two independent block diagrams in Figure 3–18 have been interrelated
e
e
to produce a single output eye velocity (˙ ).
When the target (foveal or full field) is station- ary, movement of the head results in an equiva- lent movement of the target in the opposite direction relative to the head. When both the
eye movement response is under the control of the closed-loop visuomotor system, whereas, if the head is rotated in the dark, the visual sys- tem is inoperative and the eye movement response is under the control of the vestibular system.
A few general features of this model deserve emphasis because of their relevance to clinical testing. A full-field target activates both the direct (pursuit) and indirect (velocity storage) pathways, the latter being shared with the ves- tibular system. Optokinetic after-nystagmus provides the only independent measure of the indirect pathway. In contrast, a foveal target activates predominantly the direct pathway (pursuit after-responses are minimal).236 Therefore, pursuit testing is almost exclusively a measure of the direct visuomotor pathway. At low-input frequencies and velocities (head or target), the gain of the direct pathway is an order of magnitude higher than that of the other pathways. This explains why normal sub- jects can completely inhibit the VOR when rotated with a fixation target at the low frequencies commonly used for clinical testing (i.e., 0.1 Hz) (see Fig. 1–9 in Chapter 1).
![]()
†
Retina
Direct pathway
![]()
![]()
e
![]()
e
Velocity storage
![]()
![]()
![]()
Semicircular
h canals Direct pathway
Figure 3–21. Model of visual–vestibular interaction after Cohen et al.130 ˙ , eye velocity; ˙ , head velocity; ˙ target (foveal
or full field) velocity. See text for details.
h h t
Adaptive Modification of the Vestibulo-Ocular Reflex with Vision
On the basis of psychophysical studies of Kohler,278 Gonshor and Melvill Jones279–281 began a series of experimental studies in the early 1970s that were designed to investigate the potential for adaptive plasticity within the VOR. Probably the most dramatic example of this plasticity was the complete reversal of the VOR that occurred in normal subjects after wearing optically reversing prisms.281 After about 2 weeks of wearing goggles that pro- duced continuous left–right reversal of the visual environment, the VOR measured in the dark adaptively changed such that the direction of the slow and quick phases of induced nystag- mus was the reverse of normal. The process occurred gradually over days, initially with a drop in gain, followed by a progressive change in phase (although never quite reaching the desired 180-degree phase shift). After the gog- gles were removed, the VOR gradually returned to normal somewhat faster than with the origi- nal adaptation. Subsequent studies using mag- nifying and minifying lenses in normal humans248 and a variety of animals282–285showed that the dark-measured VOR gain could be increased and decreased, respectively, almost with a machine-like precision (Fig. 3–22). Furthermore, these adaptive changes were not
restricted to a single plane. For example, if an animal was sinusoidally rotated in one plane (the horizontal) while the visual surround was simultaneously rotated in another plane (the vertical), the VOR measured with horizontal rotation in the dark developed a vertical component.286 Although the site of these induced plastic changes in the VOR remains uncertain, the cerebellum appears to play a key role. Lesions of the cerebellum in a variety of animals block adaptive plasticity of the VOR.282,283,287
Cellular Basis for Visual Vestibular Interaction
As indicated above, the vestibular nucleus is a key visual vestibular interaction center. In early studies, Waespe and Henn found that nearly every neuron in the vestibular nucleus of alert monkeys that responded to horizontal rotation of the animal in the dark also responded to horizontal rotation of the visual surround.64,65,250,288,289 During combined visual– vestibular stimulation, neurons were maximally excited (or inhibited) when the vestibular and optokinetic nystagmus were in the same direction—that is, the background moved in the opposite direction of the monkey. If the optokinetic drum was mechanically coupled to the turntable so that both rotated together,
2.0
Gain = 1.02 + 0.61 (1 – e–0.023†)
Gain = 1.02 + 0.61 (1 – e–0.023†)
Spectacles on
![]()
1.8
VOR Gain
VOR Gain
1.6
1.4
1.2
2.0
Spectacles off
![]()
1st exposure 2nd exposure 3rd exposure
Gain = 1.03 + 0.60 (1 – e–0.135†)
1.0
0 1 2 3 4 5 6 7 8
Time (days)
0 1 2 3 4 5 6
Figure 3–22. Adaptive enhancement and recovery of vestibulo-ocular reflex (VOR) gain in a monkey exposed to continuous × 2 binocular vision. The different symbols represent data from the same animal that were obtained on differ- ent occasions. The similarity of the curves they depict emphasizes the machine-like characteristics of the adaptive process. (From Miles FA, Eighmy BB. Long-term adaptive changes in primate vestibulo-ocular reflex: I. Behavioral observations. J Neurophysiol. 1980;43: 1406, with permission.)
nystagmus was reduced and neuronal activity was attenuated compared to pure vestibular stimulation in the dark.288 Later studies, how- ever, suggest that only a subgroup of secondary vestibular neurons are modified by visual adap- tation of the vestibulo-ocular reflex, the so- called floccular target neurons (FTNs).290–296 Although FTNs have similar properties to EH secondary vestibular neurons, the extent of overlap of these two neuronal populations is largely unknown.155
Lisberger and colleagues proposed a model of visual vestibular interaction that consisted of two parallel pathways: an unmodifiable path- way relayed through PVP secondary vestibular neurons and a modifiable pathway relayed through FTN secondary vestibular neurons (Fig. 3–23).290,291,296–298 In addition to primary vestibular inputs, floccular Purkinje neurons receive eye velocity and retinal slip velocity sig- nals. Consistent with the model, recordings from FTNs and PVPs after modification of the gain of the VOR found large changes in the fir- ing rate of FTNs but only slight changes in the firing rate of PVPs.290,296 Furthermore, both pathways introduce a frequency-dependent phase lag, but FTNs have a much larger phase lag than PVP neurons.291 The time delays intro- duced by the FTN cerebellar loop are consis- tent with behavioral studies that found the modifiable component of the VOR lags the
unmodifiable component by about 5 msec. Although both PVPs and FTNs make direct connections with oculomotor neurons, FTNs probably make stronger connections than PVPs.297
Comparison of Ocular and Spinal Vestibular Reflexes
It is helpful to consider the similarities and differences between the ocular and spinal ves- tibular reflexes as an introduction to the orga- nization of vestibulospinal reflexes. If a rabbit is rotated at a constant speed on a turntable and suddenly stopped (producing an impulse of acceleration to the horizontal semicircular canal), a burst of ocular nystagmus results, with the slow phase being in the direction of the rotation prior to the deceleration (in the direc- tion of endolymph flow). In addition, if the head is mobile, it deviates slowly in the same direction as the slow-phase eye deviation. In some animals, if the stimulus is large enough, quick return movements regularly interrupt the slow head deviation, resulting in head oscil- lation (“head nystagmus”). The relationship between the magnitude of reflex head
Floccular complex
Semicircular canal
P
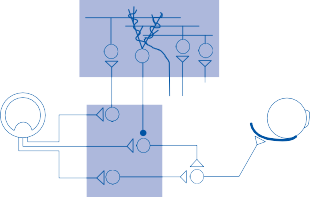
Eye velocity Retinal slip
–
Eye
Head velocity
FTN
PVP
Vestibular nucleus
Motoneuron
Figure 3–23. Vestibular nuclei neurons involved in motor learning. Head velocity signals originating in the semicircular canals are passed through the vestibular nucleus and the floccular complex to the extraocular motoneurons. Visual signals to the floccular complex (retinal slip velocity) are transmitted to the cerebellum by both mossy and climbing fibers. Eye move- ment signals (eye velocity) arrive at the floccular complex via mossy fibers. FTN, floccular target neurons; PVP, position- vestibular-pause neurons; P, Purkinje cell. (From Lisberger SG. Physiologic basis for motor learning in the vestibulo-ocular reflex. Otolaryngol Head Neck Surg. 119: 43, 1998, with permission.)
The rabbit on the turntable, if unrestrained and standing on four legs, tends to fall in the direction of the slow phase of eye and head deviation when the table is suddenly stopped. This falling tendency is counteracted by reflex activation of the antigravity muscles of the limbs on the side toward which the rabbit is falling, producing an increased extensor thrust in those limbs. At the same time, the extensor tone of the contralateral limbs is diminished and the rabbit maintains its balance. These extremity muscle reflexes are mediated via the semicircular canals and are always appropriate to prevent falling regardless of the direction of the acceleration force.302
The effector organs of the vestibulo-ocular reflexes are the extraocular muscles, while those of the vestibulospinal reflexes are the “antigravity” muscles, the extensors of the neck, trunk, and extremities. The organization of the vestibulospinal reflexes is the same as that of the vestibulo-ocular reflexes as shown in Figure 1–11 in Chapter 1. The same push– pull mechanism exists for controlling the balance between the extensor and flexor skele- tal muscles as for the agonist–antagonist extraocular muscles of the lateral and medial recti. A major difference between the organi- zation of ocular and spinal reflexes is the increased complexity of the spinal muscle response compared to the eye movement produced by an agonist and antagonist muscle acting in the horizontal plane. Even a simple movement about an extremity joint in a two- dimensional plane requires a complex pattern of contraction and relaxation in numerous muscles. Multiple agonist and antagonist mus- cles on both sides must receive appropriate signals to ensure a smooth coordinated movement. Unfortunately, a simple recording technique does not exist for quantifying this complexskeletalmuscleresponse.Furthermore,
although the vestibulospinal reflexes require a coordinated action of synergistic and antago- nistic muscles to respond to postural distur- bances, different subjects may use a different motor strategy to achieve the goal. These fac- tors have made studies of the vestibulospinal reflexes much more difficult than studies of the vestibulo-ocular reflexes.
Vestibulospinal Connections
The connections from the vestibular nuclei to the spinal cord were initially identified in cats using electrophysiological techniques.303–306 Stimulating electrodes are placed near neurons in each subnucleus and recordings are made at different levels within the spinal cord and anterior horn to identify the neurons’ termina- tions. Electromyograph (EMG) recordings were used to study the pattern of activated muscles. Stimulating electrodes have also been placed on individual canal and otolith nerves to determine the end organ input to each path- way (Fig. 3–24).307–311 Electrophysiological studies in primates have largely confirmed the findings in the cat, and in addition labeling studies of individual secondary vestibular neu- rons have traced their projections in the spinal cord.301,312–315
LATERAL VESTIBULOSPINAL TRACT
The vast majority of fibers in the lateral vestib- ulospinal tract originate from neurons in the lateral vestibular nucleus (Fig. 3–25).8 A soma- totropic pattern of projections originates in the lateral vestibular nucleus such that neurons in the rostroventral region supply the cervical cord while neurons in the dorsocaudal region innervate the lumbosacral cord. Neurons in the intermediate region supply the thoracic cord. In the spinal cord, the majority of fibers run ipsilaterally in the ventral half of the lateral funicle and the lateral part of the ventral funi- cle (Fig. 3–25). The tract terminates through- out the length of the cord, either directly on dendrites of anterior horn cells or on interneu- rons that project to anterior horn cells of the axial and proximal limb musculature.316 Some of the cells of the eighth lamina send their axons to the contralateral cord, which might account for the bilateral effects that have been observed after stimulation in the lateral
Projection Level and Pathway of Vestibulospinal Neurons
a Utriculus b Sacculus


Percentage (%)
Percentage (%)
Percentage (%)
Percentage (%)
80 80
60 60
40 40
C1/C2 C1/2
20 C3
0 T1
i-LVST MVST L3
20 C3
0 T1
i-LVST MVST L3
c-LVST c-LVST
Horizontal Canal
Anterior Canal
Posterior Canal
Percentage (%)
Percentage (%)
100
80
60
40
20
0
i-LVSTMVST c-L
C1/2 C3
T1 L3
80

Percentage (%)
Percentage (%)
60
40
20
0
i-LVST MVST
C1/2 C3
T1 L3
80

Percentage (%)
Percentage (%)
60
40
20
0
i-LVST MVST
C1/C2 C3
T1
L3
VST
c-LVST
c-LVST
Figure 3–24. Projection level and pathway of vestibulospinal neurons originating from five different vestibular end-organs. a Utricular nerve-activated vestibulospinal neurons. b Saccular nerve-activated vestibulospinal neurons. c Horizontal semicircular canal nerve-activated vestibulospinal neurons. d Anterior semicircular canal nerve-activated vestibulospinal neurons. e Posterior semicircular canal nerve-activated vestibulospinal neurons. The vertical axis in each plot indicates the relative percentage of the sum of the neurons activated from the C1/C2 junction to the total number of vestibulospinal neurons (100%). Note that, in (e), many of the ipsilateral-lateral vestibulospinal tract (i-LVST) neurons descended to or caudal to the L3 segment level. (From Kushiro K, Bai R, Kitajima N, Sugita-Kitajima A, Uchino Y. Properties and axonal trajectories of posterior semicircular canal nerve-activated vestibulospinal neurons. Exp Brain Res. 2008 Nov;191:257, with permission.)
vestibular nucleus. There may also be some crossed pathways. Activation of vestibulospinal fibers by electric stimulation in the lateral nucleus produces monosynaptic excitation of extensor motoneurons and disynaptic inhibi- tion of flexor motoneurons.317,318 The main pri- mary afferent input to the lateral vestibulospi- nal tract is from the utricule and posterior semicircular canal particularly for fibers termi- nating in the lumbar cord (Fig. 3–24).307
MEDIAL VESTIBULOSPINAL TRACT
The fibers of the medial vestibulospinal tract originate mostly from neurons in the medial vestibular nucleus and enter the spinal cord in the descending MLF (Fig. 3–25).8 The fibers travel bilaterally in the ventral funicle as far as the midthoracic level with the vast majority ending on neurons in the cervical cord. In monkey, medial vestibulospinal neurons labeled with biocytin terminate in lamina VII,
VIII, and XI of the cervical cord with the unique feature that crossed axons show a high degree of collateralization throughout the cer- vical cord, while uncrossed axons often termi- nate in a single cervical segment.301 The medial vestibulospinal tract receives afferent input from both otolith organs and all three semicir- cular canals, but the saccular input is more prominent than the utricular input and the horizontal and anterior canal input is more prominent than the posterior canal input (Fig. 3–24).
RETICULOSPINAL TRACT
The reticulospinal tract originates from neurons in the bulbar reticular formation but is heavily influenced by vestibular inputs .319 The nuclei reticularis gigantocellularis and pontis caudalis provide most of the long fibers passing into the spinal cord, although most of the neurons in the caudal reticular formation also contribute fibers.
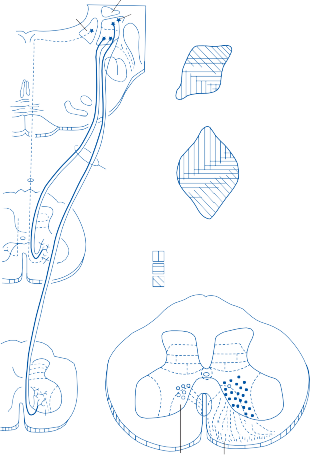
M L
CR Dorsal
Medial Lateral
Ventral Rostral
Medial vestibulo-
Lateral
Ventral Dorsal
spinal tract
C8
VII
IX
VIII
vestibulo- spinal tract
Caudal
Fibers to:
Cervical cord Thoracic cord Lumbosacral cord
I-III
L7 VII
IX
VII
IX
VIII
Medial vestibulo- spinal fibers
Lateral vestibulo- spinal fibers
Figure 3–25. Lateral and medial vestibulospinal tracts. Topographical organization within the lateral vestibular nucleus (upper right) and endings within the spinal cord (lower right). CR, Corpus Restiform; S, superior nucleus; M, medial nucleus; L, lateral nucleus. (From Brodal A. Anatomical organization of cerebello-vestibulo-spinal pathways. In: De Renck AVS, Knight J (eds). CIBA Foundation Symposium: Myotatic, Kinesthetic and Vestibular Mechanisms. Churchill, London, 1967, with permission.)
Both crossed and uncrossed fibers transverse the length of the spinal cord, terminating in the seventh and eighth laminae of the gray matter.320 Stimulation of the pontomedullary reticular formation in the regions where the long descending spinal projections originate results in inhibition of both extensor and flexor motoneurons throughout the spinal cord.321,322
The vestibular nuclei are one of many struc- tures that send fibers to the reticular formation. Axonal branches and collaterals of cells in all four main vestibular nuclei are distributed to the pontomedullary reticular formation. Only a small number of primary vestibular
fibers end in the reticular formation; the main vestibular influence on reticulospinal outflow is mediated by way of the secondary vestibular neurons. A pattern exists within the vestibu- loreticular projects such that each nucleus projects to different areas of the reticular for- mation, but no detailed somatotropic organiza- tion has been identified.8
Cerebellar–Vestibular Interaction
The “spinal” cerebellum provides a major source of input to neurons whose axons form the
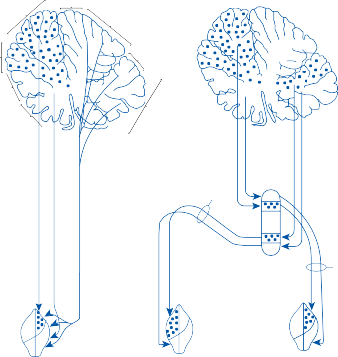
V
VII
IV VIII
III
IX
II
I X
Rostral
Hook bundle fibers
Fastigial nucleus
Caudal
Ventral
+ Dorsal
++
++
++
+
Ventral
Rostral
Dorsal
Rostral
Ventral
Dorsal
Lateral vestibular nucleus
![]()
Forelimb Hindlimb
Figure 3–26. Topographical organization of cerebellar vermian, fastigial nucleus, and lateral vestibular nucleus connec- tions. Filled circles, forelimb; +, hindlimb. (From Brodal A. Anatomy of the vestibular nuclei and their connections. In: Kornhuber HH (ed). Handbook of Sensory Physiology, Vol VI, Part I. Springer-Verlag, New York, 1974, with permission.)
lateral vestibulospinal and reticulospinal tract. A somatotopic organization of projections to the lateral nucleus occurs in both the vermian cortex and fastigial nuclei (Fig. 3–26).56,302,323 The caudal part of the fastigial nucleus gives rise to a bundle of fibers that cross the midline (Russell’s hook bundle), curving around the brachium conjunctivum before running to the contralateral lateral vestibular nucleus and dor- solateral reticular formation. In addition, direct ipsilateral outflow passes from the fastigial nucleus to areas of the reticular formation that send long fibers to the spinal cord in the reticulospinal tract. The cerebellar–reticular pathways do not appear to exhibit somatotopic organization.56 The cerebellar vermis and fasti- gial nuclei receive input from secondary vestibular neurons, the spinal cord, and the pontomedullary reticular formation. The result is a close-knit vestibular–reticular–cerebellar
functional unit for the maintenance of equilib- rium and locomotion.
Vestibulo-Collic Reflexes
The VCR stabilizes the head in space and pro- duces smooth coordinated movements to track a moving target. The head and neck represent a complex biomechanical system with a large eccentric mass located on top of a column of cervical vertebrae.324 Although theoretically there are multiple degrees of freedom about each of the eight vertebral joints, most move- ments are made about a restricted set of joint axes; for example, yaw movements are mostly
made about C1-C2 and pitch movements about C1-skull or C6-7-T1.
Both lateral and medial vestibulospinal tracts
send fibers to the cervical cord, but the medial
vestibulospinal tract is most important for coordinating neck-vestibular-ocular reflexes. As noted previously, two types of MVST fibers have been identified based on labeling studies in monkey: crossed axons with a high degree of collateralization throughout the cer- vical cord and uncrossed axons with few collat- erals that target selective cervical segments.301 The former could provide tonic input to widely distributed motor neurons to stabilize the head, while the latter could provide canal specific input to motor neurons that initiate head move- ments about selective vertebrae. Convergence of canal and otolith input onto vestibulospinal neurons is also important for generating the “spatiotemporal” convergence within the VCR.308
Cellular Mechanisms
Recordings made from identified secondary vestibulospinal neurons in squirrel monkey indicate that the neurons encode the velocity of externally applied head movement but not head velocity in space during self-generated head movements.325 So-called vestibular-only (VO) neurons, defined based on the lack of eye movement–related firing, show marked sup- pression of firing during active head move- ments compared to passive movements.326–329 Furthermore, these neurons continue to fire selectively with passive movements even dur- ing a combination of active and passive move- ments. Many of these VO neurons control the vestibulocollic reflexes through their projec- tions to the cervical spinal cord. When the goal is to make an active head movement, vestibular drive to the reflex would cause an inappropri- ate head movement in the direction opposite to the intended goal. Logically secondary neurons in these reflexes should be less responsive dur- ing active head movements. Since these neurons selectively monitor passive head move- ments even during active movements they can selectively respond to an unexpected head movement such as one that might occur while tripping over a curb while walking.Convergence of somatosensory, neck, and vestibular signals on Purkinje cells in the cerebellar vermis could provide the substrate for adaptive plasticity within the vestibulo-spinal reflexes analogous to the role of the flocculus in the vestibulo- ocular reflexes.330
SUBJECTIVE VESTIBULAR SENSATION
Unlike those sensory organs that respond to energy sources external to the body, the laby- rinths respond to self-generated forces within the head. During natural head movements, these forces are not under voluntary control and therefore the vestibular responses are more automatic than those of the other sensory modalities. For example, one can remove vision simply by closing one’s eyes, whereas one can- not suppress vestibular stimuli during head movement. The recognition of the existence of a “sixth sense” for the perception of accelera- tions (motion) is a relatively late concept that resulted from the imaginative experiments of Mach in the 1870s.331 Mach observed that the perception of motion in his different experi- ments could be altered by changing the posi- tion of the head in relation to the body, which suggested to him that the sensory organs were located in the head. His findings, along with the physiologic and histologic work of contem- poraries such as Flourens,332 Breuer,333 and Crum-Brown,334 led him to the conclusion that the semicircular canals and the otoliths were responsible for the perception of angular and linear acceleration, respectively.
Vestibulothalamocortical Connections
Microelectrode recordings from multiple dif- ferent cortical regions in multiple species have documented trisynaptic connections between the cortex and the labyrinthine end organs. The pathways from the labyrinths to the cortex have been dissected using electrophysiolocal recordings along with anterograde and retro- grade tracer techniques. Secondary neurons are located in all four vestibular nuclei and in the fastigial and anterior interposed cerebellar nuclei, and tertiary neurons are mainly located in the ventroposterior thalamus (see Fig. 1–15 in Chapter 1).335–340 Cortical regions that receive trisynaptic vestibular inputs include the parie- to-insular vestibular cortex (PIVC), the soma- tosensory cortical area 3aV, the anterior tip of the intraparietal sulcus, the medial superior temporal area, the ventral intraparietal area and the frontal eye fields and premotor/motor cortex (Fig. 3–27).341–347
2v
2v
c
Monkey 7 a,b
Galvanic (fMRI)
VTS
PIVC
Calorics right (PET)
–10 +10 +20
R L
Figure 3–27. Illustration of the normal activation–deactivation pattern during unilateral vestibular stimulation in healthy volunteers (activations in black, deactivations in blue). For comparison a schematic drawing of a monkey brain with the neu- rophysiologically determined multisensory vestibular areas 6, 3aV, 2v, 7a, b, PIVC and VTS is given (top left). Note that the locations of the activated areas during galvanic stimulation of the vestibular nerve (fMRI; top right) are similar in humans. During caloric irrigation of the right ear in healthy right-handers, activations (H215O-PET) occur in temporo-parieto-insular areas of both hemispheres, but there is a dominance of the non-dominant right hemisphere (middle: surface view of the right and left hemispheres; bottom: transverse sections Z = –10, +10, +20 mm). Deactivations are located in areas of the visual cortex bilaterally. (From Dieterich M, Brandt T. Functional brain imaging of peripheral and central vestibular disorders. Brain. 2008;131:2538, with permission)
Electrophysiological and tracer studies in rats and cats have found bilateral projections to large parts of the thalamus originating from all four vestibular subnuclei.348,349 Based on selec- tive nerve stimulation in cats it appears that
utricular-thalamic projections are mostly ipsilateral, while posterior semicircular canal and saccular-thalamic projections are mostly contralateral.350,351 Two anatomically separate graviceptive vestibulothalamic pathways have
been identified based on tracer studies in mon- key and lesion studies in patients; a crossed pathway running within the MLF and an uncrossed pathway running near to or within the medial lemniscus.352 Lesions in the former pathway result in a combination of eye and perceptual tilt, while lesions of the latter path- way cause only a perceptual tilt. In monkey the vestibulothalamic projections end mostly in the somatosensory VPL region but some also end in VPI and VPM and even in the somatomotor VL region.340,353 The largest number is at the border between VPL and VL. Different thal- amic regions project to different cortical regions. In the squirrel monkey the PIVC receives its main thalamic input from the caudal VPL and the pulvinar, while the soma- tosensory cortex (area 3aV) receives its main input from the rostral and dorsal VPL.340,343
Response Properties of Thalamic Relay Neurons
Thalamic neurons receiving vestibular input show a range of dynamic properties similar to neurons in the vestibular nuclei but with the major exception that few have eye movement sensitivity.340,353 With angular rotation most thal- amic relay neurons show a modest phase lead but unlike primary afferents, these neurons show little additional phase lead and little drop in gain at low frequencies of rotation reflecting the addi- tion of velocity storage to the primary afferent signal.337,340 For translational movements response phase and gain differed somewhat from vestibular nucleus neurons, suggesting additional processing.340,353 As in vestibular nucleus neu- rons, most thalamic neurons reflected a combi- nation of net linear acceleration originating from otolith afferents and an estimate of gravitational acceleration derived from spatially and tempo- rally transformed canal afferents. Although some cells encoded just translation or net linear accel- eration, most had intermediate properties. Most of these were more sensitive to translation than to net acceleration.
Response Properties of Vestibular Cortex Neurons
Although vestibular activity can be found in multiple cortical regions from the frontal
cortex to the parieto-occiptal cortex, a portion of the retroinsular cortex, the parieto-insular vestibular cortex (PIVC), is felt to be at the center of the vestibular cortical network.341,342 About two-thirds of neurons in PIVC respond to vestibular stimulation and almost all vestibu- lar activated neurons also respond to soma- tosensory and visual stimuli. Vestibular signals to this region mostly originate from the semi- circular canals with only rare responses to steady tilt in darkness. The optimal activation of these canal-related neurons is not in the planes of the semicircular canals but rather is distributed through all possible spatial planes through the head. Neurons with the same spa- tial plane tend to cluster in subdivisions of PIVC. Furthermore, the response to optoki- netic stimuli is best when the plane of the visual movement is in the optimal vestibular plane indicating a change to a head co-ordinate frame of reference. The gain of the vestibular and optokinetic responses is similar for low fre- quencies, but at 1 Hz the optokinetic gain is low while the vestibular gain remains strong. Overall the gain of PIVC neurons to angular acceleration is lower by a factor of four com- pared to vestibular neurons in the vestibular nuclei and in the thalamus.
Functional Brain Imaging in Normal Human Subjects
Functional magnetic resonance imaging (fMRI) and positron emission tomography (PET) studies in normal humans after galvanic stimulation of the vestibular nerve or after caloric activation of the horizontal semicircular canal have largely confirmed the multiple ves- tibulocortical projection areas identified in ani- mals (Fig. 3–27).347,354,355,356 As in monkey, the temporo-insular and temporo-parietal regions seem to be at the center of the vestibulocortical network in humans. The degree of activation in these cortical areas after vestibular stimulation is greater in the nondominant hemisphere, in the hemisphere ipsilateral to the stimulated ear, and in the hemisphere ipsilateral to the slow phase of caloric nystagmus. Interestingly, there is a deactivation of visual and somatosen- sory areas of both hemispheres after unilateral vestibular stimulation, suggesting reciprocal inhibitory interactions between the sensory systems.357 Activation of the hippocampus after
caloric stimulation is consistent with animal studies showing connections between the ves- tibular nuclei and hippocampal CA1 neurons.358,359 So-called place cells in the hippocampus encode location within the envi- ronment and are critical for normal spatial ori- entation and navigation.
Lesions of the Vestibulocortical Pathways in Patients
As discussed earlier, lesions of the vestibular nuclei or more peripheral vestibular structures lead to a combination of motor and perceptual abnormalities. For example patients with infarction of the dorsolateral medulla (Wallenberg syn- drome) typically have an ocular tilt response, spontaneous nystagmus, lateropulsion, and tilt of the perceived vertical. By contrast patients with lesions of the posterolateral thalamus or vestibular cortex have deviations of the per- ceived vertical and imbalance but without ocul- omotor signs. The imbalance associated with thalamic infarcts has been called “thalamic astasia,” a severe imbalance but without weak- ness or incoordination.360 Only lesions that involve the posterolateral thalamus cause the perception of tilt and imbalance. 362
Psychophysical Studies
SEMICIRCULAR CANALS
As Mach described,331 a subject rotated about an earth-vertical axis on a rotatory platform will perceive turning that is dependent on the per- ceived “speed of turning.” The sensation that subjects qualitatively describe as “moving” to the left or to the right progressively increases with prolonged constant acceleration, although the turning sensation increases at a lesser rate than the platform velocity. Mach noted that below a minimum or threshold angular accel- eration the subject does not perceive turning. The modern era of vestibular psychophysics began with attempts to correlate the threshold and magnitude of subjective sensation with the magnitude of angular acceleration. Although considerable difference exists in reported values, the threshold to constant angular acceleration is in the range of 0.1 to 0.5 degrees/sec2.363,364 This is approximately an
order of magnitude lower than the constant angular acceleration necessary to produce nystagmus.365 For stimulation with angular sinusoidal rotations, the perception of motion corresponds to the prediction of the pendulum model, just as the pendulum model predicts the velocity of the slow phases of nystagmus. In accordance with this model, the threshold for the perception of motion is approximately
1 degree/sec for all frequencies within the range of natural head movements.
Cupulometry developed by van Egmond and associates366 was the first quantitative test used for assessing vestibular function on the basis of subjective sensation. With this test, the subject is maintained at a constant velocity of angular rotation and then suddenly stopped. The durations of “after-turning” sensation are measured for impulses of different ampli- tude (usually 15 to 60 degrees/sec) and plotted versus the log of impulse magnitude (to obtain the so-called cupulogram). The intercept of the line with the abscissa corresponds to a sub- jective sensation threshold and the slope, a time constant of after-turning sensation. Normative data for the subjective threshold vary from 1 to 4 degrees/sec and the time con- stant of after-turning sensation, from 2 to 14 sec.365 Studies using low-frequency sinusoi- dal rotations have found identical phase shifts in the subjective sensation of turning and the VOR in normal subjects and in patients after unilateral labyrinthectomy.367 Furthermore, in patients with unilateral vestibular loss, the duration of the subjective sensation of turning during ampullofugal stimulation of the remaining intact labyrinth is shorter than the duration of the sensation of turning during ampullopetal stimulation—similar to the asym- metry in VOR amplitude seen in these patients.
Sinha et al.368 studied the perception of self- rotation with constant velocity angular rotations and found a delay of 2–5 sec before subjects perceived constant velocity and a plateau of 9–14 sec at maximum perceived velocity before they perceived a decay in velocity. Since there is a rapid rise and an immediate decay in nys- tagmus slow-phase velocity, they concluded that sensory signals from the semicircular canals must undergo additional neural process- ing beyond the contribution of the velocity storage mechanism.
Data on threshold and accuracy of estimation of earth visual vertical have been obtained with static (postural) tilt experiments.369–373 The sub- ject is strapped to a tilt platform in darkness and asked either to estimate the deviation of his head from the earth-vertical or to adjust a luminous line on a dark field to a vertical posi- tion. Normal subjects respond with an accuracy of 2 to 4 degrees for tilt angles up to 40 degrees (accuracy falls off progressively for larger angles of tilt). The subjective estimate of tilt obviously depends on the gravitational force (Fg).374 If the
subject is asked to estimate the angle of tilt
under different gravitational forces, the esti- mate will vary with Fg. For g values <1, the
angle of tilt is underestimated, whereas for g
values >1, the angle of tilt is overestimated. In experiments carried out at “zero g” in parabolic aircraft flights and in orbiting spacecraft, the subjects are unable to perceive tilt. Patients with unilateral vestibular lesions report a static tilt of the visual vertical toward the side of the lesion and have impaired estimation of the visual vertical when tilted toward the damaged ear compared to toward the intact ear.375–376 These illusionary effects are transient, disap- pearing within several weeks of the lesion.
A subject undergoing linear oscillation (e.g., on a parallel swing) reports experiencing two separate types of motion: one is a sensation of linear movement in the horizontal plane and the other is a sensation of tilt. Both sensations
vary with the changing velocity (acceleration) of the platform.365 Beginning with low ampli- tudes of oscillation, the subject initially per- ceives motion without a specific direction. This is followed by perception of the direction of linear movement and finally at higher intensi- ties of stimulation by a perception of tilting. Using dynamic stimuli, estimates of the minimal horizontal linear acceleration that normal subjects can perceive range from 5 to
15 cm/sec2.363 Interestingly, these threshold values are similar to the values obtained by Mach for the perception of vertical linear acceleration (10 to 12 cm/sec2).
During centrifugal stimulation, normal sub- jects have the illusion that a horizontal small luminous bar is roll-tilted in the direction of the resultant force by the same amount that they feel roll-tilted—the so-called oculogravic illusion (Fig. 3–28).377–379 Patients who undergo therapeutic unilateral vestibular nerve section show a marked asymmetry of this illusion: they perceive the illusion when the resultant force is directed toward their intact ear, but they per- ceive a much reduced illusion (roll-tilt) when the force is directed toward the operated ear. As with the tilt of the static visual vertical, this asymmetry in the oculogravic illusion gradually disappears over several weeks (presumably because of central compensation).377 Curthoys et al. likened this asymmetry in otolith function to the asymmetry of the semicircular canal function seen after similar operations.378
a b c
R R
g
Sees bar at rest
Sees bar during rotation
Sets bar during rotation
Figure 3–28. Oculogravic illusion experienced by normal subject seated on the arm of a centrifuge. At rest (a), the subject perceives the bar in the gravitational (g) horizontal. (b) When the centrifuge is revolving at a constant velocity (), he per- ceives himself as being roll-tilted and consequently judges that the bar has been roll-tilted in the same direction. (c) When instructed to set the rotatable bar to the gravitational horizontal, he rotates the bar through the perceived angle of roll-tilt ( in Fig. 1–1). R = resultant of the centrifugal and gravitational forces. (From Curthoys IS, et al. Human otolithic function before and after unilateral vestibular neurectomy. J Vestib Res. 1990;1: 199–209, with permission.)
Brodal A. Anatomy of the vestibular nuclei and their connections. In: Kornhuber HH, ed. Handbook of Sensory Physiology, The Vestibular System, Vol VI, Part 1. New York: Springer-Verlag; 1974.
Kappers CUA, Huber GC, Crosby ED. The Comparative Anatomy of the Nervous System of Vertebrates, Including Man. New York: Macmillan; 1936.
Fristsch B. Evolution of the vestibulo-ocular system.
response of a single utricular macule to linear acceleration.
An interesting corollary of these psychophysi- cal experiments on semicircular canal and otolithic function is that similar mechanisms operate in patients with vestibular disorders. Such patients experience an illusion of motion and tilt of the environment while stationary and have difficulty judging the relative motion of
Otolaryngol Head Neck Surg. 1998;119:182.
Mehler WR. Comparative anatomy of the vestibu- lar nuclear complex in submammalian vertebrates. In: Brodal A, Pompeiano O, eds. Basic Aspects of Central Vestibular Mechanisms. New York: Elsevier Publishing; 1972.
Honrubia V, Suarez C, Kuruvilla A, Sitko S. Central projections of primary vestibular fibers in the bull- frog. III. The anterior semicircular canal afferents. Laryngoscope. 1985;95:1526.
Ishizuka N, Sasaki S, Mannen H. Central course and
objects around them, Furthermore, the magni- tude of these illusions correlates with the mag- nitude of the velocity of the slow component of their spontaneous nystagmus. While more research needs to be conducted in this area, psychophysical studies of vestibular illusions in normal subjects provide insight into the bizarre illusions experienced by patients with vestibular lesions.
REFERENCES
Lorente de Nó R. Anatomy of the eighth nerve: the cen- tral projection of the nerve endings of the internal ear. Laryngoscope. 1933;43:1.
Gacek R. The course and central termination of first order neurons supplying vestibular endorgans in the cat. Acta Otolaryngol Suppl (Stockh). 1969; Suppl 254:1.
Carleton SC, Carpenter MB. Distribution of primary vestibular fibers in the brainstem and cerebellum of the monkey. Brain Res. 1984;294:281.
Newman A, Suarez C, Lee WS, Honrubia V. Afferent innervation of the vestibular nuclei in the chinchilla. IL Description of the vestibular nerve and nuclei. Brain Res. 1992;597:278.
Naito Y, Newman A, Lee WS, et al. Projections of the individual vestibular end-organs in the brain stem of the squirrel monkey. Hear Res. 1995;87:141.
Straka H, Dieringer N. Basic organization princi- ples of the VOR: lessons from frogs. Prog Neurobiol. 2004;73:259.
Uchino Y, Sasaki M, Sato H, Bai R, Kawamoto E. Otolith and canal integration on single vestibular neurons in cats. Exp Brain Res. 2005;164(3):271.
Barmack NH. Central Vestibular system: vestibu- lar nuclei and posterior cerebellum. Brain Res Bull. 2003;60:511.
Brodal A, Pompeiano O. The vestibular nuclei in the cat. J Anat. 1957;91:438.
terminal arborizations of single primary afferent fibers
from the horizontal canal in the cat. Neurosci Lett. 1982;33:135.
Mannen H, Sasaki S, Ishizuka N. Trajectory of primary vestibular fibers originating from the lateral, anterior and posterior semicircular canals in the cat. Proc Jpn Acad. 1982;58:237.
Sato F, Sasaki H, Sasaki S, Mannen H. Morphology of single primary vestibular afférents originating from the horizontal semicircular canal in the cat. J Comp Neurol. 1984;290:423.
Sadjadpour K, Brodal A. The vestibular nuclei in man. A morphological study in the light of experimental findings in the cat. J Hirnforsch. 1968;10:299.
Abend WF. Functional organization of the superior vestibular nucleus in the squirrel monkey. Brain Res. 1977;132:65.
Diaz C, Suarez C, Tolivia J, et al. Human vestibular nuclei: a morphometric approach. Otolaryngol Head Neck Surg. 1992;107:190.
Lopez I, Honrubia V, Baloh RW. Aging and the human vestibular nucleus. J Vestib. Res 1997;1:77.
Korte GE, Friedrich VL, Jr. The fine structure of the feline superior vestibular nucleus: identification and synaptology of the primary vestibular afferents. Brain Res. 1979;176:3.
Langer T, Fuchs AF, Chubb MC, et al. Floccular efferents in the rhesus macaque as revealed by autora- diography and horseradish peroxidase. J Comp Neurol. 1985;235:26.
Mitsacos A, Reisine H, Highstein SM. The superior vestibular nucleus: an intracellular HRP study in the cat. II. Non-vestibulo-ocular neurons. J Comp Neurol. 1983;215:92.
Mitsacos A, Reisine H, Highstein SM. The superior vestibular nucleus: an intracellular HRP study in the cat. I. Vestibulo-ocular neurons. J Comp Neurol. 1983;215:78.
Pompeiano O, Brodal A. Spino-vestibular fibers in the cat. An experimental study. J Comp Neurol. 1957;108:353.
Boyle R, Johanson C. Morphological properties of ves- tibulospinal neurons in primates. Ann NY Acad Sci. 2003;1004:183.
Carleton SC, Carpenter MB. Afferent and effer- ent connections of the medial, inferior and lateral vestibular nuclei in the cat and monkey. Brain Res. 1983;278:29.
Suarez C, Diaz C, Tolivia J, et al. Morphometric analysis of the human vestibular nuclei. Anat Rec. 1996;247:271.
McCrea RA, Strassman A, May E, Highstein SM. Anatomical and physiological characteristics of ves- tibular neurons mediating the horizontal vestibulo- ocular reflex of the squirrel monkey. J Comp Neurol. 1987;264:547.
Highstein SM, Holstein GR.The anatomy of the vestibular nuclei. Prog Brain Res. 2006;151:157.
Straka H, Vibert N, Vidal PP, Moore LE, Dutia MB. Intrinsic membrane properties of vertebrate vestibular neurons: function, development and plasticity. Prog Neurobiol. 2005;76(6):349.
Büttner-Ennever JA. The extraocular motor nuclei: organization and functional neuroanatomy. Prog Brain Res. 2006;151:95.
Smith PF, Darlington CL. Recent advances in the pharmacology of the vestibulo-ocular reflex. Trends Pharmacol Res. 1996;17:421.
de Waele C, Muhlethaler M, Vidal PP. Neurochemistry of the central vestibular pathways. Brain Res Rev. 1995;20:24.
Zanni M, Giardino L, Toschi L, Galetti G, Calza L. Distribution of neurotransmitters, neuropeptides, and receptors in the vestibular nuclei complex of the rat: an immunocytochemical, in situ hybridization and quantitative receptor autoradiographic study. Brain Res Bull. 1995;36:443.
Beitz AJ, Anderson JH. Neurochemistry of the Vestibular System. New York: CRC Press; 2000.
Sasa M, Takeshita S, Amano T, Kurisu K. Primary neurotransmitters and regulatory substances onto vestibular nucleus neurons. Biol Sci Space. 2001;15 (4):371.
Straka H, Reichenberger I, Dieringer N. Synaptic transmission by vestibular nerve afferent fibers. In: Beitz AJ, Anderson JH, eds. Neurochemistry of the Vestibular System. New York: CRC Press; 2000: 47.
Walberg F, Ottersen OP, Rinvik E. GABA, glycine, aspartate, glutamate and taurine in the vestibular nuclei: an immunocytochemical investigation in the cat. Exp Brain Res. 1990;79:547.
Lopez I, Baloh RW, Honrubia V. GABAA and glycine receptor immunoreactivity in the chinchilla vestibular nuclear complex. Assoc Res Otolaryngol Abst. 1995;15:59.
Petralia RS, Wenthold RW. Light and electron micro- scope distribution of AMPA-selective glutamate recep- tors in the rat brain. J Comp Neurol. 1992;318:329.
Petralia RS, Wenthold RW. Light and electron immu- nocytochemical localization of the NMDA recep- tor subunit NMDAR1 in the rat nervous system. J Neurosci. 1994;14:667.
Popper P, Rodrigo JP, Alvarez JC, Lopez I, Honrubia
V. Expression of the AMPA-selective receptor sub- units in the vestibular nuclei of the chinchilla. Brain Res Mol Brain Res. 1997;44:21.
Nomura I, Senba E, Kubo T, et al. Neuropeptides and -aminobutyric acid in the vestibular nuclei of the
rat: an immunohistochemical analysis. I. Distribution.
Brain Res. 1984;331:190.
Mugnaini E, Oertel WH. An atlas of the distribu- tion of GABAergic neurons and terminals in the rat CNS as revealed by GAD immunocytochemistry. In: Bjorkland A, Hökfelt T, eds. Handbook of Chemical Neuroanatomy. Vol 4. Pt 1. Amsterdam, Netherlands: Elsevier; 1985: 436.
Lopez I, Hoffman L, Honrubia V. Distribution of glutamic acid decarboxylase-like immunoreactivity in the chinchilla vestibular nuclei. Soc Neurosci Abst. 1993;137:59.
Precht W. Vestibular mechanisms. Annu Rev Neurosci. 1979;2:265.
Angelake DE, Cullen KE. Vestibular system: the many facets of a multimodal sense. Annu Rev Neurosci. 2008;31:125.
Ito M, Shiida T, Yagi N, Yamamoto M. Visual influ- ence on rabbit horizontal vestibulo-ocular reflex pre- sumably effected via the cerebellar flocculus. Brain Res. 1974;65:170.
Lisberger SG, Pavelko TA, Broussard DM. Responses during eye movements of brain stem neurons that receive monosynaptic inhibition from the flocculus and ventral paraflocculus in monkeys. J Neurophysiol. 1994;72:909.
Lisberger SG. Physiological basis for motor learning in the vestibulo-ocular reflex. Otolaryngol Head Neck Surg. 1998;119:43.
Highstein SM. Role of the flocculus of the cerebel- lum in motor learning of the vestibulo-ocular reflex. Otolaryngol Head Neck Surg. 1998;119:212.
Scudder CA, Fuchs AF. Physiological and behavioral identification of vestibular nucleus neurons mediating the horizontal vestibuloocular reflex in trained rhesus monkeys. J Neurophysiol. 1992;68:244.
Wilson VJ, Schor RH. The neural substrate of the vestibulocollic reflex. What needs to be learned. Exp Brain Res. 1999;129:483.
Pompeiano O. Cerebello-vestibular interrelations. In: Kornhuber HH ed. Handbook of Sensory Physiology. The Vestibular System. Vol VI. Pt 1. New York: Springer-Verlag; 1974.
Peterson BW. The reticulospinal system and its role in the control of movement. In: Barnes CD ed. Brainstem Control of Spinal Cord Function. New York: Academic Press; 1984: 27.
Precht W, Shimazu H. Functional connections of tonic and kinetic vestibular neurons with primary vestibular afférents. J Neurophysiol. 1965;28:1014.
Precht W, Strata P. On the pathway mediating opto- kinetic responses in the vestibular nuclear neurons. Neuroscience. 1980;5:777.
Shimazu H, Precht W. Tonic and kinetic responses of cat’s vestibular neurons to horizontal angular accelera- tion. J Neurophysiol. 1965;28:991.
Goldberg JM, Highstein SM, Moschovakis AK, Fernanandez C. Inputs from regularly and irregularly discharging vestibular nerve afferents to secondary neurons in the vestibular nuclei of the squirrel mon- key. I. An electrophysiological analysis. J Neurophysiol. 1987;58:700.
Shimazu H, Precht W. Inhibition of central vestibular neurons from the contralateral labyrinth and its medi- ating pathway. J Neurophysiol. 1966;29:467.
McCrea RA, Strassman A, May E, Highstein SM. Anatomical and physiological characteristics of ves- tibular neurons mediating the horizontal vestibulo- ocular reflex of the squirrel monkey. J Comp Neurol. 1987;264:547.
Waespe W, Henn V. Neuronal activity in the ves- tibular nuclei of the alert monkey during vestibular and optokinetic stimulation. Exp Brain Res. 1977; 27:523.
Waespe W, Henn V. Gaze stabilization in the primate: the interaction of the vestibulo-ocular reflex, opto- kinetic nystagmus, and smooth pursuit. Rev Physiol Biochem Pharmacol. 1987;106:38.
Lisberger SG, Pavelko TA, Broussard DM. Neural basis for motor learning in the vestibulo-ocular reflex of primates. I. Changes in the responses of brainstem neurons. J Neurophysiol. 1994;72:928.
Lisberger SG, Pavelko TA, Bronte-Stewart HM, et al. Neural basis for motor learning in the vestibulo-ocular reflex of primates. II. Changes in the responses of Purkinje cells in the cerebellar flocculus and ventral paraflocculus. J Neurophysiol. 1994;72:974.
Gallagher JP, Lewis MR, Shinnick-Gallagher P. An electrophysiological investigation of the rat medial vestibular nucleus in vitro. In: Correia MJ, Perachio AA, eds. Progress in Clinical and Biological Research. Vol 176. New York: Alan R. Liss; 1985: 293.
Lewis MR, Phelan KD, Shinnick-Gallagher P, Gallagher JP. Primary afferent excitatory transmis- sion recorded intracellularly in vitro from rat medial vestibular neurons. Synapse. 1989;3:149.
Babalian AL, Vibert N, Assié G, Serafin M, Mühlethaler M, Vidal PP. Central vestibular networks in the guinea- pig: functional characterization in the isolated whole brain in vitro. Neuroscience. 1997;81:405.
Him A, Dutia MB. Intrinsic excitability changes in vestibular nucleus neurons after unilateral deafferen- tation. Brain Res. 2001;908:58.
Ris L, Godaux L. Neuronal activity in the vestibular nuclei after contralateral or bilateral labyrinthectomy in the alert guinea pig. J Neurophysiol. 1998;80:2352.
Serafin M, de Waele C, Khateb A, Vidal PP, Mühlethaler M. Medial vestibular nucleus in the guin- ea-pig. I. Intrinsic membrane properties in brainstem slices. Exp Brain Res. 1991;84:417.
Serafin M, de Waele C, Khateb A, Vidal PP, Mühlethaler M. Medial vestibular nucleus in the guinea-pig. II. Ionic basis of the intrinsic mem- brane properties in brainstem slices. Exp Brain Res. 1991;84:426.
Johnston AR, MacLeod NK, Dutia MB. Ionic conduc- tances contributing to spike repolarization and after- potentials in rat medial vestibular nucleus neurons. J Physiol. 1994;481:61.
Smith MR, Nelson AB, du Lac S. Regulation of fir- ing response gain by calcium-dependent mecha- nisms in vestibular nucleus neurons. J Neurophysiol. 2002;87:2031.
Takazawa T, Saito Y, Tsuzuki K, Ozawa S. Membrane and firing properties of glutamatergic and GABAergic neurons in the rat medial vestibular nucleus J Neurophysiol. 2204;92:3106.
Precht W, Shimazu H, Markham CH. A mechanism of central compensation of vestibular function following hemilabyrinthectomy. J Neurophysiol. 1966;29:996.
Ris L, Godeaux E. Neuronal activity in the vestibular nuclei after contralateral or bilateral labyrinthectomy in the alert guinea pig. J Neurophysiol. 1998;80:2352.
Vibert N, Babalian A, Serafin M, Case J-P, Mühlethaler M, Vidal P-P Plastic changes underlying vestibular com- pensation in the guinea-pig persist in isolated, in vitro whole brain preparations. Neuroscience. 1999;93:413.
Precht W, Dieringer N. Neuronal events paralleling functional recovery (compensation) following periph- eral vestibular lesions. In: Berthoz A, Melvill Jones G, eds. Adaptive Mechanisms in Gaze Control: Facts and Theories. Amsterdam, Netherlands: Elsevier; 1985: 251.
Ried S, Maioli C, Precht W. Vestibular nuclear neuron activity in chronically hemilabyrinthectomized cats. Acta Otolaryngol (Stockh). 1984;98:1.
Sirkin DW, Precht W, Courjon JH. Initial, rapid phase of recovery from unilateral vestibular nerve lesion not dependent on survival of central portion of vestibular nerve. Brain Res. 1984;302:245.
Yagi T, Markham CH. Neural correlates of com- pensation after hemilabyrinthectomy. Exp Neurol. 1984;84:98.
Bergquist F, Ludwig M, Dutia MB. Role of the com- missural inhibitory system in vestibular compensation in the rat. J Physiol. 2008;586(pt 18):4441.
Yamanaka T, Him A, Cameron SA, Dutia MB. Rapid compensatory changes in GABA receptor efficacy in rat vestibular neurones after unilateral labyrinthec- tomy. J Physiol. 2000;523(pt 2):413.
Curthoys IS, Halmagyi GM. Vestibular compensation.
Adv Otorhinolaryngol. 1999;55:82.
Darlington CL, Dutia MB, Smith PF. The contribu- tion of the intrinsic excitability of vestibular nucleus neurons to recovery from vestibular damage. Eur J Neurosci. 2002;15:1719.
Dieringer N. Activity-related postlesional vestibular reorganization. Ann NY Acad Sci. 2003;1004:50.
Ris L, Godaux E. Voltage-gated calcium channels con- tribute to the pattern of the resting discharge in guinea pig medial vestibular nucleus neurons. Neurosci Lett. 2001;297:142.
Beraneck M, Hachemaoui M, Idoux E, et al. Long- term plasticity of ipsilesional medial vestibular nucleus neurons after unilateral labyrinthectomy. J Neurophysiol. 2003;90(1):184.
Beraneck M, Idoux E, Uno A, Vidal PP, Moore LE, Vibert N. Unilateral labyrinthectomy modifies the membrane properties of contralesional vestibular neu- rons. J Neurophysiol. 2004;92:1668.
Llinás R, Walton K. Vestibular compensation: a dis- tributed property of the central nervous system. In: Asanuma H, Wilson VJ, eds. Integration in the Nervous System. Tokyo, Japan: Igaku Shoin; 1979: 145.
Paterson JM, Short D, Flatman PW, Seckl JR, Aitken A, Dutia MB. Changes in protein expression in the rat medial vestibular nuclei during vestibular compensa- tion. J Physiol. 2006;575(pt 3):777.
Kaufman GD, Anderson JH, Beitz AJ. Brainstem Fos expression following acute unilateral labyrinthectomy in the rat. NeuroReport. 1992;3:829.
Cirelli C, Pompeiano M, D’Ascanio P, Arrighi P, Pompeiano O. c-fos Expression in the rat brain after unilateral labyrinthectomy and its relation to the uncompensated and compensated stages. Neuroscience. 1996;70:515.
Darlington CL, Smith PF. Molecular mechanisms of recovery from vestibular damage in mammals: recent advances. Prog Neurobiol. 2000;62:313.
Li H, Dokas LA, Godfrey DA, Rubin AM. Remodeling of synaptic connections in the deafferented ves- tibular nuclear complex. J Vestib Res. 2002;12: 167.
Gacek RR, Khetarpal U. Neurotrophin 3, not brain- derived neurotrophic factor or neurotrophin 4, knockout mice have delay in vestibular compensa- tion after unilateral labyrinthectomy. Laryngoscope. 1998;108:671.
Cohen YE, Andersen RA. A common reference frame for movement plans in the posterior parietal cortex. Nat Rev Neurosci. 2002;3(7):553.
Avillac M, Denève S, Olivier E, Pouget A, Duhamel JR. Reference frames for representing visual and tactile locations in parietal cortex. Nat Neurosci. 2005;8(7):941.
Cohen B, Wearne S, Dai M, Raphan T. Spatial orien- tation of the angular vestibulo-ocular reflex. J Vestib Res. 1999;9(3):163.
Wearne S, Raphan T, Cohen B. Effects of tilt of the gravito-inertial acceleration vector on the angu- lar vestibuloocular reflex during centrifugation. J Neurophysiol. 1999;81(5):2175.
Paige GD, Tomko DL. Eye movement responses to linear head motion in the squirrel monkey. II. Visual- vestibular interactions and kinematic considerations. J Neurophysiol. 1991;65(5):1183.
Green AM, Angelaki DE. Resolution of sensory ambi- guities for gaze stabilization requires a second neural integrator. J Neurosci. 2003;23(28):9265.
Angelaki DE, Cullen KE. Vestibular system: the many facets of a multimodal sense. Annu Rev Neurosci. 2008;31:125.
Merfeld DM, Park S, Gianna-Poulin C, Black FO, Wood S. Vestibular perception and action employ qualitatively different mechanisms. I. Frequency response of VOR and perceptual responses during Translation and Tilt. J Neurophysiol. 2005;94(1): 186.
Merfeld DM, Park S, Gianna-Poulin C, Black FO, Wood S. Vestibular perception and action employ qualitatively different mechanisms. II. VOR and per- ceptual responses during combined Tilt & Translation. J Neurophysiol. 2005;94(1):199.
von Holst E, Mittelstaedt H. Das reafferenzprinzip [in German]. Naturwissenschaften. 1950;37:464.
Lorente de Nó R. Vestibulo-ocular reflex arc. Arch Neurol Psychiatry. 1933;30:245.
Raphan T, Matsuo V, Cohen B. Velocity storage in the vestibulo-ocular reflex arc (VOR). Exp Brain Res. 1979;35:229.
Robinson DA. The use of control systems analysis in the neurophysiology of eye movements. Annu Rev Neurosci. 1981;4:463.
Holstein GR, Martinelli GP Wearne S, Cohen
B. Ultrastructure of vestibular commissural neu- ron related to velocity storage in the monkey. Neuroscience. 1999;93:155.
Holstein GR, Martinelli GP, Cohen B. The ultra- structure of GABA-immunoreactive vestibular com- missural neurons related to velocity storage in the monkey. Neuroscience. 1999;93:171.
Holstein GR, Martinelli GP, Cohen B. The ultra- structure features of non-commissural GABAergic neurons in the medial vestibular nucleus of the mon- key. Neuroscience. 1999;93:183.
Lorente de Nó R. The regulation of eye positions and movements induced by the labyrinth. Laryngoscope. 1932;42:233.
Ito M. The vestibulo-cerebellar relationships: ves- tibulo-ocular reflex arc and flocculus. In: Naunton RF, ed. The Vestibular System. New York: Academic Press; 1975.
Cohen B. The vestibulo-ocular reflex arc. In: Kornhuber HH, ed. Handbook of Sensory Physiology, The Vestibular System. Vol 6. Pt 1. New York: Springer-Verlag; 1974.
Uchino Y, Suzuki S. Axon collaterals to the extraocu- lar motoneuron pools of inhibitory vestibulo-ocular activated from the anterior, posterior and horizon- tal semicircular canals in the cat. Neurosci Lett. 1983;37:129.
Uchino Y, Hirai N, Suzuki S. Branching pattern and properties of vertical- and horizontal-related excitatory vestibuloocular neurons in the cat. J Neurophysiol. 1982;48:891.
Ohgaki T, Curthoys IS, Markham CH. Morphology of physiologically identified second order vestibular neurons in cat, using intracellularly injected HRP. J Comp Neurol. 1988;276:389.
Fukushima K, Perlmutter SI, Baker JF, Peterson BW. Spatial properties of second-order vestibulo- ocular relay neurons in the alert cat. Exp Brain Res. 1990;81:462.
Peterson BW, Baker JF, Perlmutter SI, Iwamoto
Y. Neuronal substrates of spatial transformations in vestibuloocular and vestibulocollic reflexes. Ann NY Acad Sci. 1992;656:485.
Skavenski AA, Robinson DA. Role of abducens neurons in vestibulo-ocular reflex. J Neurophysiol. 1973;36:724.
Cannon SC, Robinson DA. Neural integrator failure from brain stem lesions in monkey. Invest Ophthalmol Vis Sci. 1985;26(suppl 3):47.
Cheron G, Godaux E. Disabling of the oculomo- tor neural integrator by kainic acid injections in the prepositus-vestibular complex of the cat. J Physiol. 1987;394:267.
Carpenter RHS. Cerebellectomy and the transfer function of the vestibulo-ocular reflex in the decer- ebrate cat. Proc R Soc B. 1972;181:353.
Galiana HL, Outerbridge JS. A bilateral model for central neural pathways in vestibulo-ocular reflex. J Neurophysiol. 1984;51:210.
Chan WW, Galiana HL. A non-linear model of the neural integrator in oculomotor control. Conf Proc IEEE Eng Med Biol Soc. 2007;2007:1156.
McCrea RA, Strassman A, May E, Highstein SM. Anatomical and physiological characteristics of ves- tibular neurons mediating the horizontal vestibulo- ocular reflex of the squirrel monkey. J Comp Neurol. 1987;264:547.
Scudder CA, Fuchs AF. Physiological and behav- ioral identification of vestibular nucleus neurons mediating the horizontal vestibuloocular reflex in trained rhesus monkeys. J Neurophysiol. 1992;86: 244.
Chen-Huang C, McCrea RA. Viewing distance related sensory processing in the ascending tract of Deiters vestibulo-ocular reflex pathway. J Vestib Res. 1998;8:175.
Maeda M, Shimazu H, Shinoda Y. Nature of synap- tic events in cat abducens motoneurons at slow and quick phase of vestibular nystagmus. J Neurophysiol. 1972;35:279.
Baker R, Berthoz A. Organization of vestibular nys- tagmus in oblique oculomotor system. J Neurophysiol. 1974;37:195.
Honrubia V, Reingold DB, Lau CGY, Ward PH. Neural correlates of nystagmus in abducens nerve. J Neurophysiol. 1979;42:1282.
Robinson DA. Oculomotor unit behavior in the mon- key. J Neurophysiol. 1970;33:393.
Cullen KE, Galiana HL, Sylvestre PA. Comparing extraocular motoneuron discharges during head-re- strained saccades and head-unrestrained gaze shifts. J Neurophysiol. 2000;83:630.
Cohen B, Henn V. The origin of quick phases of nystagmus in the horizontal plane. Bibl Ophthalmol. 1972;82:36.
Keller EL. Participation of medial pontine reticular formation in eye movement generation in monkey. J Neurophysiol. 1974;37:316.
Henn V, Hepp K, Buttner-Ennever JA. The pri- mate oculomotor system. II. Premotor system. Hum Neurobiol. 1982;1:87.
Kato I, Nakamura T, Kanayama R, Aoyagi M. Slow saccades and quick phases of nystagmus after pon- tine lesions. Acta Otolaryngol Suppl (Stockh). 1994;511:95.
Hikosaka O, Kawakami T. Inhibitory interneurons in the reticular formation and their relation to vestibular nystagmus. Brain Res. 1976;117:513.
Melvill Jones G. Predominance of anti-compensatory oculomotor responses during rapid head rotation. Aerospace Med. 1964;35:965.
Honrubia V, Baloh RW, Lau CG, Sills AW. The pat- terns of eye movements during physiologic vestibu- lar nystagmus in man. Trans Am Acad Ophthalmol Otolaryngol. 1977;84:339.
Markham CH. How does the brain generate horizon- tal vestibular nystagmus? In: Baloh RW, Halmagyi GM, eds. Disorders of the Vestibular System. New York: Oxford University Press; 1996.
Uemura T, Cohen B. Effects of vestibular nuclei lesions on vestibulo-ocular reflexes and posture in monkeys. Acta Otolaryngol Suppl (Stockh). 1973;315:1.
Cohen B, Komatsuzaki A, Bender MB. Electrooculographic syndrome in monkeys after pontine reticular formation lesions. Arch Neurol. 1968;18:78.
Henn V, Lang W, Hepp K, Reisine H. Experimental gaze palsies in monkeys and their relation to human pathology. Brain. 1984;107:619.
Bender MB. Brain control of conjugate horizontal and vertical eye movements. A survey of the struc- tural and functional correlates. Brain. 1970;103:23.
Blanks RHI, Anderson JH, Precht W. Response char- acteristics of semicircular canal and otolith systems in cat. II. Responses of trochlear motoneurons. Exp Brain Res. 1978;32:509.
Collins WE. Arousal and vestibular habituation. In: Kornhuber HH, ed. Handbook of Sensory Physiology, The Vestibular System. Vol 6. Pt 2. New York: Springer-Verlag; 1974.
Collins WE. Manipulation of arousal and its effects upon human vestibular nystagmus induced by caloric irrigation and angular accelerations. Aerospace Med. 1963;34:124.
Jaeger J, Henn V. Habituation of the vestibulo-ocular reflex (VOR) in the monkey during sinusoidal rotation in the dark. Exp Brain Res. 1981;41:108.
Baloh RW, Henn V, Jaeger J. Habituation of the human vestibulo-ocular reflex by low frequency har- monic acceleration. Am J Otolaryngol. 1982;3:235.
Angelaki DE. Eyes on target: what neurons must do for the vestibuloocular reflex during linear motion. J Neurophysiol. 2004;92:20.
Hess BJM, Angelaki DE. Vestibular contributions to gaze stability during transient forward and backward motion. J Neurophysiol. 2003;90:1996.
Paige GD, Telford L, Seidman SH, Barnes GR. Human vestibuloocular reflex and its interactions with vision and fixation distance during linear and angular head movement. J Neurophysiol. 1998;80:2391.
Buizza A, Avanzini P, Schmid R. Visual-vestibular interaction during angular and linear body accel- eration: modeling and simulation. In: Fedina L, Kanyar B, Kocsis B, Kollai M, eds. Mathematical and Computational Methods in Physiology. Oxford, England: Pergamon; 1981.
Viirre E, Tweed D, Milner K, Vilis T. A reexami- nation of the gain of the vestibulo-ocular reflex. J Neurophysiol. 1986;56:439.
Crane BT, Demer JL. Human gaze stabilization dur- ing natural activities: translation, rotation, magnifi- cation and target distance effects. J Neurophysiol. 1997;78:2129.
Schwindt PC, Richter A, Precht W. Short latency utricular and canal input to ipsilateral abducens motoneurons. Brain Res. 1973;60:259.
Blanks RHI, Anderson JH, Precht W. Response char- acteristics of semicircular canal and otolith systems in cat. II. Responses of trochlear motoneurons. Exp Brain Res. 1978;32:509.
Eckmiller R. Concerning the linear acceleration input to the neural oculomotor control system in primates. In: Roucoux A, Crommelinck M, eds. Physiological and Pathological Aspects of Eye Movements. The Hague, Netherlands: Junk; 1982.
Suzuki J-I, Tokumasu K, Goto K. Eye movements from single utricular nerve stimulation in the cat. Acta Otolaryngol (Stockh). 1969;68:350.
Fluur E, Mellström A. The otolith organs and their influence on oculomotor movements. Exp Neurol. 1971;30:139.
Fluur E, Mellström A. Utricular stimulation and ocul- omotor reactions. Laryngoscope. 1970;80:1701.
Fluur E, Mellström A. Saccular stimulation and ocul- omotor reactions. Laryngoscope. 1970;80:1713.
Imagawa M, Isu N, Sasaki M, Endo K, Ikegami H, Uchino Y. Axonal projections of utricular afferents to the vestibular nuclei and the abducens nucleus in cats. Neurosci Lett. 1995;186:87.
Uchino Y, Ikegami H, Sasaki M, Endo K, Imagawa M, Isu N. Monosynaptic and disynaptic connections in the
utriculo-ocular reflex arc of the cat. J Neurophysiol. 1994;71:950.
Uchino Y, Sasaki M, Sato H, Imagawa M, Suwa H, Isu
N. Utriculoocular reflex arc of the cat. J Neurophysiol. 1996;76:1896.
Uchino Y, Sato H, Sasaki M, et al. Sacculocollic reflex arcs in cats. J Neurophysiol. 1997;77:3003.
Uchino Y, Sato H, Zakir M, et al. Commissural effects in the otolith system. Exp Brain Res. 2001;136:421.
Bai R, Meng H, Sato H, Imagawa M, Sasaki M, Uchino Y. Properties of utricular-activated vestibular neurons that project to the contralateral vestibular nuclei in the cat. Exp Brain Res. 2002;147:419.
Uchino Y, Sato H, Suwa H. Excitatory and inhibitory inputs from saccular afferents to single vestibular neurons in the cat. J Neurophysiol. 1997;78:2186.
Ogawa Y, Kushiro K, Zakir M, Sato H, Uchino Y. Neuronal organization of the utricular macula con- cerned with innervation of single vestibular neurons in the cat. Neurosci Lett. 2000;278:89.
Green AM, Galiana HL. Hypothesis for shared central processing of canal and otolith signals. J Neurophysiol. 1998;80:2222.
Mussallum WS, Tomlinson RD. Model for the trans- lational vestibuloocular reflex (VOR). J Neurophysiol. 1999;82:2010.
Paige GD, Tomko DL. Eye movement responses to linear head motion in the squirrel monkey. I. Basic characteristics. J Neurophysiol. 1991;65:1170.
Paige GD, Tomko DL. Eye movement responses to linear head motion in the squirrel monkey. II. Visual- vestibular interactions and kinematic considerations. J Neurophysiol. 1991;65:1183.
Lempert T, Gianna C, Brookes G, Bronstein A, Gresty M. Horizontal otolith-ocular responses in humans after unilateral vestibular deafferentation. Exp Brain Res. 1998;118:533.
Crane BT, Tian J, Wiest G, Demer JL. Initiation of the human heave linear vestibulo-ocular reflex. Exp Brain Res. 2003;148:247.
Baarsma EA, Collewijn H. Eye movements due to linear accelerations in the rabbit. J Physiol. 1975;245:227.
Miller EF, II. Counterrolling of the human eye produced by head tilt with respect to gravity. Acta Otolaryngol (Stockh). 1962;54:479.
Diamond SO, Markham CH. Binocular counter- rolling in humans with unilateral labyrinthectomy and in normal controls. Ann NY Acad Sci. 1981;374:69.
Curthoys IS, Dai MJ, Halmagyi CM. Human ocular torsional position before and after unilateral vestibu- lar neurectomy. Exp Brain Res. 1991;85:218.
Diamond SG, Markham CH. Ocular counterrolling as an indicator of vestibular otolith function. Neurology. 1983;3:1460.
Markham CH, Diamond SG. Ocular counterrolling in response to static and dynamic tilting: implications for human otolith function. J Vestib Res. 2002-2003;12(2- 3):127.
Angelaki DE, Anderson JH. The horizontal vestibulo- ocular reflex during linear acceleration in the frontal plane of the cat. Exp Brain Res. 1991;86:40.
Angelaki DE, Anderson JH. The vestibulo-ocular reflex in the cat during linear acceleration in the sagittal plane. Brain Res. 1991;543:347.
Curthoys IS, Wearne SL, Dai MJ, Halmagyi GM, Holden JR. Linear acceleration modulates the nys- tagmus induced by angular acceleration stimulation of the horizontal canal. Ann NY Acad Sci. 1992;656: 716.
Merfeld DM, Young LR. The vestibulo-ocular reflex of the squirrel monkey during eccentric rotation and roll tilt. Exp Brain Res. 1995;106:111.
Sargent EW, Paige GD. The primate vestibulo-ocu- lar reflex during combined linear and angular head motion. Exp Brain Res. 1991;87:75.
Wearne S, Raphan T, Cohen B. Effects of tilt of the gravito-inertial acceleration vector on the angu- lar vestibuloocular reflex during centrifugation. J Neurophysiol. 1999;81(5):2175.
Waespe W, Henn V. Gaze stabilization in the primate: the interaction of the vestibulo-ocular reflex, optoki- netic nystagmus, and smooth pursuit. Rev Physiol Biochem Pharmacol. 1987;106:38.
Raphan T, Cohen B. Velocity storage and the ocu- lar response to multidimensional vestibular stimuli. In: Berthoz A, Melville Jones G, eds. Adaptive Mechanisms in Gaze Control. Amsterdam, Netherlands: Elsevier; 1985: 123.
Raphan T, Cohen B. Multidimensional modeling of the vestibulo-ocular reflex. In: Keller E, Zee D, eds. Adaptive Processes in Visual and Oculomotor Systems. New York: Pergamon Press; 1986.
Cohen B, Maruta J, Raphan T. Orientation of the eyes to gravitoinertial acceleration. Ann NY Acad Sci. 2001;942:241.
De Kleyn A. Recherches quantitatives sur les posi- tions compensatories l’oeil chez de lapin [in French]. Arch Neerl Physiol. 1922;7:138.
Gresty MA. A reexamination of “neck reflex” eye movements in the rabbit. Acta Otolaryngol (Stockh). 1976;81:386.
Brandt T. Cervical vertigo—reality or fiction? Audiol Neurootol. 1996;1:187.
McCouch GP, Deering ID, Ling TH. Location of receptors for tonic neck reflexes. J Neurophysiol. 1951;14:191.
Igarashi M, Miyata H, Alford BR, Wright WK. Nystagmus after experimental cervical lesions. Laryngoscope. 1972;82:1609.
De Jong PT, de Jong JM, Cohen B, Jongkees LB. Ataxia and nystagmus induced by injection of local anesthetics in the neck. Ann Neurol. 1977;1:240.
Rubin AM, Young JH, Milne AC, et al. Vestibular- neck integration in the vestibular nuclei. Brain Res. 1975;96:99.
Gdowski GT, McCrea RA. Neck proprioceptive inputs to primate vestibular nucleus neurons. Exp Brain Res. 2000;135:511.
Gdowski GT, Belton T, McCrea RA. The neuro- physiological substrate for the cervico-ocular reflex in the squirrel monkey. Exp Brain Res. 2001;140: 253.
Roy JE, Cullen KE. Vestibuloocular reflex signal modulation during voluntary and passive head move- ments. J Neurophysiol. 2002;87:2337.
Roy JE, Cullen KE. Brain stem pursuit pathways: dissociating visual, vestibular, and proprioceptive inputs during combined eye-head gaze tracking. J Neurophysiol. 2003;90(1):271.
Takemori S, Suzuki J-I. Eye deviations from neck torsion in humans. Ann Otol. 1971;80:439.
Meiry JL. Vestibular and proprioceptive stabilization of eye movements. In: Bach-Y-Rita P, Collins CC, Hyde JE, eds. The Control of Eye Movements. New York: Academic Press; 1971.
Barnes CR, Forbat LN. Cervical and vestibular affer- ent control of oculomotor response in man. Acta Otolaryngol. 1979;88:79.
Barlow D, Freeman W. Cervico-ocular reflex in the normal adult. Acta Otolaryngol. 1980;89:487.
Sawyer RN, Jr., Thurston SE, Becker KR, et al. The cervico-ocular reflex of normal human subjects in response to transient and sinusoidal trunk rotations. J Vestib Res. 1994;4:245.
Leigh RJ, Zee DS. Neurology of Eye Movements. 4th ed. Contemporary Neurology Series. Toronto, Ontario: Oxford Univeristy Press; 2006.
Shinoda Y, Sugiuchi Y, Izawa Y, Takahashi M. Neural circuits for triggering saccades in the brainstem. Prog Brain Res. 2008;171:79.
Baarsma EA, Collewijn H. Vestibulo-ocular and opto- kinetic reactions to rotation and their interaction in the rabbit. J Physiol. 1974;238:603.
Collewijn H. Direction-selective units in the rabbit’s nucleus of the optic tract. Brain Res. 1975;100:489.
Robinson DA. Control of eye movements. In: Brooks VB, ed. Handbook of Physiology. The Nervous System
Washington DC: American Physiological Society 1981: 1275.
Dubois MFW, Collewijn H. Optokinetic reactions in man elicited by localized retinal motion stimuli. Vis Res. 1979;19:1105.
Van Die G, Collewijn H. Optokinetic nystagmus in man. Hum Neurobiol. 1982;1:111.
Naegele JR, Held R. The postnatal development of monocular optokinetic nystagmus in infants. Vis Res. 1982;22:391.
Distler C, Hoffmann KP. Development of the opto- kinetic response in macaques: a comparison with cat and man. Ann NY Acad Sci. 2003;1004:10.
Baloh RW, Yee RD, Honrubia V. Optokinetic asym- metry in patients with maldeveloped foveas. Brain Res. 1980;186:211.
Atkinson J. Development of optokinetic nystagmus in the human infant and monkey infant: an ana- logue to development in kittens. In: Freeman RD, ed. Developmental Neurobiology of Vision. NAFO Advanced Study Institute Series: Series A, Life Sciences. New York: Plenum Press; 1979.
Cohen B, Matsuo V, Raphan TH. Quantitative analy- sis of the velocity characteristics of optokinetic nys- tagmus and optokinetic after-nystagmus. J Physiol. 1977;270:321.
Paige CD. Vestibulo-ocular reflex and its interactions with visual following mechanisms in the squirrel mon- key, I. Response characteristics in normal animals. J Neurophysiol. 1983;49:134.
Boyle R, Buttner U, Markert G. Vestibular nuclei activity and eye movements in the alert monkey dur- ing sinusoidal optokinetic stimulation. Exp Brain Res. 1985;57:362.
Baloh RW, Yee RD, Honrubia V. Optokinetic nystagmus and parietal lobe lesions. Ann Neurol. 1980;7:269.
Baloh RW, Yee RD, Honrubia V. Late cortical cer- ebellar atrophy: clinical and oculographic features. Brain. 1986;109:159.
Honrubia V, Downey WL, Mitchell DP, Ward PH. Experimental studies on optokinetic nystagmus.
Normal humans. Acta Otolaryngol (Stockh). 1968;65:441.
Koenig E, Dichgans J. Aftereffects of vestibular and optokinetic stimulation and their interaction. Ann NY Acad Sci. 1981;374:434.
Segal BN, Liben S. Modulation of human velocity storage sampled during intermittently illuminated optokinetic stimulation. Exp Brain Res. 1985;59:515.
Thier P, Ilg UJ. The neural basis of smooth-pursuit eye movements. Curr Opin Neurobiol. 2005;15(6):645
Barnes GR, Benson AJ, Prior ARJ. Visual-vestibular interaction in the control of eye movement. Aviat Space Environ Med. 1978;49:557.
Meyer CH, Lasker AG, Robinson DA. The upper limit of human smooth pursuit velocity. Vis Res. 1985;25:561.
Lisberger SG, Evinger C, Johanson GW, et al. Relationship between eye acceleration and retinal image velocity during foveal smooth pursuit in man and monkey. J Neurophysiol. 1981;46:229.
Steinbach MJ. Pursuing the perceptual rather than the retinal stimulus. Vis Res. 1976;16:1371.
Kommerell G, Taumer R. Investigations of the eye tracking system through stabilized retinal images. Bibl Ophthalmol. 1972;82:288.
Pola J, Wyatt HJ. Target position and velocity: the stimuli for smooth pursuit eye movements. Vis Res. 1980;20:523.
Lencer R, Trillenberg P. Neurophysiology and neu- roanatomy of smooth pursuit in humans. Brain Cogn. 2008;68(3):219.
Cohen B, Henn V, Raphan T, Dennett D. Velocity storage, nystagmus, and visual-vestibular interactions in humans. Ann NY Acad Sci. 1981;374:421.
Ter Braak JWG. Investigations on optokinetic nys- tagmus. In: Collewijn H, ed. The Oculomotor System of the Rabbit and its Plasticity. Studies of Brain Function. Vol 5. Berlin: Springer-Verlag; 1981.
Hobbelen JF, Collewijn H. Effect of cerebrocortical and collicular ablations upon the optokinetic reac- tions in the rabbit. Doc Ophthalmol. 1971;30:227.
Zee DS, Tusa RJ, Herdman SJ, et al. Effects of occip- ital lobectomy upon eye movements in primate. J Neurophysiol. 1987;58:883.
Leh SE, Johansen-Berg H, Ptito A. Unconscious vision: new insights into the neuronal correlate of blindsight using diffusion tractography. Brain. 2006;129(pt 7):1822.
Ter Braak JWG, Schenk VWD, Van Vliet AGM. Visual reactions in a case of long-lasting cortical blindness. J Neurol Neurosurg Psychiatry. 1971;34:140.
Baloh RW, Yee RD, Honrubia V, Jacobson K. A comparison of the dynamics of horizontal and verti- cal smooth pursuit in normal human subjects. Aviat Space Environ Med. 1988;59:121.
Melvill Jones G. Adaptive modulation of VOR param- eters by vision. In: Berthoz A, Melvill Jones G, eds. Adaptive Mechanisms in Gaze Control: Reviews in Oculomotor Research. Amsterdam, Netherlands: Elsevier; 1985: 21.
Dichgans J, Schmidt CL, Graf W. Visual input improves the speedometer function of the vestibular nuclei in the goldfish. Exp Brain Res. 1973;18:319.
Waespe W, Henn V. Motion information in the vestibular nuclei of alert monkeys: visual and ves- tibular input vs. optomotor output. Prog Brain Res. 1979;50:693.
Simpson JI. The accessory optic system. Annu Rev Neurosci. 1984;7:13.
Lázár G. Role of the accessory optic system in the optokinetic nystagmus of the frog. Brain Behav Evol. 1973;5:443.
Ghelarducci B, Ito M, Yagi N. Impulse discharges from flocculus Purkinje cells of alert rabbits during visual stimulation combined with horizontal head rotation in the rabbit. Brain Res. 1975;87:66.
Maekawa K, Takeda T. Mossy fiber responses evoked in the cerebellar flocculus of rabbits by stimulation of the optic pathway. Brain Res. 1975;98:590.
Maekawa K, Takeda T. Electrophysiological iden- tification of the climbing and mossy fiber pathways from the rabbit’s retina to the contralateral cerebellar flocculus. Brain Res. 1976;109:169.
Hoffman KP, Schoppman A. Retinal input to direc- tion selective cells in the nucleus tractus opticus of the cat. Brain Res. 1975;99:359.
Branth SE, Karten HJ. Direct accessory optic pro- jections to the vestibulocerebellum: a possible chan- nel for oculomotor control systems. Exp Brain Res. 1977;28:73.
Winfield JA, Hendrickson A, Kimm J. Anatomical evi- dence that the medial terminal nucleus of the acces- sory optic tract in mammals provides a visual mossy fiber input to the flocculus. Brain Res. 1978;151:175.
Hoffman KP, Distler C, Erickson RG, Mader W. Physiological and anatomical identification of the nucleus of the optic tract and dorsal terminal nucleus of the accessory optic tract in monkeys. Exp Brain Res. 1988;69:635.
Schiff D, Cohen B, Raphan T Nystagmus induced by stimulation of the nucleus of the optic tract in the monkey. Exp Brain Res. 1988;70:1.
Dow B. Functional classes of cells and their laminar distribution in monkey visual cortex. J Neurophysiol. 1974;37:927.
Zeki SM. The responses of cells in the anterior bank of the superior temporal sulcus in macaque monkeys. J Physiol (Lond). 1980;308:85P.
Maunsell JHR, Van Essen DC. Functional proper- ties of neurons in middle temporal visual area of the macaque monkey. I. Selectivity for stimulus direction, speed and orientation. J Neurophysiol. 1983;49:1127.
Albright TD. Direction and orientation selectiv- ity of neurons in visual area MT of the macaque. J Neurophysiol. 1984;52:1106.
Tanaka K, Hikosaka K, Saito H, et al. Analysis of local and wide-field movements in the superior tempo- ral visual areas of the macaque monkey. J Neurosci. 1986;6:134.
Ilg UJ. The role of areas MT and MST in coding of visual motion underlying the execution of smooth pursuit. Vision Res. 2008;48(20):2062.
Robinson DA, Goldberg ME, Stanton GB. Parietal association cortex in the primate: sensory mecha- nisms and behavioral modulation. J Neurophysiol. 1978;41:910.
Sakata H, Sibutani H, Kawano K. Functional prop- erties of visual tracking neurons in posterior parietal association cortex of the monkey. J Neurophysiol. 1983;49:1364.
Fukushima J, Akao T, Kurkin S, Kaneko CR, Fukushima K. The vestibular-related frontal cor- tex and its role in smooth-pursuit eye movements and vestibular-pursuit interactions. J Vestib Res. 2006;16(1-2):1.
Lencer R, Trillenberg P. Neurophysiology and neu- roanatomy of smooth pursuit in humans. Brain Cogn. 2008;68(3):219.
May JG, Anderson RA. Different patterns ofcortico- pontine projections from separate cortical fields within the inferior parietal lobule and dorsal prelunate gyrus of the macaque. Exp Brain Res. 1986;63:265.
May JG, Keller EL, Suzuki DA. Smooth pursuit eye movement deficits with chemical lesions in the dorso- lateral pontine nucleus of the monkey. J Neurophysiol. 1988;59:952.
Thier P, Ilg UJ. The neural basis of smooth-pursuit eye movements. Curr Opin Neurobiol. 2005;15(6):645.
Thier P, Möck M. The oculomotor role of the pontine nuclei and the nucleus reticularis tegmenti pontis. Prog Brain Res. 2006;151:293.
Lynch JC, McLaren JW. The contribution of parieto- occipital association cortex to the control of slow eye movements. In: Lennerstrand G, Zee DS, Keller EL, eds. Functional Basis of Ocular Motility Disorders. Oxford, England: Pergamon Press; 1982.
Zee DS, Yamazaki A, Butler PH, et al. Effects of abla- tion of flocculus and paraflocculus on eye movements in primate. J Neurophysiol. 1981;46:878.
Kato I, Harada K, Hasegawa T, et al. Role of the nucleus of the optic tract in monkeys in relation to optokinetic nystagmus. Brain Res. 1986;364:12.
Kohler I. Experiments with goggles. Sci Am. 1962;206:62.
Gonshor A, Melvill Jones G. Plasticity in the adult human vestibulo-ocular reflex arc. Proc Can Fed Biol Soc. 1971;14:11.
Gonshor A, Melvill Jones G. Short-term adaptive changes in the human vestibulo-ocular reflex arc. J Physiol. 1976;256:361.
Gonshor A, Melvill Jones G. Extreme vestibulo-ocu- lar adaptation induced by prolonged optical reversal of vision. J Physiol. 1976;256:381.
Robinson DA. Adaptive gain control of vestibulo- ocular reflex by the cerebellum. J Neurophysiol. 1976;39:954.
Miles FA, Braitman DJ, Dow BM. Long term adap- tive changes in primate vestibulo-ocular reflexes:
IV. Electrophysiological observations in flocculus of adapted monkeys. J Neurophysiol. 1980;43:1477.
Mandl G, Melvill Jones G, Cynader M. Adaptability of the vestibulo-ocular reflex to vision reversal in strobe reared cats. Brain Res. 1981;209:35.
Wallman J, Velez J, Weinstein B, et al. Avian vestib- ulo-ocular reflex: adaptive plasticity and developmen- tal changes. J Neurophysiol. 1982;48:952.
Ito M, Shiida T, Yagi N, et al. The cerebellar modification of rabbit’s horizontal vestibulo- ocular reflex induced by sustained head rotation combined with visual stimulation. Proc Jpn Acad. 1974;50:85.
Schultheis LW, Robinson DA. Directional plasticity of the vestibulo-ocular reflex in the cat. Ann NY Acad Sci. 1981;374:504.
Waespe W, Henn V. Conflicting visual-vestibular stimulation and vestibular nucleus activity in alert monkeys. Exp Brain Res. 1978;133:203.
Waespe W, Henn V. Vestibular nuclei activity dur- ing optokinetic after-nystagmus (OKAN) in the alert monkey. Exp Brain Res. 1977;30:323.
Lisberger SG, Pavelko TA. Brain stem neurons in modified pathways for motor learning in the primate vestibulo-ocular reflex. Science. 1988;242:771.
Lisberger SG, Pavelko TA, Broussard DM. Responses during eye movements of brain stem neurons that receive monosynaptic inhibition from the flocculus and ventral paraflocculus in monkeys. J Neurophysiol. 1994;72:909.
Partsalis AM, Zhang Y, Highstein SM. Dorsal Y group in the squirrel monkey. I. Neuronal responses dur- ing rapid and long-term modifications of the vertical VOR. J Neurophysiol. 1995;73:615.
Partsalis AM, Zhang Y, Highstein SM. Dorsal Y group in the squirrel monkey. II. Contribution of the cer- ebellar flocculus to neuronal responses in normal and adapted animals. J Neurophysiol. 1995;73:632.
Zhang Y, Partsalis AM, Highstein SM. Properties of superior vestibular nucleus flocculus target neurons in the squirrel monkey: II Signal components revealed by reversible flocculus inactivation. J Neurophysiol. 1995;73:2279.
Zhang Y, Partsalis AM, Highstein SM. Properties of superior vestibular nucleus flocculus target neu- rons in the squirrel monkey. I General properties in comparison with flocculus projecting neurons. J Neurophysiol. 1995;73:2261.
Ramachandran R, Lisberger SG. Normal perfor- mance and expression of learning in the vestibulo-oc- ular reflex (VOR) at high frequencies. J Neurophysiol. 2005;93(4):2028.
Ramachandran R, Lisberger SG. Transformation of vestibular signals into motor commands in the vestib- uloocular reflex pathways of monkeys. J Neurophysiol. 2006;96(3):1061.
Ramachandran R, Lisberger SG. Neural substrate of modified and unmodified pathways for learning in monkey vestibuloocular reflex. J Neurophysiol. 2008;100(4):1868.
Baker J, Goldberg JM, Peterson B. Spatial and tem- poral response properties of the vestibulo-collic reflex in decerebrate cats. J Neurophysiol. 1985;54: 735.
Perlmutter SI, Iwamoto Y, Baker JF, Peterson BW. Interdependence of spatial properties and projection patterns of medial vestibulospinal tract neurons in the cat. J Neurophysiol. 1998;79(1):270.
Boyle R. Vestibulospinal control of reflex and voluntary head movement. Ann NY Acad Sci. 2001;942:364.
Roberts TDM. Neurophysiology of Postural Mechanisms. New York: Plenum Press; 1967.
Rapoport S, Susswein A, Uchino Y, Wilson VJ. Properties of vestibular neurones projecting to neck segments of the cat spinal cord. J Physiol. 1977;268(2):493.
Hongo T, Kudo N, Tanaka R. The vestibulospinal tract: crossed and uncrossed effects on hindlimb motoneu- rones in the cat. Exp Brain Res. 1975;24(1):37.
Abzug C, Maeda M, Peterson BW, Wilson VJ. Cervical branching of lumbar vestibulospinal axons. J Physiol. 1974;243(2):499.
Akaike T. Neuronal organization of the vestibulospi- nal system in the cat. Brain Res. 1983;259(2):217.
Kushiro K, Bai R, Kitajima N, Sugita-Kitajima A, Uchino Y. Properties and axonal trajectories of posterior semicircular canal nerve-activated ves- tibulospinal neurons. Exp Brain Res. 2008;191(3): 257.
Uchino Y, Sasaki M, Sato H, Bai R, Kawamoto E. Otolith and canal integration on single vestibular neu- rons in cats. Exp Brain Res. 2005;164(3):271.
Sugita A, Bai R, Imagawa M, et al. Properties of horizontal semicircular canal nerve-activated vestibu- lospinal neurons in cats. Exp Brain Res. 2004;156(4): 478.
Sato H, Imagawa M, Meng H, Zhang X, Bai R, Uchino Y. Convergence of ipsilateral semicircular canal inputs onto single vestibular nucleus neurons in cats. Exp Brain Res. 2002;145(3):351.
Perlmutter SI, Iwamoto Y, Baker JF, Peterson BW. Spatial alignment of rotational and static tilt responses of vestibulospinal neurons in the cat. J Neurophysiol. 1999;82:855.
Boyle R, Johanson C. Morphological properties of vestibulospinal neurons in primates. Ann NY Acad Sci. 2003;1004:183.
Boyle R. Morphology of lumbar-projecting lateral vestibulospinal neurons in the brainstem and cervi- cal spinal cord in the squirrel monkey. Arch Ital Biol. 2000;138(2):107.
Boyle R, Belton T, McCrea RA. Responses of iden- tified vestibulospinal neurons to voluntary eye and head movements in the squirrel monkey. Ann NY Acad Sci. 1996;781:244.
Shinoda Y, Sugiuchi Y, Izawa Y, Hata Y. Long descending motor tract axons and their control of neck and axial muscles. Prog Brain Res. 2006;151: 527.
Nyberg-Hansen R. Sites and mode of termination of fibers of the vestibulo-spinal tract in the cat. An experimental study with silver impregnation meth- ods. J Comp Neurol. 1964;122:369.
Lund S, Pompeiano O. Descending pathways with monosynaptic action on motoneurons. Experientia. 1965;21:602.
Erulkar SD, Sprague JM, Whitsel BL, et al. Organization of the vestibular projection to the spinal cord of the cat. J Neurophysiol. 1966;29:626.
Peterson BW. The reticulospinal system and its role in the control of movement. In: Barnes CD, ed. Brainstem Control of Spinal Cord Function. New York: Academic Press; 1984: 27.
Nyberg-Hansen R. Sites and mode of termination of reticulospinal fibers in the cat. An experimental study with silver impregnation methods. J Comp Neurol. 1965;124:71.
Llinás R, Terzuolo CA. Mechanisms of supraspinal actions upon spinal cord activities. Reticular inhibi- tory mechanisms on alpha-extensor motoneurons. J Neurophysiol. 1964;27:579.
Llinás R, Terzuolo CA. Mechanisms of supraspi- nal actions upon spinal cord activities. Reticular inhibitory mechanisms upon flexor motoneurons. J Neurophysiol. 1965;28:413.
Brodal A. Anatomical organization of cerebello-ves- tibulo-spinal pathways. In: De Renck AVS, Knight J, eds. CIBA Foundation Symposium: Myotatic, Kinesthetic and Vestibular Mechanisms. London: Churchill; 1967.
Vasavada AN, Li S, Delp SL. Influence of muscle morphometry and moment arms on the moment- generating capacity of human neck muscles. Spine. 1998;23:412.
Angelaki DE, Shaikh AG, Green AM, Dickman JD. Neurons compute internal models of the physical laws of motion. Nature. 2004;430:560.
McCrea RA, Gdowski GT, Boyle R, Belton T. Firing behavior of vestibular neurons during active and passive head movements: vestibulo-spinal and other noneye-movement related neurons. J Neurophysiol. 1999;82:416.
Roy JE, Cullen KE. Vestibuloocular reflex signal modulation during voluntary and passive head move- ments. J Neurophysiol. 2002;87:2337.
Roy JE, Cullen KE. Brain stem pursuit pathways: dissociating visual, vestibular, and proprioceptive inputs during combined eye-head gaze tracking. J Neurophysiol. 2003;90:271.
Roy JE, Cullen KE. Dissociating self-generated from passively applied head motion: neural mechanisms in the vestibular nuclei. J Neurosci. 2004;24:2102.
Bruschini L, Andre P, Pompeiano O, Manzoni D. Responses of Purkinje-cells of the cerebellar anterior vermis to stimulation of vestibular and somatosensory receptors. Neuroscience. 2006;142(1):235.
Mach E. Grundlinien der Lehre von der Bewegungsempfundungen. [in German] Amsterdam, Netherlands: Bonset; 1967, reprint of 1815 edition.
Flourens P. Recherches Expérimentais sur les Propriétés et les Fonctions du Systéme Nerveux dans les Animaux Vertébrés. Paris: Crevot; 1842.
Breuer J. Über die Funktion der Bogengänge des Ohrlabyrinthes [in German]. Wien Med Jahrb. 1874;4:72.
Crum-Brown A. On the sense of rotation and the anatomy and physiology of the semicircular canals of the internal ear. J Anat Physiol. 1874;8:327.
Deecke L, Schwarz DWF, Fredrickson JM. Nucleus ventroposterior inferior (VPI) as the thalamic relay in the rhesus monkey. I. Field potential investigation. Exp Brain Res. 1974;20:88.
Deecke L, Schwarz DW, Fredrickson JM. Vestibular responses in the rhesus monkey ventroposterior thal- amus. II. Vestibulo-proprioceptive convergence at thalamic neurons. Exp Brain Res. 1977;30:219.
Büttner U, Henn V. Thalamic unit activity in the alert monkey during natural vestibular stimulation. Brain Res. 1976;103:127.
Lang W, Büttner-Ennever JA, Büttner U. Vestibular projections to the monkey thalamus: an autoradio- graphic study. Brain Res. 1979;177:3.
Maciewicz R, Phipps BS, Bry J, Highstein SM. The vestibulothalamic pathway: contribution of the ascending tract of Deiters. Brain Res. 1982;252:1.
Meng H, May PJ, Dickman JD, Angelaki DE. Vestibular signals in primate thalamus: properties and origins. J Neurosci. 2007;27:13590.
Grüsser OJ, Pause M, Schreiter U. Localization and responses of neurons in the parieto-insular cortex of awake monkeys (Macaca fascicularis). J Physiol (Lond). 1990;430:537.
Grüsser OJ, Pause M, Schreiter U. Vestibular neu- rones in the parieto-insular cortex of monkeys (Macaca fascicularis): visual and neck receptor responses. J Physiol (Lond). 1990;430:559.
Akbarian S, Grüsser OJ, Guldin WO. Thalamic connec- tions of the vestibular cortical fields in the squirrel mon- key (Saimiri sciureus). J Comp Neurol. 1992;326:423.
Ebata S, Sugiuchi Y, Izawa Y, Shinomiya K, Shinoda
Y. Vestibular projection to the periarcuate cortex in the monkey. Neurosci Res. 2004;49:55.
De Waele C, Baudonniere PM, Lepecq JC, Tran Ba HP, Vidal PP. Vestibular projections in the human cortex. Exp Brain Res. 2001;141:541.
Bense S, Stephan T, Yousry TA, Brandt T, Dieterich
M. Multisensory cortical signal increases and decreases during vestibular galvanic stimulation (fMRI). J Neurophysiol. 2001;85:886.
Dieterich M, Brandt T. Functional brain imaging of peripheral and central vestibular disorders. Brain. 2008;131(pt 10):2538.
Shiroyama T, Kayahara T, Yasui Y, Nomura J, Nakano
K. Projections of the vestibular nuclei to the thalamus in the rat: a Phaseolus vulgaris leucoagglutinin study. J Comp Neurol. 1999;407(3):318.
Kotchabhakdi N, Rinvik E, Walberg F, Yingchareon
K. The vestibulothalamic projections in the cat stud- ied by retrograde axonal transport of horseradish per- oxidase. Exp Brain Res. 1980;40:405.
Meng H, Bai RS, Sato H, Imagawa M, Sasaki M, Uchino Y. Otolith-activated vestibulothalamic neu- rons in cats. Exp Brain Res. 2001;141:415.
Matsuo S, Hosogai M, Matsui H, Ikoma H. Posterior canal-activated vestibulocortical pathways in cats. Neurosci Lett. 1995;183(1-2):131.
Zwergal A, Büttner-Ennever J, Brandt T, Strupp
M. An ipsilateral vestibulothalamic tract adjacent to the medial lemniscus in humans. Brain. 2008;131(pt 11):2928.
Marlinski V, McCrea RA. Activity of ventroposterior thalamus neurons during rotation and translation in the horizontal plane in the alert squirrel monkey. J Neurophysiol. 2008;99(5):2533.
Bottini G, Sterzi R, Paulesu E, et al. Identification of the central vestibular projections in man: a positron emission tomography activation study. Exp Brain Res. 1994;99(1):164.
355 Bottini G, Karnath HO, Vallar G, et al. Cerebral representations for egocentric space: Functional- anatomical evidence from caloric vestibular stimula- tion and neck vibration. Brain. 2001;124(pt 6):1182.
Fasold O, von Brevern M, Kuhberg M, Ploner CJ, Villringer A, Lempert T, Wenzel R. Human ves- tibulat cortex as identified with caloric stimulation in functional magnetic resonance imaging. Neuroimage. 2002;17:1384.
Deutschländer A, Hüfner K, Kalla R, et al. Unilateral vestibular failure suppresses cortical visual motion processing. Brain. 2008;131(pt 4):1025.
Vitte E, Derosier C, Caritu Y, Berthoz A, Hasboun D, Soulie D. Activation of the hippocampal forma- tion by vestibular stimulation: a functional magnetic resonance imaging study. Exp Brain Res. 1996;112: 523.
Horii A, Russell NA, Smith PF, Darlington CL, Bilkey DK. Vestibular influences on CA1 neurons in the rat hippocampus: an electrophysiological study in vivo. Exp Brain Res. 2004;155:245.
Masdeu JC, Gorelick PB. Thalamic astasia: inability to stand after unilateral thalamic lesions. Ann Neurol. 1988;23:596.
Dieterich M, Brandt T. Thalamic infarctions: differ- ential effects on vestibular function in the roll plane (35 patients). Neurology. 1993;43(9):1732.
Dieterich M, Bartenstein P, Spiegel S, Bense S, Schwaiger M, Brandt T. Thalamic infarctions cause side-specific suppression of vestibular cortex activa- tions. Brain. 2005;128(pt 9):2052.
Guedry FT. Psychophysics of vestibular sensation. In: Kornhuber HH, ed. Handbook of Sensory Physiology, The Vestibular System. Vol 6. Part 2. New York: Springer-Verlag; 1974.
Clark B. Thresholds for the perception of angular acceleration in man. Aerospace Med. 1967;38:443.
Jongkees LBW, Groen JJ. The nature of the vestibu- lar stimulus. J Laryngol. 1946;61:529.
van Egmond AAJ, Groen JJ, Jongkees LBW. The turning test with small regulable stimuli. J Laryngol Otol. 1948;62:63.
Honrubia V, Jenkins HA, Baloh RW, Konrad HR, Yee RD, Ward PH. Comparison of vestibular subjec- tive sensation and nystagmus responses during the computerized harmonic acceleration tests. Ann Otol Rhinol Laryngol. 1982;91:493.
Sinha N, Zaher N, Shaikh AG, Lasker AG, Zee DS, Tarnutzer AA.Perception of self motion during and after passive rotation of the body around an earth- vertical axis. Prog Brain Res. 2008;171:277.
Mittelstaedt H. New diagnostic tests for the function of the utricles, saccules and somatic gravi-ceptors. Acta Otolaryngol Suppl (Stockh). 1995;520:188.
Clark B. The vestibular system. Annu Rev Psychol. 1970;21:273.
Böhmer A, Mast F. Asessing otolith function by sybjec- tive visual vertical. Ann NY Acad Sci. 1999;871:221.
Bauermeister M. Effect of body tilt on apparent ver- ticality, apparent body position, and their relation. J Exp Psychol. 1964;67:142.
Graybiel A. Measurement of otolith function in man. In: Kornhuber HH, ed. Handbook of Sensory Physiology, The Vestibular System. Vol 6. Part 2. New York: Springer-Verlag; 1974.
Ormsby CC, Young LR. Perception of static orienta- tion in a constant gravitoinertial environment. Aviat Space Environ Med. 1976;47:159.
Böhmer A. The subjective visual vertical as a clinical parameter for acute and chronic vestibular (otolith) disorders. Acta Otolaryngol. 1999;119:126.
Tribukait A, Bergenius J, Brantberg K. Subjective visual horizontal during follow-up after unilateral vestibular deafferentation with gentamicin. Acta Otolaryngol (Stockh). 1998;118:479.
Dai MJ, Curthoys IS, Halmagyi GM. Linear accelera- tion in the roll plane before and after unilateral ves- tibular neurectomy. Exp Brain Res. 1989;77:315.
Curthoys IS, Dai MJ, Halmagyi GM. Human otolithic function before and after unilateral vestibular neurec- tomy. J Vestib Res. 1990;I:199.
Böhmer A, Mast F. Chronic unilateral loss of otolith function revealed by the subjective visual vertical during off center yaw rotation. J Vestib Res. 1999; 9:413.
This page intentionally left blank
![]()
![]()
This page intentionally left blank
![]()
SPECIFIC DISORDERS BURDEN ON PATIENTS HEALTH CARE UTILIZATION SUMMARY
Dizziness consistently ranks among the most common symptoms experienced by patients, and it is also among the most common reasons that patients seek medical care.1,2 Accurate epi- demiological information is important for understanding the impact of a symptom or disorder on patients and on the health-care system. The prevalence of disorders can be defined and the information can also be used to identify associations with the disorder, predic- tors of the disorder, and also links to outcomes. This information informs health-care policy and also care of individual patients. Knowledge about prevalence is needed when a physician formulates a differential diagnosis and also helps guide the interpretation of tests and use of treatments by enabling accurate assessments of the pretest probability of a disorder. But obtaining accurate epidemiologic data about dizziness is difficult because of ambiguity regarding the term dizziness3 and also selection and information bias inherent to this type of research.4 For accurate estimates, required elements include a valid sampling method of a large population, valid tools for classifying participants, and a robust response rate.
The most detailed information about dizzi- ness in the general population comes from a cross-sectional telephone survey of subjects age 18 years in Germany.5 A two-stage ran- dom sampling method was used. The first stage was part of a general health survey in Germany.
Nearly 16,000 households were called using random digit dialing and 52% (8318 subjects) of those contacted agreed to participate. All subjects were asked the following question, “Did you ever experience moderate or severe dizziness or vertigo?” Nearly 30% (29.3%, 95% CI 27.8%–30.9%) of participants responded “yes” to this question (Fig. 4–1).6 Other epide- miological studies have found similar preva- lence rates.7–9 The prevalence of dizziness was higher in women compared to men (35.9%, 95% CI 33.7%–38.3% versus 22.6%, 95% CI
20.6%–24.7%, respectively).6
Next, a random subsample of participants reporting “yes” to the dizziness question were invited to complete a detailed neurotologic phone interview (response rate 87%, 1003/1212). The neurotologic phone interview was validated by the authors. Of the partici- pants that experienced dizziness at some point, 24% (n = 243) reported “vestibular vertigo” as the type of dizziness. Vestibular vertigo was defined as either rotational vertigo, positional vertigo, or recurrent dizziness with nausea and either oscillopsia or episodic imbalance. Rotational vertigo was defined as an illusion of self-motion or object motion, and positional vertigo was defined as vertigo or dizziness pre- cipitated by changes of head position, such as lying down or turning in bed. Applying this information to the broader general population sample, the lifetime prevalence of vestibular
121
Population Prevalence
Population Prevalence
20%
10%
“Moderate or severe dizziness
“Moderate or severe dizziness
or vertigo.” German Telephone
or vertigo.” German Telephone
“Dizziness in which respondent seems
“Dizziness in which respondent seems
“Dizziness in which things
“Dizziness in which things
around”, Scotland mail survey^
around”, Scotland mail survey^
“Unsteadiness, lightheadeness or
“Unsteadiness, lightheadeness or
feeling faint”, Scotland mail survey^
feeling faint”, Scotland mail survey^
“Vertigo associated with a moving
“Vertigo associated with a moving
sensation”, Finland mail survey
sensation”, Finland mail survey
†
†
spin
spin
0%
Survey*
Survey*
to move”, Scotland mail survey^
to move”, Scotland mail survey^
*German National Telephone Health Survey 2003, response rate 8,318/15,996 (52%), Neuhauser HK, et. al., Neurology 2005;65:898–904.
^Scotland mail survey, response rate 7,244/12,100 households (60%). Hannaford PC, et. al., Family Practice 2005;22:227–233.
†Finland mail survey, response rate 3,138/5,000 (63%).
Havia M, et. al., Otolaryngology–Head and Neck Surgery 2005;133:762–768.
Figure 4–1. Population prevalence of dizziness symptoms.
vertigo in the general population was estimated to be 7.4% (95% CI, 6.5%–8.3%) (Fig. 4–2).
The remainder of the subjects reporting both- ersome dizziness were classified as either a nonvestibular type of dizziness (n = 742) or a dizziness that could not be classified as either vestibular or nonvestibular (n = 18).6
The proportion of vestibular vertigo among participants with moderate to severe dizziness increased with age, so that vestibular vertigo was the type of dizziness reported by 37% of those age 60 years.5 Vestibular vertigo was recurrent (at least two attacks) in 89% of sub- jects. Two-thirds of subjects who reported ves- tibular vertigo experienced it within the past 12 months—a rate suggesting recall bias. Of sub- jects with rotational vertigo (as opposed to diz- ziness with nausea and either oscillopsia or imbalance), approximately half reported spon- taneous attacks and half reported positionally triggered attacks. The duration of attacks was
less than 1 minute in approximately half of the participants (mostly positionally triggered group), between 1 and 60 minutes in a quarter of participants, and an hour to several days in the remaining quarter of participants. Among subjects with vestibular vertigo, 80% reported a history of headaches and 62% met criteria for migraine headaches or probable migraine headaches. In a multivariable regression model examining the association of vestibular vertigo with sociodemographic factors and comorbid conditions among patients with vestibular ver- tigo in the last 12 months (163 subjects) com- pared with a dizzy-free comparison group (2816 subjects), the following factors demon- strated a significant association: Female gender (OR 3.2, 95% CI, 2.2–4.7), self-reported depression in past year (OR 2.5, 95% CI, 1.5–4.0), bothersome tinnitus in the past 7 days (OR 3.7, 95% CI 2.4–5.6), hypertension (OR
2.1, 95% CI 1.5–3.1), and increased blood
Population Prevalence of Moderate or Severe Dizziness or Vertigo
None (69.5%)
Non-vestibular dizziness or undifferentiated (22.1%)
Vestibular vertigo (7.4%)
Position triggered rotational vertigo (43%)
Spontaneous rotational vertigo (40%)
Vestibular dizziness* (17%)
*Vestibular dizziness = recurrent dizziness with nausea and either oscillopsia or episodic imbalance.
From German National Telephone Health Interview Survey 2003. Neuhauser HK. Neurology. 2005; 65:898–904.
Figure 4–2. Lifetime prevalence of moderate or severe dizziness or vertigo.
lipids (OR 1.7, 95% CI, 1.2–2.4). Increasing age and many other covariates had a significant bivariate association with vestibular vertigo but did not retain a significant association in the multivariable model. The association of migraine with dizziness could not be tested in the model because a migraine history was not taken in the general survey portion, so that the migraine status of the comparison group was not known.
The German epidemiological study also sought to classify subjects with dizziness into specific causes of dizziness.5 A structured interview that had been validated in a specialty clinic set- ting against the expert diagnosis of clinicians
was used. Based on responses to the phone interview, subjects were classified into benign paroxysmal positional vertigo (BPPV), migrain- ous vertigo, and Meniere’s disease.
The most common specific cause of dizzi- ness was BPPV. The lifetime prevalence of BPPV was estimated to be 2.4% of the general population. BPPV accounted for 8% (80/1003) of all subjects with a history of moderate or severe dizziness and 33% (80/243) of all sub- jects with vestibular vertigo. In this survey study, the criteria for BPPV classification included (a) recurrent vestibular vertigo, (b) attack duration always <1 min, (c) symptoms invariably provoked by changes of head posi- tion (specifically, lying down, turning over in the supine position, reclining the head, rising up from supine position, or bending forward), and (d) symptoms not attributable to another disorder. The prevalence of BPPV increased
with age. In subjects age 80 years or greater, nearly 10% reported BPPV. Of those with BPPV, almost two-thirds reported attacks dur- ing the past year and 25% reported BPPV dur- ing the past 4 weeks. The mean age of onset was 49.4 ± 13.8 years. The median duration of the last bout of BPPV was 2 weeks (range 0.5 days to 104 weeks). One-third of subjects reported that the episodes lasted longer than
1 month. More than half (56%) reported recurrent bouts of dizziness. The type of dizzi- ness was rotational vertigo in most patients (86%) with the remainder reporting the symptom as dizziness with nausea and oscillop- sia rather than rotational vertigo. About half of the group also reported imbalance during attacks. All participants reported BPPV attacks triggered by either turning over in bed (85%) or getting in or out of bed. In a multi- variable logistic regression model (54 BPPV patients, 6136 general population controls), age, hypertension, increased blood lipids, stroke, and migraine were associated with BPPV diagnosis, though overfitting of the model is a concern.
The lifetime prevalence of migrainous ver- tigo was estimated to be 0.98% (95% CI, 0.70%–1.37%).10 The criteria for migrainous vertigo were vestibular vertigo plus the follow- ing: (1) a history of migraine according to the criteria of the International Headache Society,
(2) at least one migrainous symptom during at least two vertiginous attacks (i.e., migrainous headache, photophobia, phonophobia, or visual aura), and (3) symptoms not attributable to another disorder. Of those patients reporting vestibular vertigo (243), 33 (14%) were classi- fied as migrainous vertigo. Two-thirds of the migrainous vertigo subjects reported spontane- ous rotational vertigo and 24% reported posi- tional vertigo (overlap with BPPV was not reported). The remainder (9%) reported ves- tibular dizziness rather than rotational vertigo. Headache always accompanied vertigo attacks in only 24%. Duration of attacks was <1 minute in 25%, 1–59 minutes in 44%, 1–24 hours in 28%, and >24 hours in 3%. Cochlear symptoms (i.e., tinnitus, aural fullness, or hearing loss) during vertigo attacks were reported by 12 sub- jects (36%) with migrainous vertigo but none had progressive hearing loss. Typical migraine triggers were reported in 61% of those with migrainous vertigo. In a multivariable model,
only a history of coronary artery disease was significantly associated with migrainous ver- tigo, but overfitting the model is a concern (tested 11 independent variables, but only 30 outcome positive subjects).
In this study, very few subjects were classi- fied with Meniere’s disease as the cause of diz- ziness.11 Of the 243 subjects with vestibular dizziness, only 4 (1.6%) met the criteria for Meniere’s disease. This translates to a popula- tion prevalence of 0.12% for Meniere’s disease. The criteria for Meniere’s disease included at least two vertigo attacks lasting at least 20 min- utes, interictal unilateral hearing loss, and at least one cochlear symptom (i.e., tinnitus, hear- ing loss, or aural fullness) during at least two vertigo attacks.
Many studies show that dizziness symptoms and particularly vertigo and vestibular disor- ders have an adverse effect on quality of life.12–22 In the German epidemiological study, one-fifth (18.8%) of subjects with dizziness reported an interruption in daily activities and 12.2% reported avoiding leaving the house because of the dizziness symptoms.6 Of working subjects with dizziness, one-fifth (20.7%) reported taking sick leave as a result of the dizziness. The burden was greater in the subgroup with vestibular vertigo compared to those with nonvestibular dizziness. More than 40% of those with vestibular vertigo reported an interruption in daily activities. And 80% of subjects with vestibular vertigo reported at least one of the following (as the result of the vertigo): interruption of daily activities, medical consultation, or taking sick leave. The rate was 57% in subjects with nonvestibular dizziness and 63% in the overall dizziness group. Quality of life, measured by the SF-8, was reduced in both vestibular vertigo subjects and nonvestibular dizziness subjects when compared to control subjects in the general population without dizziness, adjusting for age and sex.6 For BPPV subjects, 69% reported restricting head movements in order to avoid attacks.23 And during episodes, 24% reported giving up driving a car with 18% avoiding leaving their home.
From the German epidemiological study, 58% of the subjects who reported bothersome dizzi- ness sought out a medical evaluation.6 This rate was lower in the Scotland postal survey (23%).9 A previous hospitalization because of dizziness was reported by 7% of those with dizziness.6 Both medical consultations and hospital visits were more common for subjects with vestibu- lar vertigo than for subjects with nonvestibular dizziness. Most of the subjects with a medical consultation reported seeing a general practi- tioner (52%), followed by a neurologist (16%) or an otolaryngologist (14%).6 About a quarter (24%) of subjects seeking a medical consulta- tion saw more than one type of physician. Subjects with BPPV had the highest rate of medical consultation (78%), followed by sub- jects with migrainous vertigo (67%), other ves- tibular vertigo (67%), other nonvestibular diz- ziness (61%), and orthostatic dizziness (46%).6 Diagnostic tests were commonly performed in dizziness patients but were not reported at the aggregate level of dizziness patients from the German study. For patients with migrain- ous vertigo who saw a physician, most under- went one or more diagnostic test, including an audiogram (27%), electroencephalogram (27%), brainstem auditory evoked potentials (21%), cranial computed tomography (CT) or magnetic resonance imaging (MRI) (21%), caloric test (18%), or ultrasound of carotid or vertebral arteries (12%).10 In subjects with BPPV, most also underwent diagnostic testing (77%), but only 27% reported diagnostic positional testing (i.e., Dix-Hallpike testing).23 Furthermore, most BPPV subjects who presented to a physician received either no treatment (45%) or medica- tion treatment (27%) for vertigo.23 Only 10% of BPPV subjects who presented to a physician underwent a particle repositioning maneuver.23 Most of the repositioning maneuvers were Brandt-Daroff exercises, rather than the most
effective Epley or Semont maneuvers.
Diagnostic testing among patients with dizzi- ness in the emergency room in the United States has been reported using the National Hospital Ambulatory Medical Care Survey (NHAMCS).2 NHMACS is an annual four-stage probability sampling study where patients are selected and medical records are reviewed. This ongoing study can be used to study trends over time.
For patients reporting vertigo or dizziness as a reason for visiting the emergency room, about 10% underwent a CT scan in 1995 and this rate increased by 169% to nearly 30% in 2004.
Dizziness is common among the general popu- lation and leads to substantial burden on patients and the health-care system. Dizziness that is vestibular in origin accounts for about a quarter of patients with dizziness symptoms, and the most common cause of vestibular ori- gin dizziness is BPPV. Migrainous vertigo also appears to be common. Meniere’s disease, on the other hand, was relatively uncommon in the general population and also as a proportion of vestibular origin dizziness. Most patients with vestibular origin dizziness present for a medical evaluation. Diagnostic tests are com- monly ordered and in the United States rates of use of imaging studies have dramatically increased. Positional testing and particle repo- sitioning seem to be underutilized.
Though the German study is a large and detailed epidemiological study on dizziness and vertigo, more epidemiological studies are needed in other populations. Remaining needs from epidemiologic studies include more data from other populations and prospective longitudinal cohorts with relevant outcome measures so we can see how processes of care impact important patient outcomes in real-world settings.
REFERENCES
Kroenke K, Jackson JL. Outcome in general medical patients presenting with common symptoms: a prospec- tive study with a 2-week and a 3-month follow-up. Fam Pract. 1998;15:398.
Kerber KA, Meurer WJ, West BT, Fendrick AM. Dizziness presentations in U.S. emergency depart- ments, 1995-2004. Acad Emerg Med. 2008;15:744.
Newman-Toker DE, Cannon LM, Stofferahn ME, Rothman RE, Hsieh YH, Zee DS. Imprecision in patient reports of dizziness symptom quality: a cross- sectional study conducted in an acute care setting. Mayo Clin Proc. 2007;82:1329.
Nallamothu BK, Hayward RA, Bates ER. Beyond the randomized clinical trial: the role of effectiveness stud- ies in evaluating cardiovascular therapies. Circulation. 2008;118:1294.
Neuhauser HK, von Brevern M, Radtke A, et al. Epidemiology of vestibular vertigo: a neurotologic survey of the general population. Neurology. 2005;65: 898.
Neuhauser HK, Radtke A, von Brevern M, Lezius F, Feldmann M, Lempert T. Burden of dizziness and vertigo in the community. Arch Intern Med. 2008;168:2118.
Colledge NR, Wilson JA, Macintyre CC, MacLennan WJ. The prevalence and characteristics of dizziness in an elderly community. Age Ageing. 1994;23:117.
Havia M, Kentala E, Pyykko I. Prevalence of Meniere’s disease in general population of Southern Finland. Otolaryngol Head Neck Surg. 2005;133:762.
Hannaford PC, Simpson JA, Bisset AF, Davis A, McKerrow W, Mills R. The prevalence of ear, nose and throat problems in the community: results from a national cross-sectional postal survey in Scotland. Fam Pract. 2005;22:227.
Neuhauser HK, Radtke A, von Brevern M, et al. Migrainous vertigo: prevalence and impact on quality of life. Neurology. 2006;67:1028.
Radtke A, von Brevern M, Feldmann M, et al. Screening for Meniere’s disease in the general popu- lation - the needle in the haystack. Acta Otolaryngol. 2008;128:272.
Tinetti ME, Williams CS, Gill TM. Health, functional, and psychological outcomes among older persons with chronic dizziness. J Am Geriatr Soc. 2000;48: 417.
Aggarwal NT, Bennett DA, Bienias JL, Mendes de Leon CF, Morris MC, Evans DA. The prevalence of dizziness and its association with functional disability in a biracial community population. J Gerontol A Biol Sci Med Sci. 2000;55:M288.
Grimby A, Rosenhall U. Health-related quality of life and dizziness in old age. Gerontology. 1995;41: 286.
Hsu LC, Hu HH, Wong WJ, Wang SJ, Luk YO, Chern CM. Quality of life in elderly patients with dizziness: analysis of the Short-Form Health Survey in 197 patients. Acta Otolaryngol. 2005;125:55.
Nazareth I, Landau S, Yardley L, Luxon L. Patterns of presentations of dizziness in primary care–a cross- sectional cluster analysis study. J Psychosom Res. 2006;60:395.
Lopez-Escamez JA, Gamiz MJ, Fernandez-Perez A, Gomez-Finana M. Long-term outcome and health- related quality of life in benign paroxysmal posi- tional vertigo. Eur Arch Otorhinolaryngol. 2005;262: 507.
Fife D, FitzGerald JE. Do patients with benign par- oxysmal positional vertigo receive prompt treatment? Analysis of waiting times and human and financial costs associated with current practice. Int J Audiol. 2005;44:50.
Kinney SE, Sandridge SA, Newman CW. Long-term effects of Meniere’s disease on hearing and quality of life. Am J Otol. 1997;18:67.
Soderman AC, Bagger-Sjoback D, Bergenius J, Langius
Factors influencing quality of life in patients with Meniere’s disease, identified by a multidimensional approach. Otol Neurotol. 2002;23:941.
Yardley L, Owen N, Nazareth I, Luxon L. Prevalence and presentation of dizziness in a general practice community sample of working age people. Br J Gen Pract. 1998;48:1131.
Nazareth I, Yardley L, Owen N, Luxon L. Outcome of symptoms of dizziness in a general practice com- munity sample. Fam Pract. 1999;16:616.
von Brevern M, Radtke A, Lezius F, et al. Epidemiology of benign paroxysmal positional vertigo: a popula- tion based study. J Neurol Neurosurg Psychiatry. 2007;78:710.
![]()
VERTIGO
Central versus Peripheral Causes Time Course
Precipitating Factors Associated Symptoms Compensation Predisposing Factors Family History
Diagnosis and Management
NEAR-FAINT DIZZINESS
Orthostatic Hypotension
Postural Tachycardia Syndrome (POTS) Vasovagal Attacks
Hyperventilation
PSYCHOPHYSIOLOGIC DIZZINESS (CHRONIC SUBJECTIVE DIZZINESS)
Panic Disorder Phobic Dizziness Chronic Anxiety Pathophysiology
Diagnosis and Management
DRUG-INDUCED DIZZINESS HYPOGLYCEMIA DISEQUILIBRIUM
Common Causes
Gait Disorders in Older People Falls in Older People Diagnosis and Management OCULAR DIZZINESS
Common Causes Oscillopsia Management
MULTISENSORY DIZZINESS
Management
PHYSIOLOGIC DIZZINESS
Motion Sickness Space Sickness Height Vertigo
Mal de Debarquement Syndrome
SUMMARY: DISTINGUISHING BETWEEN VESTIBULAR AND NONVESTIBULAR TYPES OF DIZZINESS
Dizziness is a nonspecific term that patients use to refer to some sort of an ill feeling. The variety of symptoms that patients label as “diz- ziness” is large and includes visualized spinning of the environment, other types of movement of the environment (e.g., bouncing, rocking), “internal” movement sensations (i.e., move- ment sensation but no visualized movement of the environment), light-headedness, near-faint, unsteadiness standing or walking, disorienta- tion or confusion, or even anxiety. The initial task of the clinician is to obtain a description of
what the patient means by dizziness. Patients should be encouraged to use their own words to describe the sensation and how the sensa- tion interferes with their daily activities. Some of these descriptions can be highly suggestive of a lesion location or even a specific etiology, but a patient’s description of the type of dizziness can also be unclear or even inconsis- tent.1 Because the dizziness description can be problematic, other important characteristics, including the circumstances, duration, associ- ated symptoms, and triggers, become equally
127
Table 5–1 Mechanisms of Common Types of Dizziness
![]()
Type Mechanism
![]()
Vertigo Imbalance in tonic vestibular signals Near-faint dizziness Diffusely decreased cerebral blood flow
Psychophysiologic dizziness
Impaired central integration of sensory signals
Hypoglycemic dizziness Inadequate brain glucose; increased circulatory catecholamines Disequilibrium Loss of vestibulospinal, proprioceptive, cerebellar, or motor function Ocular dizziness Visual–vestibular mismatch due to impaired vision
Multisensory dizziness Partial loss of multiple sensory system function
Physiologic dizziness Sensory conflict due to unusual combination of sensory signals
Drug-induced dizziness CNS depression, cerebellar toxicity; change in cupula’s specific gravity (alcohol)
![]()
important to detail—particularly when the symptom is vague or mild. Because the diagnostic evaluation and management differ markedly depending on the features of the diz- ziness, it is critical that the examining physician determine the type and characteristics of the dizziness (Table 5–1) before proceeding with exhaustive diagnostic studies.
Vertigo is an illusion of movement, usually that of rotation, although patients occasionally describe a sensation of linear displacement or tilt. The afferent nerves from the otoliths and semicircular canals of each labyrinth maintain a balanced tonic rate of firing into the vestibular nuclei. Asymmetric involvement of this baseline activity anywhere in the periph- eral and central vestibular pathways leads to an illusion of movement. For example, damage to a semicircular canal or its afferent nerve produces a sensation of angular rotation in the plane of that canal similar to the sensation experienced during physiologic stimulation. More typically, lesions affect the afferent input from all the canals and otoliths of one labyrinth, producing a sensation of rotation in a plane determined by the balance of afferent signals from the contralateral labyrinth (usually near the horizontal plane, inasmuch as the ver- tical canal and otolith signals partially cancel out). If a patient with a unilateral vestibular lesion attempts to fixate on an object, it will appear blurred and seem to be moving in the direction opposite that of the slow phase of the
patient’s spontaneous nystagmus (i.e., away from the side of the lesion). This illusion of movement occurs because the brain interprets the target displacement on the retina as object movement rather than as eye movement. An illusion of linear movement or tilting suggests isolated involvement of an otolith or its central connections.
Vertigo is strongly suggestive of an imbal- ance within the vestibular system. The main caveat is that a symptom is subjective and subjective symptoms—including vertigo—can be inconsistent and unreliable.1 A description of vertigo is probably a less valid indicator of vestibular imbalance when it is mild or an “internal” (i.e., no visualized movement of the environment) sensation. It may also be less valid when the patient acknowledges vertigo only after being specifically asked about it rather than providing a spontaneous descrip- tion of it.
Central versus Peripheral Causes
The symptom of vertigo does not indicate where in the system a vestibular imbalance originates. The same sensation can result from lesions in such diverse locations as the inner ear, the deep paravertebral stretch receptors of the neck, the visual–vestibular interaction cen- ters in the brain stem and cerebellum, or in the subjective sensation pathways of the thalamus or cortex. Distinction between peripheral and central causes of vertigo can usually be made, however, on the basis of other features in the history (Table 5–2).
![]()
Table 5–2 Differentiation between Peripheral (End-organ and Nerve) and Central Causes of Vertigo
Nausea and Vomiting
Imbalance Hearing
Loss
Oscillopsia Neurologic
Symptoms
Compensation
![]()
Peripheral Severe Mild Common Mild Rare Rapid
Central Moderate Severe Rare Severe Common Slow
![]()
Well-documented lesions within the vestib- ular pathways sometimes produce only a non- specific sensation of disorientation without a clearly defined illusion of movement. Normal subjects undergoing caloric stimulation (i.e., a physiologic imbalance in the vestibular system) occasionally describe the experience with terms such as floating or even giddiness. For these reasons one must not be too restrictive in clas- sifying dizziness on the basis of subjective description alone.
Time Course
Vertigo invariably occurs in episodes, usually abrupt in onset, followed by decreasing inten- sity as the inciting factor dissipates or as com- pensation occurs. Continuous dizziness without fluctuation for long periods of time (e.g., weeks or longer) is not typical of vestibular disorders. Durations associated with several of the more common causes of vertigo are outlined in Table 5–3. Episodes lasting seconds suggest the diag- nosis of benign positional vertigo.2 During the acute phase, such patients may report a non- specific feeling of disorientation and imbalance along with nausea and vomiting that lasts for hours to days, but on careful questioning one can identify recurrent brief attacks of positional
Table 5–3 Duration of Common Causes of Vertigo
![]()
Seconds Benign positional vertigo Minutes Vertebrobasilar insufficiency,
migraine*
Hours Meniere’s syndrome
Days Vestibular neuritis, infarction of
labyrinth
![]()
*Though migraine vertigo is commonly minutes in dura- tion, it can also last seconds, hours, or days.
vertigo interspersed with a more persistent nonspecific dizziness. An episode of vertigo lasting minutes suggests a transient vascular ischemic attack.3 This is the typical duration of vertigo with transient ischemia within the basi- lar vertebral circulation. With a typical bout of Meniere’s syndrome, the vertigo reaches a peak within minutes, remains severe for an hour or two, and then gradually resolves over the next few hours.4 Vertigo gradually resolving over several days occurs with viral vestibular neuri- tis, labyrinthine trauma, infarction of the laby- rinth, or any lesion that produces permanent damage to the inner ear or vestibular nerve.5 Even with a complete unilateral loss of vestibu- lar function, the vertigo will gradually resolve as central compensation occurs. The onset is abrupt with trauma and vascular occlusion, whereas it is typically more gradual in onset (over hours) with viral vestibular neuritis.
Precipitating Factors
The events just prior to an episode of vertigo are important in determining the cause. Rapid head movements commonly induce vertigo because they accentuate any imbalance within the vestibular pathways. Even after compensa- tion has occurred, head movements or change in position can lead to a brief sensation of ver- tigo and disorientation. Positional vertigo is commonly induced by turning over in bed, sit- ting up from a lying position, extending the neck to look upward (so-called top shelf ver- tigo), or bending over and straightening up. Patients with a fistula in the bony labyrinth develop brief episodes of vertigo precipitated by changes in middle ear pressure (coughing, sneezing).6 The pressure change in the middle ear is transferred directly to the inner ear (usu- ally the horizontal semicircular canal) through the fistula. Occasionally, loud noises induce transient vertigo in patients with inner ear
Associated Symptoms
Autonomic symptoms such as sweating, pallor, nausea, and vomiting commonly accompany dizziness caused by vestibular lesions, but such symptoms are uncommon with other types of dizziness. Typically, the autonomic symptoms are more pronounced when the vertigo has a peripheral origin, although there are frequent exceptions to the rule. Occasionally, vegetative symptoms are the only manifestation of a vestibular lesion. Numerous interconnecting pathways between brainstem vestibular and autonomic centers account for this close asso- ciation of vestibular and autonomic symptoms. The site of the lesion determines the symp- toms that accompany vertigo (Table 5–4). In addition to vertigo, lesions of the labyrinth or eighth nerve commonly produce auditory symptoms such as hearing loss, tinnitus, a sen- sation of pressure or fullness in the ear, or pain in the ear. Lesions of the internal auditory canal also produce hearing loss and tinnitus and may be associated with ipsilateral facial weakness, whereas those in the cerebellar- pontine angle may be associated with ipsilateral
facial numbness and weakness and ipsilateral extremity ataxia. As with vertigo, the time course of an associated hearing loss can help determine the cause. Fluctuating hearing loss and tinnitus are characteristic of Meniere’s syndrome. Patients with this disorder usually notice a buildup of pressure in the ear just prior to the onset of hearing loss, tinnitus, and ver- tigo. Complete unilateral deafness and vertigo occur with viral involvement of the labyrinth and or eighth nerve and with vascular occlusion to the inner ear. A slow, progressive unilateral hearing loss over months suggests the existence of an acoustic neuroma or other cerebellopon- tine angle tumor.
Because of the proximity of other neuronal centers and fiber tracts in the brain stem and cerebellum, it is unusual to find lesions in these areas that produce isolated vestibular symptoms. Lesions of the brain stem invariably are associated with other cranial nerve and long-tract symptoms. For example, vertigo caused by transient vertebrobasilar insuffi- ciency is associated with other brainstem and occipital lobe symptoms such as diplopia, hemi- anoptic field defects, drop attacks, weakness, numbness, dysarthria, and ataxia. Lesions of the cerebellum (e.g., infarction or hemorrhage) may be relatively silent but are typically associ- ated with extremity or truncal ataxia in addition to vertigo. Hearing loss for pure tones is unusual with central lesions, even in the late stages.
Vertigo can occur as part of an aura of an epileptic seizure.8 The cortical projections of
Table 5–4 Symptoms Associated with Vertigo Due to Lesions at Different Anatomical Locations
![]()
Inner Ear Hearing loss Tinnitus Pressure Pain
Internal Auditory Canal
Hearing loss Tinnitus
Facial weakness
Cerebellopontine angle
Hearing loss Tinnitus
Facial weakness and numbness Extremity incoordination
Brain stem Diplopia Dysarthria
Perioral numbness
Extremity weakness and numbness Drop attacks
Cerebellum Imbalance Incoordination
Temporal lobe
Absence spells
Visual (formed), olfactory or gustatory hallucinations
Occipital lobe
Visual field loss
Visual hallucinations (unformed)
![]()
Compensation
The severity of symptoms following a vestibu- lar lesion depends on (1) the extent of the lesion, (2) whether the lesion is unilateral or bilateral, and (3) the rapidity with which the functional loss occurs. Patients who slowly lose vestibular function bilaterally (e.g., secondary to ototoxic drugs) often do not complain of ver- tigo but will report oscillopsia with head move- ments and instability when walking (due to loss of vestibulo-ocular and vestibulospinal reflexes, respectively). If a patient slowly loses vestibu- lar function on one side over a period of months to years (e.g., with an acoustic neuroma), symp- toms and signs may be absent. A sudden unilat- eral loss of vestibular function, by contrast, is a dramatic and debilitating event. Patients com- plain of severe vertigo and nausea, are pale and perspiring, and usually vomit repeatedly. They prefer to lie quietly in a dark room but can walk if forced to (falling toward the side of the lesion). A brisk spontaneous nystagmus inter- feres with vision. These symptoms and signs are transient, however, and the process of compensation begins almost immediately. Within 1 week of the lesion, a young patient can walk without difficulty and with fixation can inhibit the spontaneous nystagmus. Within 1 month most patients return to work with little if any residual symptoms. Occasionally patients will have difficulty compensating for unilateral loss of vestibular function. The presence of migraine, anxiety, or depression seems to cor- relate with persistence of symptoms.9
Predisposing Factors
The patient’s general state of health just prior to the onset of dizziness should be carefully investigated. Most severe systemic disorders
are associated with dizziness due to either par- tial involvement of all the body-orienting sys- tems or a decreased capacity of the central ner- vous system (CNS) to deal with information from these systems (a type of multisensory dizziness). Some systemic disorders such as vasculitis, bacterial endocarditis, and septice- mia can selectively damage the vestibular sys- tem by interfering with its blood supply. Such patients may develop severe vertigo and vomit- ing typical of an acute peripheral vestibular loss. Patients with viral vestibular neuritis fre- quently report an upper respiratory tract illness either within 2 or 3 weeks before or at the time of onset of vertigo. Chronic middle ear infec- tions may lead to bacterial labyrinthitis or serous labyrinthopathy, and patients with bacterial meningitis may develop bacterial lab- yrinthitis through the direct cerebrospinal fluid–perilymph connections. Patients with Meniere’s syndrome may have an attack of ver- tigo precipitated by foods high in salt content.
Head injury can damage the delicate laby- rinthine membranes with or without associated bone fracture. Labyrinthine trauma may result in a single prolonged episode of vertigo or, more commonly, recurrent episodes of posi- tional vertigo. The more common nonspecific light-headed dizziness following head trauma is probably not related to vestibular damage, inasmuch as common associated symptoms and signs are absent. Surgery in or about the ear is a major cause of trauma to the labyrinthine membranes. Vertigo not infrequently follows surgery confined to the middle ear. Past medi- cal history should focus on past or chronic medical illnesses, such as diabetes mellitus, atherosclerotic vascular disease, syphilis (con- genital or acquired), and major allergies, that might predispose the patient to vestibular sys- tem damage. Viral illnesses that damage the inner ear in utero or in infancy (e.g., rubella, mumps, rubeola) may be followed years later by recurrent episodes of vertigo.10 This so- called delayed endolymphatic hydrops may not be associated with auditory symptoms such as hearing loss, tinnitus, or ear pressure because the patient may be deaf in the damaged ear.
Family History
Common vestibular disorders with a genetic predisposition include migraine, Meniere’s
syndrome, otosclerosis, neurofibromatosis, and spinocerebellar degeneration. Migraine can present as isolated episodes of vertigo (a migraine equivalent) in some members of a family, whereas other members have classic migraine headaches.11 Some varieties of “famil- ial Meniere’s syndrome” are likely due to migraine. Patients with otosclerosis usually present with a conductive hearing loss, although sensorineural hearing loss and vertigo may result if the otic capsule becomes involved. Neurofibromatosis type 1 (NF1) is manifested by the combination of pigmented skin lesions, multiple tumors of the spinal and cranial nerves, tumors of the skin, and intracranial gliomas. Neurofibromatosis type 2 (NF2) is commonly manifested by bilateral acoustic neuromas and meningiomas without periph- eral manifestations. Any young patient (<30 years old) with an acoustic neuroma should be followed carefully for development of a tumor on the opposite side (due to NF2). Central varieties of positional vertigo are commonly seen in patients with the inherited ataxia syn- dromes (particularly common with spinocere- bellar atrophy type 6[SCA-6]).12 Positional vertigo and ataxia are also commonly seen with Hippel-Lindau disease. This autosomal domi- nant disorder is characterized by hemangio- blastomas of the cerebellar hemispheres, angiomas of the retina, and cystic changes in the kidney and pancreas. The diagnosis should be considered in any patient with a cerebellar tumor or hemorrhage who manifests an ele- vated hematocrit.
Congenital deformities of the inner ear may result from abnormal genes or from abnormal development in utero. Most of the inherited malformations of the inner ear are associated with multiple malformations in other organs producing a characteristic clinical profile (e.g., Alport’s and Waardenburg’s syndromes). Progressive atrophy of the cochlear and ves- tibular nerves may be seen as part of a more diffuse degenerative disorder (e.g., with
Friedreich’s ataxia or olivopontocerebellar atrophy) or may occur as an isolated phenom- enon (both autosomal dominant and recessive inheritance). Although uncommon, the syn- drome of familial periodic ataxia and vertigo (episodic ataxia type 2[EA-2]) is important to recognize because it often responds dramati- cally to acetazolamide. With this dominantly inherited disorder, episodes of vertigo and ataxia recur throughout the patient’s life sometimes without objective findings between episodes.
Diagnosis and Management
The reader is referred to Chapters 9–18 for discussion in detail of the diagnosis and man- agement of common causes of vertigo.
Near-faint dizziness or presyncope can best be described as the lightheaded sensation of an impending faint. It is often associated with a feeling of unsteadiness or even of falling. Near- faint dizziness results from pancerebral isch- emia.13 Common causes are summarized in Table 5–5. We emphasize that near-faint dizzi- ness is not a symptom of focal occlusive cere- brovascular disease (i.e., not a symptom of impending stroke).
Orthostatic Hypotension
All of us have experienced light-headedness after rapidly assuming the standing position from the supine or sitting position. This symp- tom is transient and of little consequence. Also, not uncommonly, susceptible subjects may develop presyncopal lightheadedness and may even faint after standing for a prolonged period
Table 5–5 Common Causes of Near-Faint Dizziness
![]()
Cause Precipitating Factors
![]()
Orthostatic hypotension Reduced blood volume, hypotensive drugs, autonomic dysfunction Vasovagal attack Prolonged standing in hot sun, fear, severe pain, acute vertigo
Hyperventilation Anxiety, stress, panic attacks
Decreased cardiac output Arrhythmia, valvular disease, heart failure
![]()
in the hot sun. Recurrent symptoms of postural hypotension, however, can usually be traced to either reduced blood volume, the chronic use of hypotensive drugs, or autonomic dysfunc- tion.14,15 Nearly all of the antihypertensive drugs, a large number of antidepressants and major tranquilizers, and long-term bed rest will pre- dispose a patient to orthostatic hypotension.
DIAGNOSIS AND MANAGEMENT
Near-faint dizziness with orthostatic hypoten- sion can develop immediately on standing or insidiously after several minutes of standing. The diagnosis is made by documenting an acute or progressive decline in mean blood pressure of more than 10 to 15 mmHg with a corre- sponding increase in pulse rate while the patient is in the erect position. In patients with autonomic insufficiency the pulse rate will remain unchanged despite the hypotension. Autonomic impairment can be documented at the bedside by taking the pulse while the supine patient performs a vigorous Valsalva maneuver. Normally, the pulse slows and the mean blood pressure increases by 10 to 30 mmHg in the immediate post-Valsalva period. Orthostatic hypotension can often be eliminated by remov- ing offending drugs or by correcting the causes of blood-volume depletion. In patients with autonomic insufficiency, increased salt intake can increase blood volume, and elastic stock- ings can prevent pooling of blood in the lower extremities.16,17 In severe cases the salt- retaining steroid fluorocortisone can aid in expanding blood volume. Orthostatic lighthead- edness is particularly common in the elderly and in many cases may be an effect of arterio- sclerosis (manifest by white matter hyperinten- sities on magnetic resonance imaging [MRI]) leading to reduced cerebral blood flow as opposed to the typical causes of orthostatic hypotension. Patients with arteriosclerosis have reduced cerebral blood flow even though blood pressure measured at the arm is normal. 18
Postural Tachycardia Syndrome (POTS)
POTS is associated with orthostatic dizziness but without orthostatic hypotension.19 The pulse typically increases above 120 beats per minute on standing and there are associated
autonomic symptoms. It most commonly occurs in young women (ages 20–40 years). Multiple pathophysiological mechanisms have been associated with POTS, including periph- eral denernation, hypovolemia, beta receptor hypersensitivity, deconditioning, and psycho- logical factors.
DIAGNOSIS AND MANAGEMENT
The diagnosis of POTS is made by documenting a heart rate increase of >30 beats per minute in a patient who develops orthostatic symptoms on standing. Treatment begins with a high-salt diet, copious fluids, and postural training. Some patients benefit from low doses of beta-receptor antagonists or vasoconstrictors.
Vasovagal Attacks
Prior to a common faint, one experiences sen- sations of light-headedness, giddiness, nausea, and an abdominal sinking sensation. Typically the subject is pale and there are associated signs of parasympathetic hyperactivity, includ- ing piloerection and sweating. These symptoms are induced when emotions such as fear and anxiety, initiated in the forebrain limbic sys- tem, activate the medullary vasodepressor cen- ters.13,20 The consequences are a fall in heart rate and blood pressure and a decline in car- diac output, leading to a decrease in cerebral blood flow. Parasympathetic hyperactivity accounts for the slowing of heart rate, and diminished sympathetic tone leads to vasodila- tion. Normal cardiovascular reflexes are rein- stated if the subject lies supine or if there is loss of consciousness with a common faint.
Vasodepressor light-headedness commonly occurs when a subject has fasted for a long period of time, is exposed to hot, moist weather, and/or has stood for a prolonged period of time. Some individuals are clearly more susceptible to presyncopal light-headedness and the common faint than others, and occasionally one can find a family history with members in several gener- ations who are susceptible. Family pedigrees are most consistent with an autosomal dominant transmission with incomplete penetrance but so far no definite genetic mutation has been identi- fied.21 Vasodepressor episodes can also be pre- cipitated by acute visceral pain or by a sudden severe attack of vertigo. This explains the
occasional patient with an acute peripheral ves- tibular lesion who will present with a history of syncope. In this case, it is important to obtain a history of severe vertigo and autonomic symp- toms preceding the loss of consciousness.
DIAGNOSIS AND MANAGEMENT
The first and most important step in the evalu- ation of a vasovagal event is to assess the risk for a serious cause (i.e., myocardial infarction, arrhythmia, stroke) or a future serious event. A clinical decision rule—the San Francisco Syncope Rule—was developed and validated for this purpose.22 The probability of a serious cause or future event (outcome at 30 days) is very low (<0.5%) when the patient has none of the following: history of congestive heart fail- ure, hematocrit less than 30%, abnormal electrocardiogram (EKG) (new changes or non-sinus rhythm), complaint of shortness of breath, or a systolic blood pressure less than 90 mmHg at triage.
Patients found to be a low risk for severe causes or events can be treated symptomatically rather than requiring an extensive evaluation.
Hyperventilation
Chronic anxiety with associated hyperventila- tion is a common cause of persistent near-faint dizziness.23 Patients typically describe sensa- tions of light-headedness, faintness, and giddi- ness along with other sensations described later in the section “Psychophysiologic Dizziness.” Associated symptoms typically include fre- quent sighing, air hunger, perioral numbness, paresthesias of the extremities, lump in the throat, and tightness in the chest. Patients often report being unable to obtain the satis- faction of a full deep breath and they will sigh frequently as though they were trying to catch their breath. Studies in patients who hyperven- tilate with medical procedures such as blood drawing or injections showed excessively deep and irregular breathing which was associated with symptoms of dizziness and fainting.24 Hyperventilation causes presyncopal light- headedness by lowering the carbon dioxide content of the blood, thus producing constriction of the cerebrovasculature. In most subjects only a moderate increase in respiratory rate can drop the PaCO2 levels to
25 mm of mercury or less in a few minutes. Once this level is achieved, the subject does not have to breathe excessively to maintain
the low PaCO2, so it is possible to be chronically hypocapnic without appearing to
hyperventilate.25
DIAGNOSIS AND MANAGEMENT
The diagnosis rests on identifying the charac- teristic associated symptoms in the setting of anxiety and dyspnea. It is usually helpful to have the patient voluntarily overbreathe to reproduce his or her symptoms and to provide insight into the mechanism.23 In addition to educating the patient, treatment must be directed at the underlying anxiety. Behavioral interventions directed at dysfunctional breath- ing can benefit susceptible patients.24 Long- term use of tranquilizers should be avoided because increased tolerance and dependency commonly occur.
PSYCHOPHYSIOLOGIC DIZZINESS (CHRONIC SUBJECTIVE DIZZINESS)
A wide range of dizzy sensations are associated with psychiatric illnesses.26,27 Feelings of disso- ciation, as though one has left one’s own body, are common. Patients use terms such as “float- ing,” “swimming,” and “giddiness” to describe the dizzy sensation. They may report a feeling of imbalance (commonly a rocking or falling sensation) or even of spinning inside the head— sensations that can usually be differentiated from vertigo because they are not associated with an illusion of movement of the environ- ment or with nystagmus.28 Psychophysiologic dizziness may be constant or occur in attacks and is typically associated with symptoms of anxiety. Common associated somatic com- plaints include tension headache, heart palpi- tations, gastric distress, urinary frequency, backache, and a generalized feeling of weak- ness and fatigue. Attacks may be provoked by sensory stimuli (driving on a freeway, walking on a brightly polished floor, watching a train go by) or by social situations (eating in a restau- rant, shopping in a department store, attending a reception). Symptoms often begin after a period of stress, especially after the death of a
loved one or after a patient has been through an illness, and may continue for months or years.
Panic Disorder
Common causes of anxiety in daily life are cir- cumstances in which one must make a decision that could have major implications for future social and economic status. The symptoms associated with this type of anxiety are usually transitory and completely reversible.29 Anxiety can also be associated with a number of neuro- logic and psychiatric disorders. For example, the first sign of dementia or manic depressive illness can be an attack of severe anxiety with- out obvious cause.
Panic attacks are a distinct form of anxiety that typically occur in a background of persis- tent apprehension but at times when there appears to be no obviously threatening circum- stance.30,31 Such attacks often occur when it would be difficult for one to make a rapid exit (e.g., traveling in an airplane or train, driving in the fast lane of the freeway, shopping in a crowded store, or waiting in a supermarket line). The condition typically builds up over 10 to 15 min with progressively increasing anxiety associated with dizziness, shortness of breath, sweating, flushing, trembling or shaking, heart palpitations, paresthesias, and a generalized feeling of weakness (Table 5–6). The dizziness can take several forms, from a giddy, unsteady sensation to a progressing, presyncopal light- headedness. Patients may experience a tight- ness in the chest as though the lungs cannot be adequately filled. Hyperventilation and enhanced CO2 sensitivity are also common.32
The person may try to flee and in the future
avoid the situation in which the panic attack occurred. There is a clear genetic predisposi- tion to panic disorders, distinguishing them from the more common anxieties that are a response to specific life situations.33
Phobic Dizziness
Agoraphobia, defined as a morbid fear and avoidance of being in public places, is closely linked with anxiety disorders and panic attacks.29,34 Often agoraphobia is secondary to panic attacks; the patient restricts outside
Table 5–6 Common Symptoms during Panic Attacks
![]()
Shortness of breath, smothering, choking Palpitations, accelerated heart rate Chest pain or discomfort
Sweating
Dizziness, unsteady feeling, sensory illusions Nausea or abdominal distress Depersonalization or derealization
Numbness or tingling sensations (paresthesias) Flushes (hot flashes) or chills
Trembling or shaking Fear of dying
Fear of going crazy or doing something uncontrolled
![]()
activities to the point of becoming housebound for fear of having a panic attack. The multiple symptoms of panic attacks (see Table 5–6), including dizziness, are commonly reported by patients with agoraphobia. By contrast, simple phobias, such as fear of flying, heights, and snakes, are usually associated with generalized anxiety rather than panic episodes.
Phobic postural vertigo is characterized by a frightening feeling of dizziness with subjective postural and gait instability.35 Although patients have multiple symptoms of panic attacks, often with a steadily mounting fear of impending death, they feel physically ill and the associated symptoms of anxiety are brought out only after appropriate questions have been asked. They describe their dizziness as a perception of illu- sory body motion that can occur in brief bouts lasting seconds or be prolonged over hours and days. Typically patients have a fear of falling when sitting or standing, and active body move- ments provoke unpleasant illusions of body acceleration along with simultaneous illusory movement of a stationary environment. With the attack, patients experience anxiety, psycho- motor restlessness with escape reactions, a sudden desire to flee from the place where the attack is provoked, aimless walking, and, if seated, a rigid grasp of the arms of the chair. Anticipatory anxiety leads to further attacks of dizziness despite the discrepancy between the subjective fear of falling and the absence of objective unsteadiness. Although some patients develop typical symptoms of agoraphobia, oth- ers are able to continue their social and work habits despite symptoms they feel are dominat- ing their lives.34 The outlook for these patients
is good with most having improvement or even complete resolution of symptoms.36
Some patients develop a profound fear of falling in open spaces where a visuospatial reference is absent.37 Unlike the fear of public places found with agoraphobia, patients with space phobia fear open spaces where there is no “visual” support nearby. They will crawl on the floor to cross a room or walk close to walls or hedges in streets. The average age of onset of space phobia is later than that of agorapho- bia (55 years compared with 24 years), and the former is rarely associated with depression or free-floating anxiety, as typically seen with ago- raphobia. Marks suggested that many of these patients have an underlying organic disorder of balance, because they are resistant to the expo- sure treatments that are often successful with agoraphobics.37 A variant of space phobia (the so-called motorist’s disorientation syndrome) is an illusion of falling to the side or that the car is turning to the side when driving in open spaces, on featureless roads, or on the brows of hills.38 These abnormal sensations are typically accom- panied by a panic reaction. Typical of phobias, these patients develop avoidance behavior, either driving at very low speeds in restricted areas or completely stopping driving.
Chronic Anxiety
Unlike acute anxiety, chronic anxiety is often difficult to ascribe to a specific inciting factor.29 Symptoms are less intense although qualita- tively similar to those of acute anxiety. The patient may complain of dizziness and giddi- ness that persist for years, present from morn- ing to night. The patient appears tense and on edge, and there are often symptoms of associ- ated chronic depression. As with acute anxiety, there are typically associated somatic com- plaints, and on examination there may be several physical signs of chronic tension mani- fested by a fine tremor of the extended hands, very brisk deep tendon reflexes, chronic tachy- cardia, and pupillary dilatation.
Pathophysiology
The pathophysiologic mechanism of psy- chophysiologic dizziness is poorly understood. Although hyperventilation with its concomitant
cerebrovascular vasospasm can explain the presyncopal light-headed sensation, it cannot explain the many complex sensory distortions such as feelings of dissociation, illusions of body movement, imbalance, and fear of fall- ing.39 Abnormalities within the autonomic ner- vous system may account for some of these symptoms.40 In susceptible patients, panic attacks can be precipitated, by a large number of substances, including carbon dioxide, lac- tate, caffeine, isoproterenol, yohimbine, and benzodiazepine receptor antagonists.33 All these agents interact with the central nora- drenergic neuronal system. A popular hypoth- esis is that panic attacks result from central dyscontrol of the locus ceruleus, leading to the episodic release of catecholamines. Studies using positron-emission tomography (PET) scanning in patients with panic disorder dem- onstrated an asymmetry of blood flow and oxy- gen utilization in the parahippocampal gyrus (increased on the right side), 41,42 one of the major projection areas of the locus ceruleus. The parahippocampal region is closely interre- lated with the hippocampus, a key multimodal sensory integrative center that receives projec- tions from the association areas of all sensory modalities and projects to other limbic struc- tures and to autonomic centers in the hypo- thalamus and brain stem. Abnormalities in the hypothalamic orexin/hypocretin neuronal sys- tem may also explain the breathing and sleep dysfunction seen with panic disorder.43
Diagnosis and Management
The diagnosis of psychophysiologic dizziness rests on finding the characteristic associated symptoms of acute and chronic anxiety discussed previously. One must keep in mind that vestibular disorders can also cause anxiety and fear of further attacks of vertigo.44,45 A classic vicious cycle may develop whereby the vestibular disturbance causes anxiety, which in turn causes chronic dizziness that may persist after the vestibular imbalance has been compensated. A negative examination in the face of obvious signs of acute and chronic anxi- ety will help support the presumed diagnosis based on the history. It can sometimes be difficult to recognize panic attacks because patients will focus on the somatic symptoms, especially the dizziness and autonomic
symptoms, rather than the intense anxiety asso- ciated with the attack.
The first step in management of patients with psychophysiologic dizziness is to acknowl- edge their symptoms as “real,” due to physio- logic changes occurring in their bodies and that the pattern of symptoms is commonly reported by other patients. Patients are often convinced that they have a severe neurologic disorder and that the anxiety, which they have recognized, is secondary to the physical disorder. It is impor- tant to them that the physician understands that they are suffering from “physical symp- toms.” An explanation for how the release of catecholamines can produce symptoms such as tachycardia, chest pain, paresthesias, and dizzi- ness may improve their acceptance and provide the groundwork for therapeutic considerations. Three general classes of medications are com- monly used in the treatment of panic attacks:
(1) the tricyclic amines (e.g., imipramine and desipramine), (2) benzodiazepines (e.g., alpra- zolam or clonzapam), and (3) the selective serotonin reuptake inhibitors (SSRIs) (e.g., paroxetine and fluoxetine).31 The SSRIs typi- cally have an acceptable side effect profile and do not have the tolerance and dependency that occurs with benzodiazepines. These medica- tions should be used in conjunction with sup- portive psychotherapy. Patients with phobic dizziness may respond to cognitive behavioral therapy, although long-term benefit has not been demonstrated.46 We strongly encourage patients to enter a progressive exercise program with the goal of gradually improving their diminished physical fitness. It is very important that patients feel responsible for their therapy program.
A careful history of medications is critical in evaluating any patient complaining of dizziness (Table 5–7). Antihypertensive and antidepres- sant medications are common causes of near faint dizziness and falls in the elderly.47,48 Ototoxic drugs such as the aminoglycosides and cisplatin can cause vertigo if hair cell loss is asymmetrical, but more often they cause dis- equilibrium and oscillopsia from bilateral sym- metricalend-organdamage.49,50Carbamazepine, phenytoin, primidone, and alcohol can cause acute reversible disequilibrium and chronic irreversible disequilibrium from cerebellar dysfunction. Sedating drugs cause a nonspe- cific dizziness typically described as a fogginess, cloudiness, or giddiness that is presumably due to diffuse depression of the central nervous system. A number of commonly used drugs produce a characteristic drug intoxication syn- drome that might be confused with other types of dizziness. The associated confusion, disori- entation, memory and cognitive deficits, gaze- evoked nystagmus, and gait and extremity ataxia indicate combined cortical and cerebel- lar dysfunction.51 These drugs affect multiple neurotransmitters within the central nervous system (CNS), but the cause of the drug intoxi- cation syndrome is unknown.
The most commonly recognized syndrome is that associated with alcohol ingestion. A light- headed, swimming sensation is typically associ- ated with slowing of cognitive functions and motor responses. Gaze-evoked nystagmus with horizontal gaze deviation and gait ataxia are early reliable signs of the syndrome. With increased intoxication, gaze-evoked nystagmus occurs with
Table 5–7 Type and Mechanism of Dizziness Associated with Commonly Used Drugs
![]()
Drug Type of Dizziness Mechanism
![]()
Aminoglycosides, cisplatin Vertigo, disequilibrium Damage to vestibular hair cells
Antiepileptic: carbamazepine, phenytoin, primidone
Tranquilizers: barbiturates, benzodiazepines, tricyclic amines, marijuana
Disequilibrium, intoxication Cerebellar toxicity, CNS depression Intoxication CNS depression
Antihypertensives, diuretics Near-faint Postural hypotension, reduced cere- bral blood flow
Alcohol Intoxication, disequilibrium, positional vertigo
CNS depression, cerebellar toxicity, change in cupula-specific gravity
![]()
vertical gaze deviation, and the upper extremi- ties are ataxic. The alcohol concentration in the blood can be reasonably well predicted by the degree of gaze-evoked nystagmus.
The diagnosis rests on finding the character- istic combination of symptoms and signs in a patient taking one of the offending drugs. Blood levels can now be routinely obtained on most of these drugs, so a specific diagnosis is possible.
Hypoglycemia may lead to behavioral changes, light-headedness, lethargy, confusion, amnesia, seizures, weakness, shakiness, fatigue, and diaphoresis.52 It usually is a complication of insulin or sulfonylurea treatment in diabetic patients, but it may occur with insulinomas or as a fasting or postprandial phenomenon. Postprandial symptoms of shakiness, palpita- tions, fatigue, and dizziness have been termed functional hypoglycemia because most such cases are not associated with significantly low plasma glucose levels. The history in patients suspected of having hypoglycemia should focus on whether insulin or sulfonylu- reas have been taken. Diagnosis rests on mea- suring plasma glucose, insulin, and c-peptide. c-Peptide is the connecting peptide that is cleaved from proinsulin to form insulin.52 Elevation of c-peptide and insulin suggests excessive endogenous insulin (such as from an insulinoma). An elevated insulin level with nor- mal or suppressed c-peptide level indicates excessive exogenous insulin, since c-peptide is not present in pharmaceutical insulin and is in fact suppressed by it.
Common Causes
Patients often use the term “dizziness” to describe a sensation of imbalance or disequi- librium that occurs only when they are stand- ing or walking and is unrelated to an abnormal head sensation. Imbalance is common with acute unilateral peripheral vestibular lesions, but it is transient and invariably associated with
subjective vertigo. Both the vertigo and imbal- ance are compensated for within a few days. Patients who slowly lose vestibular function on one side, such as with an acoustic neuroma, may not experience vertigo but often describe a vague feeling of imbalance and unsteadiness on their feet. Bilateral symmetrical vestibular loss results in a more pronounced and persis- tent unsteadiness, which may be incapacitating in elderly patients.53,54 The imbalance due to loss of vestibulospinal and proprioceptive func- tion is typically worse in the dark, when the patient is unable to use vision to compensate for the loss (Fig. 5–1). Patients with a severe bilateral vestibular loss will report movement induced oscillopsia, often described as a feel- ing of looking through the lens of a video cam- era. Patients with cerebellar lesions, on the other hand, show little change in their balance with and without vision (the basis for the Romberg test). Disequilibrium may be the presenting symptom of lesions involving the motor centers of the basal ganglia and fron- tal lobes such as with Parkinson’s disease, hydrocephalus, and the multiple lacunar infarct syndrome.
Gait Disorders in Older People
The gradual loss of cells in the sensory and motor centers of the brain with aging is usually a very subtle process that parallels similar slight changes in memory and other cognitive functions, generally considered the normal aging process.55 The gait of normal elderly men is characterized by slight anteroflexion of the upper torso with flexion of the arms and knees, diminished arm swing, and shorter step length;56 the gait of older women tends to be more narrow based, with a waddling quality.57 When minor, these changes are not likely to lead to a specific medical complaint. However, a small number of older patients develop a progressive deterioration of gait, beginning in the eighth and ninth decades. Their steps shorten and the base widens until their gait is reduced to a shuffle. They turn en bloc rather than with a normal pivot, and upon arising they have great difficulty in initiating the first step. Once they begin, their arms are held rigidly at their sides, and they exhibit a characteristic stooped posture. Walking in tandem is impossible.
Unilateral peripheral vestibular lesion
Unilateral peripheral vestibular lesion
Associated with acute vertigo
Yes
No
Much worse in dark
Yes
Oscillopsia
+/– hearing loss
Yes
Cerebellar lesion
Cerebellar lesion
Yes
No
Incoordination of extremities
No
No
Bilateral vestibular lesion
Bilateral vestibular lesion
Associated numbness, weakness, bowel and bladder dysfunction
Yes
Proprioceptive loss
Proprioceptive loss
Slow, loss of associated movement
Yes
Frontal lobe, basal ganglia
Frontal lobe, basal ganglia
Figure 5–1. Logic for distinguishing between different causes of imbalance.
On examination, patients are unable to relax their limbs voluntarily. This phenomenon has been described as Gegenhalten or paratonic rigidity. Cortical release signs commonly accompany the diffuse rigidity. The patients attribute their difficulty in walking to a lack of confidence or a fear of falling, and, not surpris- ingly, major falls frequently occur. In the late stages, patients cannot walk unassisted and may have great difficulty sitting down from a stand- ing position. They land on the edge or side of the chair and fall off. Ultimately they are con- fined to bed. The neuropathologic basis of this gait disorder is poorly understood. Postmortem examinations typically show diffuse cortical atrophy and subcortical small-vessel ischemic changes.58–60
60, and it is greater in women than in men. Most falls in the elderly result from an acciden- tal slip or trip (Table 5–8). The cause can often be traced to decreased sensory input, slowing of responses, and weakness of support.53 Medications are a common contributing fac- tor.47 Falls can be directly traced to an acute attack of dizziness in less than 10% of patients.64 This low incidence probably can be attributed to the fact that most types of dizziness, includ- ing attacks of vertigo, begin slowly enough to
Table 5–8 Common Causes of Falls in Older People64
![]()
Cause %
Accidental (falling on stairs, slips, trips) 40–50
Falls in Older People
Falls in the elderly are a common source of morbidity and mortality.61–63 The risk of falls
Neurologic (drop attacks, weakness, ataxia)
Dizziness (orthostatic hypotension, arrhythmia, vertigo)
20–30
5–10
increases linearly with age beyond the age of
Uncertain 5–10
![]()
allow the patient to sit down or to grab onto a support to avoid falling.
Diagnosis and Management
Considering the many possible loci of dysfunc- tion, the examination of a patient complaining of disequilibrium must include a careful assess- ment of gait, strength, coordination, reflexes, and sensory function (particularly of the lower extremities).53,55 The broad-based ataxic gait of cerebellar disorders is readily distinguished from the milder gait disorders seen with ves- tibular or sensory loss. Furthermore, other cer- ebellar signs (e.g., dysmetria, dysarthria, inten- tion tremor, pathologically impaired smooth pursuit, and gaze-evoked nystagmus) usually accompany the gait ataxia. Bilateral vestibular loss may or may not be associated with hearing loss. The diagnosis rests on finding reduced vestibular function based on a positive head thrust test (bilateral) or decreased or absent response to caloric and rotational stimulation (see Chapter 7).
The deterioration of gait that occurs with aging must be distinguished from that associ- ated with lesions of the cortical and subcortical motor centers.55 The shuffling, flexed, steppage gait of Parkinson’s disease resembles the nor- mal gait of older males. The diagnosis of Parkinson’s disease rests on finding associated signs, including bradykinesia, cogwheel rigid- ity, and the characteristic “pill-rolling” tremor. Apraxia of gait, characterized by slow, halting, sliding steps as if the patient’s feet were adher- ing to the floor, is caused by bilateral frontal lobe dysfunction. Common causes include multiple subcortical infarcts, infiltrating tumors, and hydrocephalus. These abnormali- ties are easily identified with computerized tomography (CT) and MRI.
With the exception of normal pressure hydrocephalus, which can be dramatically reversed with placement of a shunt, most gait disorders in the elderly are not reversible. Some can be helped by improving support with canes or a walker. Optimizing footwear is very important.65 Tranquilizing medications and polypharmacy should be scrupulously avoided, inasmuch as they can further impair the central integration of sensory information.66 Physical therapy programs consisting of gait and balance training and strengthening
exercises help slow the progression. Exercise intervention has clearly been shown to reduce risk and rate of falls.67 The greatest benefit of exercise on fall rates was seen with programs that included challenging balance exercises and long duration (>50 hours).68
Common Causes
Many patients complain of a vague dizziness when they first wear glasses. They describe a feeling of disorientation, often accompanied by headache. The dizziness most frequently accompanies correction of astigmatism but also occurs after a change in magnification. It is nearly always mild and short-lived. A more per- sistent and distressing dizziness may occur in patients who are required to use high magnifi- cation or who have had a lens implant after cataract removal to correct severe visual loss. In these cases, the vestibulo-ocular reflex must adapt if visual objects are to be stabilized dur- ing head movements. This compensation pro- cess may be slow or inadequate in elderly patients or in subjects who require magnifica- tion so high that it is beyond the adaptive range of the vestibulo-ocular reflex.69 Multifocal glasses may predispose to dizziness and falls in older people.70
Dizziness also can result from an imbalance in the extraocular muscles. After an acute ocu- lar muscle paralysis, looking in the direction of the paralyzed muscle causes dizziness (in addi- tion to diplopia). This dizziness results from a mismatch between where the brain “thinks” the eye is, based on its efferent innervation, and where it actually is, based on the visual sig- nal.71 As with other types of ocular dizziness, the nervous system usually adapts to this altered spatial information and the dizziness is rarely severe or prolonged.
Oscillopsia
The optic illusion that stationary objects are moving back and forth or up and down is called oscillopsia. It is usually a sign of vestibular, brainstem, or cerebellar involvement, although rarely it can result from paralysis of the eye
Spontaneous nystagmus
Unilateral peripheral vestibular lesion
Unilateral peripheral vestibular lesion
No Yes
Bilateral peripheral vestibular or cerebellar lesion
Bilateral peripheral vestibular or cerebellar lesion
Yes
Head movement induced
No
Transient with vertigo
No
Yes
Visual association cortex
Central vestibular or cerebellar lesion
Visual association cortex
Central vestibular or cerebellar lesion
Figure 5–2. Logic for distinguishing between different causes of oscillopsia.
muscles or from a lesion in the visual associa- tion areas in the cortex (Fig. 5–2). Oscillopsia can be either a constant symptom or a move- ment-induced symptom. Not surprisingly, con- stant oscillopsia is associated with acquired spontaneous nystagmus. If a patient attempts to fixate on an object after an acute unilateral peripheral vestibular lesion, it will appear blurred and seem to be moving in the opposite direction of the slow phase of the spontaneous nystagmus. Some patients will report a flicking back and forth associated with the fast compo- nent of nystagmus. The oscillopsia associated with unilateral peripheral vestibular lesions is usually transient, disappearing as the acute ver- tigo and spontaneous nystagmus disappear. Patients with spontaneous nystagmus due to lesions of the central vestibular pathways report severe persistent oscillopsia, invariably associ- ated with other symptoms and signs of brain- stem dysfunction.
Oscillopsia that occurs only with head move- ment suggests some abnormality of the vestib- ulo-ocular reflexes. Patients with symmetrical loss of vestibulo-ocular reflex function (e.g., due to ototoxic drugs) are unable to fixate on objects when walking because the surround- ings appear to be bouncing up and down.72 The head oscillates in the vertical plane in the fre- quency range of 2 to 3 Hz. The visual pursuit system cannot compensate for the loss of ves- tibular function in this frequency range. In order to see the faces of passersby, patients learn to stop and hold their heads still. When reading, they learn to stabilize the head by
placing their hand on their chin to prevent even the slightest movements associated with pulsatile cerebral blood flow. Patients with cer- ebellar lesions cannot suppress their vestibulo- ocular reflex with fixation. They experience a brief sensation of oscillopsia after each rapid head movement, owing to a transient, unwanted vestibular nystagmus. These patients typically have gaze-evoked nystagmus on lateral or verti- cal gaze, so they may experience oscillopsia with both head and eye movements.
Management
Ocular dizziness due to changes in refraction is rarely severe and usually disappears spontane- ously as the patient adjusts to the altered visual environment. Patients should be encouraged to return to normal activities even though the diz- ziness is initially worse. In the long term this will accelerate the central compensation process. By contrast, oscillopsia is often a severe, persistent symptom that can be disabling. The most both- ersome type is that associated with an acquired central spontaneous nystagmus (see “Spontaneous Nystagmus” in Chapter 6). With one exception (Baclofen for periodic alternating nystagmus), drugs are not effective in suppress- ing oscillopsia due to central spontaneous nys- tagmus.73 If the nystagmus has a clearly defined null region, prisms fitted in glasses or eye mus- cle surgery can sometimes be helpful. In patients with head movement–induced oscillopsia due to bilateral peripheral vestibular loss, the visual
and neck ocular reflexes can compensate for the loss during low-frequency head movements but not during high-frequency movements.74,75 When walking, patients learn to stop and hold their head still to see clearly. Unlike the widely available devices used to augment an impaired auditory system, there are no current devices that augment the vestibular system. A number of devices that aim to improve the overall bal- ance function in patients with bilateral vestibul- opathy are in the development phase or early clinical testing phase.76–83 These devices gener- ally encode head or body movements into tactile cues that are delivered to the trunk and tongue.
Occasionally, one can trace dizziness to disease involving multiple sensory systems, particularly in elderly patients and in patients with systemic disorders, such as diabetes mellitus.84 A typical combination might include peripheral neurop- athy resulting in diminished touch and proprio- ceptive input, decreased visual acuity (cataracts, glaucoma), and impaired hearing (as in presby- cusis). In such patients an added vestibular impairment (from ototoxic drugs, for example) can be devastating, making it impossible for them to walk without assistance. Patients with multisensory dizziness may be unable to adapt to unfamiliar surroundings, such as in the hospital. Not infrequently, their complaint of dizziness will improve when they return to familiar surroundings at home.
Management
Treatment is directed at increasing sensory input wherever possible. This might include improved diabetic control, surgery for cataracts or glaucoma, amplification for presbycusis, and the use of a cane or walker to improve support and increase somatosensory signals. As with other balance disorders, sedating medications should be avoided.
Physiologic dizziness refers to a group of phe- nomena that occur in normal subjects with
physiologic stimulation of the vestibular, visual, or somatosensory systems. It typically results from a mismatch in sensory signals, resulting in a feeling of disorientation, imbalance, and veg- etative symptoms.
Motion Sickness
Motion sickness refers to the syndrome of diz- ziness, perspiration, nausea, vomiting, increased salivation, yawning, and generalized malaise induced by motion.85 It is usually produced by vestibular stimulation but also can occur with visual stimulation (e.g., with prolonged optokinetic stimulation). Both linear and angular head acceleration induce motion sick- ness if applied for long periods in susceptible subjects. Combinations of linear and angular acceleration or multiplanar angular accelera- tions are particularly effective. Rotation about the vertical axis, along with either voluntary or involuntary nodding movements in the sagittal plane, rapidly produce motion sickness in nearly everybody. This movement combines linear and angular acceleration (Coriolis effect).
Autonomic symptoms are usually the initial manifestation of motion sickness.86 Sensitive sweat detectors can identify increased sweating as soon as 5 sec after onset of motion, and grossly detectable sweating is usually apparent before any noticeable nausea. Increased saliva- tion and frequent swallowing movements occur early. Gastric motility is reduced and digestion is impaired. Hyperventilation is almost always present, and the resulting hypocapnia leads to changes in blood volume with pooling in the lower parts of the body, predisposing the sub- ject to postural hypotension. Motion sickness affects the appetite so that even the sight or smell of food is distressing.
Other symptoms of increased sensitivity may suggest a general hypersensitivity syndrome in these patients. Other hypersensitivity symp- toms include sensitivity to light, sound, and other pain syndromes, dry eyes, and irritable bowel syndrome.
Some people are sensitive to development of motion sickness, but others are highly resistant. Most will adapt to prolonged vestibular stimula- tion, whereas some never adapt (the chronically seasick ocean voyager). For unknown reasons, babies are highly resistant to motion sickness.
Unfortunately, there is no reliable way to predict who will develop motion sickness. Thresholds for vestibular stimulation (rotational or caloric) and the rate of habi- tuation to vestibular stimulation are no differ- ent in susceptible and resistant subjects.87 Patients whose labyrinths have been inacti- vated by congenital or acquired disease are resistant to motion sickness, whether induced by visual or vestibular stimuli. Such patients can withstand prolonged exposure to wave motion during a heavy storm at sea that would lead to motion sickness in even the most hard- ened seaman.
Genetic factors clearly are important in sus- ceptibility to motion sickness. Twin studies indicate heritability for motion sickness in the range of 55% to 70%.88 Families with migraine have increased sensitivity to developing motion sickness.89 A functional polymorphism in the alpha2-adrenergic receptor gene was signifi- cantly more common in subjects susceptible to motion sickness compared to those who were not sensitive to motion sickness.90 Since this receptor mediates central and peripheral auto- nomic responses, alleles that affect the level of expression could explain the varied autonomic responses to provocative motion. However, the findings need to be replicated in other populations.
Motion sickness seems to result from a visual–vestibular conflict.91 This theory is sup- ported by the fact that visual influences during body motion have a clear effect on the develop- ment of motion sickness. The symptoms are aggravated if one sits in an enclosed cabin on a ship or in the back seat of a moving vehicle. Because the environment is moving with the subject, visual–vestibular conflict occurs. The vestibular system signals movement while the visual system signals a stationary environ- ment. Motion sickness can be alleviated by improving the match between visual and ves- tibular signals. This can be accomplished on a ship by standing on deck and focusing on the distant horizon or on land, if possible. When riding in a car, the susceptible subject should sit in the front seat to allow ample peripheral vision of the stationary surround. Motion sick- ness suppressants such as scopolamine and dimenhydrinate are effective, presumably by diminishing activity at the vestibular nucleus and thereby diminishing the potential for visual–vestibular conflict.85,92
Space Sickness
Space sickness is a kind of motion sickness that is induced by active head movements in space.93,94 It has occurred in approximately 50% of the astronauts and cosmonauts who have entered space. Most adapt within 2 to 3 days. Because active head movements do not elicit motion sickness within the gravitational condi- tions on earth, the absence of gravity appears to be a key factor. The leading theory at pres- ent is that the symptoms are generated by a mismatch between otolith and semicircular canal signals as well as between otolith and visual signals.94 On earth, the semicircular canals and otoliths work together, sensing the angular and linear acceleration components of active head movements, but in space the oto- liths fail to signal orientation of the head in the absence of gravity. Thus, the afferent signals generated by head movements in space are dif- ferent from the signals expected from prior calibration on earth. The vestibular system must recalibrate to account for the absence of gravity; presumably this recalibration takes about 3 days. Supporting this notion, some astronauts develop transient motion sickness when they return to earth, although it is usually of shorter duration than in space.
Height Vertigo
Height vertigo refers to the subjective sensa- tion of instability and imbalance along with a fear of falling and vegetative symptoms that normal subjects experience in high places. More appropriate terms might be “height diz- ziness” or “height sickness,” inasmuch as there is usually no illusion of movement. Height ver- tigo occurs when the distance between the observer and visible stationary contrasting objects in the environment becomes critically large.95 Presumably, the normal lateral and fore–aft body sway sensed by the vestibular system conflicts with the visual information of no sway (the greater the distance between the eyes and the nearest stationary object, the smaller the angular displacement on the ret- ina). The symptoms can be reduced by having the subject sit or lie down to increase soma- tosensory input or by having a nearby station- ary object in the visual periphery, such as a railing or window frame. Some subjects develop
Mal de Debarquement Syndrome
Most of us have experienced the persistent rocking sensation after disembarking from a boat, particularly after a long voyage. This usu- ally subsides gradually over a few hours and seldom is of major significance. Rarely, patients report the persistent rocking sensation of a boat long after returning to land (months to years).97,98 These patients often report that their symptoms are less bothersome when they are in motion such as driving, swimming, or going back on a boat. The syndrome typically devel- ops in middle age and is more common in women than in men.98 Both motion-triggered and spontaneous episodes occur with the latter more common in patients with migraine. The cause is unknown but presumably it represents maladaptation within the central vestibular pathways.
SUMMARY: DISTINGUISHING BETWEEN VESTIBULAR AND NONVESTIBULAR TYPES OF DIZZINESS
Although the description alone does not distin- guish between vestibular and nonvestibular causes of dizziness, certain words are commonly
used to describe each type of dizziness (Table 5–9). A sensation of “spinning” nearly always indicates a vestibular disorder, particularly if it is moderate to severe in intensity and when the environment is seen moving (as opposed to an “internal” sensation). Patients with nonvestibu- lar dizziness occasionally will report a sensation of spinning inside the head, but the environ- ment remains still and they do not have nystag- mus. Patients with vestibular lesions often liken the sensation to that of being “drunk” or “motion sick.” They describe feelings of “imbalance,” as though they are “falling” or “tilting” to one side. Illusions of motion of the environment are rare but illusions of self-motion are common in patients with nonvestibular dizziness. These patients typically use terms such as “light- headed,” “floating,” “rocking,” “giddy,” or “swimming.” The sensation that one has left one’s body is characteristic of psychophysiologic dizziness.
Vertigo is an episodic phenomenon, whereas nonvestibular dizziness is often continuous. An exception would be presyncopal light-headed- ness caused by postural hypotension or cardiac arrhythmia. Patients with psychophysiologic dizziness often report being dizzy from morn- ing to night without changes for months to years at a time. Vertigo is typically aggravated by head movements, whereas nonvestibular dizziness is often aggravated by movement of visual targets. Episodes of dizziness induced by position change suggest a vestibular lesion if postural hypotension has been ruled out. Although stress can aggravate both vestibular and nonvestibular dizziness, dizziness that is reliably precipitated by stress suggests a non- vestibular cause. Finally, episodes of dizziness
Table 5–9 Distinguishing between Vestibular and Nonvestibular Types of Dizziness
![]()
![]()
Factor Vestibular Nonvestibular
Common descriptive terms
Spinning (environment moves), merry-go-round, drunkenness, tilting, motion sickness,
off-balance
Light-headed, floating, dissociated from body, swimming, giddy, “internal” spinning (environment stationary)
Course Episodic Constant
Common precipitating or aggravating factors
Commonly associated symptoms
Head movements, position change
Nausea, vomiting, unsteadiness, tinnitus, hearing loss, impaired vision, oscillopsia
Stress, hyperventilation, cardiac arrhythmias, situations
Paresthesias, syncope, difficulty concentrating, tension headache
![]()
Mathias CJ. Orthostatic hypotension: causes, mechanisms, and influencing factors. Neurology. 1995;45(suppl 5):S5.
Freeman R. Current pharmacologic treatment for
nonvestibular cause.
The presence of associated symptoms can also help one distinguish between vestibu- lar and nonvestibular causes of dizziness. Nausea and vomiting are usual with vertigo but uncommon with other types of dizziness. Associated auditory or neurologic symptoms suggest a vestibular disorder, presyncopal symptoms and syncope, a nonvestibular disor- der. Multiple symptoms of acute and chronic anxiety commonly accompany psychophysio- logic dizziness.
orthostatic hypotension. Clin Auton Res. 2008;18 (suppl 1):14–8.
Robertson D, Davis TL. Recent advances in the treatment of orthostatic hypotension. Neurology. 1995;45(suppl 5):S26.
Low PA, Singer W. Management of neurogenic orthostatic hypotension: an update. Lancet Neurol. 2008;7(5):451.
Marstrand JR, Garde E, Rostrup E, et al. Cerebral perfusion and cerebrovascular reactivity are reduced in white matter hyperintesities. Stroke. 2002;33: 972.
Low PA, Sandroni P, Joyner M, Shen WK. Postural tachycardia syndrome (POTS). J Cardiovasc Electrophysiol. 2009;20(3):352.
Kaufman H. Neurally mediated syncope: pathogen- esis, diagnosis, and treatment. Neurology. 1995;45
REFERENCES
(suppl 5):S12.
Bizios AS, Sheldon
RS. Vasovagal syncope: state or
Newman-Toker DE, Cannon LM, Stofferahn ME, Rothman RE, Hsieh YH, Zee DS. Imprecision in patient reports of dizziness symptom quality: a cross sectional study conducted in an acute care setting. Mayo Clin Proc. 2007;82:1329.
Baloh RW, Honrubia V, Jacobson K. Benign positional vertigo. Clinical and oculographic features in 240 cases. Neurology. 1987;37:371.
Grad A, Baloh RW. Vertigo of vascular origin. Clinical and oculographic features. Arch Neurol. 1989;46:281.
Weber PC, Adkins WY, Jr. The differential diagno- sis of Meniere’s disease. Otolaryngol Clin North Am. 1997;30:977.
Baloh RW. Clinical practice. Vestibular neuritis.
N Engl J Med. 2003;348(11):1027.
Fitzgerald DC, Getson P, Brasseux CO. Perilymphatic fistula: a Washington, DC, experience. Ann Otol Rhinol Laryngol. 1997;106:830.
Watson SRD, Halmagyi GM, Colebatch JG. Vestibular hypersensitivity to sound (Tulio Phenomenon). Neurology. 2000;54:722.
Kluge M, Beyenburg S, Fernández G, Elger CE. Epileptic vertigo: evidence for vestibular rep- resentation in human frontal cortex. Neurology. 2000;55(12):1906.
Best C, Eckhardt-Henn A, Tschan R, Dieterich M. Why do subjective vertigo and dizziness persist over one year after a vestibular vertigo syndrome? Ann NY Acad Sci. 2009;1164:334.
Nadol JB, Weiss AD, Parker. SW. Vertigo of delayed onset after sudden deafness. Ann Otol Rhinol Laryngol. 1975;84:841.
Cha YH, Lee H, Santell LS, Baloh RW. Association of benign recurrent vertigo and migraine in 208 patients. Cephalalgia. 2009;29(5):550.
Jen JC, Yue Q, Karrim J, Nelson SF, Baloh RW. Spinocerebellar ataxia type 6 with positional vertigo and acetazolamide-responsive episodic ataxia. J Neurol Neurosurg Psychiatry. 1998;65:565.
Weimer LH, Zadeh P. Neurological aspects of syn- cope and orthostatic intolerance. Med Clin North Am. 2009;93(2):427, ix.
trait? Curr Opin Cardiol. 2009;24(1):68.
Quinn J, McDermott D, Steill I, Kohn M, Wells G. Prospective validation of the San Francisco syncope rule to predict patients with serious outcomes. Ann Emerg Med. 2006;47:448.
Maganan GJ. Hyperventilation syndromes: infre- quently recognized common expressions of anxiety and stress. Medicine. 1982;61:219.
Ritz T, Wilhelm FH, Meuret AE, Gerlach AL, Roth WT. Do blood phobia patients hyperventilate during exposure by breathing faster, deeper, or both? Depress Anxiety. 2009;26(2):E60.
Bass C, Gardner WN. Respiratory and psychiatric abnormalities in chronic symptomatic hyperventila- tion. BMJ. 1985;290:1387.
Ruckenstein MJ, Staab JP. Chronic subjective dizzi- ness. Otolaryngol Clin North Am. 2009;42(1):71, ix.
Wiltink J, Tschan R, Michal M, et al. Dizziness: anxiety, health care utilization and health behavior— results from a representative German community sur- vey. J Psychosom Res. 2009;66(5):417.
Furman JM, Jacob R. Psychiatric dizziness. Neurology. 1997;48:1161.
Schiffer RB. Psychiatric disorders in medical practice. In: Goldman L, Ausiello D, eds. Cecil Textbook of Medicine. 23rd ed. Philadelphia: WB Saunders; 2008: 2628.
Johnson MR, Lydiard IB, Ballenger JC. Panic dis- order: pathophysiology and drug treatment. Drugs. 1995;49:328.
Gorman JM. A 28 year old woman with panic disorder.
JAMA. 2001;286:450.
Nardi AE, Freire RC, Zin WA. Panic disorder and control of breathing. Respir Physiol Neurobiol. 2009;167(1):133.
Ballenger JC. Biological aspects of panic disorder. Am J Psychiatry. 1986;143:516.
Jacob RG, Furman JM, Calaban CD. Psychiatricaspects of vestibular disorders. In: Baloh RW, Halmagyi GM, eds. Disorders of the Vestibular System. New York: Oxford University Press; 1996: 509.
Brandt T, Huppert D, Dieterich M. Phobic postural vertigo: a first follow-up. J Neurol. 1994;241:191.
Huppert D. Strupp M, Rettinger H, Hecht J, Brandt
T. Phobic postural vertigo – long term follow-up (5 to 15 years) of 106 patients. J Neurol. 2005;252: 564.
Marks I. Space “phobia”: a pseudo-agoraphobic syn- drome. J Neurol Neurosurg Psychiatry. 1981;44:387.
Page NGR, Gresty MA. Motorist’s vestibular disori- entation syndrome. J Neurol Neurosurg Psychiatry. 1985;48:729.
Garssen B, Buikhusen M, van Dyck R. Hyperventilation and panic attacks. Am J Psychiatry. 1996;153:513.
Staab JP, Ruckenstein MJ. Autonomic nervous sys- tem function in chronic dizziness. Otol Neurotol. 2007;28(6):854.
Reiman EM, Raichle ME, Robins E, et al. The appli- cation of positron emission tomography to the study of panic disorder. Am J Psychiatry. 1986;143:469.
Graeff F, Del-Ben CM. Neurobiology of panic dis- order: from animal models to brain neuroimaging. Neurosci Biobehav Rev. 2008;32:1326.
Williams RH, Burdakov D. Hypothalamic orexins/ hypocretins as regulators of breathing. Expert Rev Mol Med. 2008;10:e28.
Jacob RG, Furman JM, Durrant JD, Turner SM. Panic, agoraphobia, and vestibular dysfunction. Am J Psychiatry. 1996;153:503.
Asmundson GJ, Larsen DK, Stein MB. Panic disorder and vestibular disturbance: an overview of empirical findings and clinical implications. J Psychosom Res. 1998;44:107.
Holmberg J, Karlberg M, Harlacher U, Magnusson
M. One-year follow-up of cognitive behavioral therapy for phobic postural vertigo. J Neurol. 2007;254(9): 1189.
Rhalimi M, Helou R, Jaecker P. Medication use and increased risk of falls in hospitalized elderly patients: a retrospective, case-control study. Drugs Aging. 2009;26(10):847.
Darowski A, Chambers SA, Chambers DJ. Antidepressants and falls in the elderly. Drugs Aging. 2009;26(5):381.
Halmagyi GM, Fattore CM, Curthoys IS, et al. Gentamicin vestibulotoxicity. Otolaryngol Head Neck Surg. 1994;111:571.
Ishiyama G, Ishiyama A, Kerber K, Baloh RW. Gentamicin ototoxicity: clinical features and the effect on the human vestibulo-ocular reflex. Acta Otolaryngol. 2006 Oct;126(10):1057.
Gallagher BB, Baumel IP, Mattson RH, Woodbury SG. Primidone, diphenylhydantoin and phenobaribtal: aspects of cute and chronic toxicity. Neurology. 1973;23:145.
Service FJ. Hypoglycemia. Med Clin North Am. 1995;79:1.
Fife TD, Baloh RW. Disequilibrium of unknown cause in older people. Ann Neurol. 1993;43:694.
Agrawal Y, Carey JP, Della Santina CC, Schubert MC, Minor LB. Disorders of balance and vestibular function in US adults: data from the National Health and Nutrition Examination Survey, 2001-2004. Arch Intern Med. 2009;169(10):938.
Baloh RW. Disequilibrium and gait disorders in older people. Rev Clin Gerontol. 1996;6:41.
Murray MP, Kory RC, Clarkson BH. Walking patterns in healthy old men. J Gerontol. 1969;24:169.
Finley FR, Cody KA, Finizie RV. Locomotion pat- terns in elderly women. Arch Phys Med Rehabil. 1969;50:140.
Baloh RW, Vinters HV. White matter lesions and dis- equilibrium in older people: II. Clinical-pathological correlation. Arch Neurol. 1995;52:975.
Whitman GT, DiPatre PL, Lopez IA, et al. Neuropathology in older people with disequilibrium of unknown cause. Neurology. 1999;53:34.
Masdeu JC, Wolfson L. White matter lesions predis- pose to falls in older people. Stroke. 2009;40(9):e546.
Tinetti ME, Speechley M, Ginter SF. Risk factors for falls among elderly persons living in the community. N Engl J Med. 1988;319:1701.
Gribbin J, Hubbard R, Smith C, Gladman J, Lewis
S. Incidence and mortality of falls amongst older people in primary care in the United Kingdom. QJM. 2009;102(7):477.
Shumway-Cook A, Ciol MA, Hoffman J, Dudgeon BJ, Yorkston K, Chan L. Falls in the Medicare population: incidence, associated factors, and impact on health care. Phys Ther. 2009;89(4):324. ePub ahead of print, Feb 19, 2009.
Rubenstein LZ, Robbins AS, Schulman BL, et al. Falls and instability in the elderly. J Am Geriatr. 1988;36:266.
Menant JC, Steele JR, Menz HB, Munro BJ, Lord SR. Optimizing footwear for older people at risk of falls. J Rehabil Res Dev. 2008;45(8):1167.
Guideline for the prevention of falls in older people.
J Am Geriatr Soc. 2001;49:664.
Gillespie LD, Robertson MC, Gillespie WJ, et al. Interventions for preventing falls in older people liv- ing in the community. Cochrane Database Sys Rev. 2009;2:CD007146.
Sherrington C, Whitney JC, Lord SR, Herbert RD, Cumming RG, Close JC. Effective exercise for the prevention of falls: a systematic review and meta- analysis. J Am Geriatr Soc. 2008;56(12):2234.
Demer JL, Porter FI, Goldberg J, et al. Dynamic visual acuity with telescopic spectacles: improve- ment with adaptation. Invest Ophthalmol Vis Sci. 1988;29:1184.
Menant JC, St George RJ, Sandery B, Fitzpatrick RC, Lord SR. Older people contact more obstacles when wearing multifocal glasses and performing a secondary visual task. J Am Geriatr Soc. 2009;57:1833.
Brandt T Daroff R. The multisensory physiologi- cal and pathological vertigo syndromes. Ann Neurol. 1980;7:195.
Crawford J. Living without a balancing mechanism.
N Engl J Med. 1952;246:458.
Halmagyi GM, Rudge P, Gresty MA, et al. Treatment of periodic alternating nystagmus. Ann Neurol. 1980;8:609.
Chambers LTX, Mai M, Barber HO. Bilateral ves- tibular loss, oscillopsia, and the cervicoocular reflex. Otolaryngol Head Neck Surg. 1985;93:403.
Bronstein AM, Hood JD. Oscillopsia of periph- eral vestibular origin. Central and cervical com- pensatory mechanisms. Acta Otolaryngol (Stockh). 1987;104:307.
Wall C, III, Weinberg MS. Balance prostheses for postural control. IEEE Eng Med Biol Mag. 2003;22: 84.
Wall C, III, Kentala E. Control of Sway using vibrot- actile feedback of body tilt in patients with moder- ate and severe postural control deficits. J Vestib Res. 2005;15:313.
Peterka RJ, Wall C, III, Kentala E. Determining the effectiveness of a vibrotactile balance prosthesis. J Vestib Res. 2006;16:45.
Danilov YP, Tyler ME, Bach-y-Rita P. Spectral analy- sis of head based stabilogram in normal and bilateral vestibular subjects. J Vestib Res. 2004;14:126.
Danilov YP, Tyler ME, Bach-y-Rita P. Effects of electrotactile head-based feedback on subjects with bilateral vestibular dysfunction [abstract]. J Vestib Res. 2004;14:187.
Tyler ME, Danilov YP, Bach-y-Rita P. Closing an open-loop control system: vestibular substitution through the tongue. J Integr Neurosci. 2003;2:159.
Bach-y-Rita P, Kaczmarek KA, Tyler ME, Garcia- Lara J. Form perception with a 49-point electrotac- tile stimulus array on the tongue. J Rehabil Res Dev. 1998;35:472.
Goebel JA, Sinks BC, Parker BE, et al. Effectiveness of head-mounted vibritactile stimulation in subjects with bilateral vestibular loss: a phase 1 clinical trial. Otol Neurotol. 2009;30:210.
Gassmann KG, Rupprecht R, IZG Study Group. Dizziness in an older community dwelling popula- tion: a multifactorial syndrome. J Nutr Health Aging. 2009;13(3):278.
Shupak A, Gordon CR. Motion sickness: advances in pathogenesis, prediction, prevention, and treatment. Aviat Space Environ Med. 2006;77(12):1213.
Muth ER. Motion and space sickness: intestinal and autonomic correlates. Auton Neurosci. 2006;129 (1-2):58.
Golding JF. Motion sickness susceptibility. Auton Neurosci. 2006;129(1-2):67.
Reavley CM, Golding JF, Cherkas LF, Spector TD, MacGregor AJ. Genetic influences on motion sickness susceptibility in adult women: a classical twin study. Aviat Space Environ Med. 2006;77(11):1148.
Neuhauser H, Lempert T. Vestibular migraine. Neurol Clin. 2009;27(2):379.
Finley JC, Jr., O’Leary M, Wester D, et al. A genetic polymorphism of the alpha2-adrenergic recep- tor increases autonomic responses to stress. J Appl Physiol. 2004;96(6):2231.
Money KE. Motion sickness. Physiol Rev. 1970;50:1.
Spinks AB, Wasiak J, Villanueva EV, Bernath V. Scopolamine (hyoscine) for preventing and treat- ing motion sickness. Cochrane Database Sys Rev. 2007;3:CD002851.
Oman CM, Lichtenberg BK, Money KE, McCoy RK. MIT/Canadian vestibular experiments on the Spacelab-I mission 4. Space motion sickness: symp- toms, stimuli, and predictability. Exp Brain Res. 1986;64:316.
Lackner JR, Dizio P. Space motion sickness. Exp Brain Res. 2006;175(3):377.
Brandt T, Arnold F, Bless W, Kapteyn TS. The mech- anism of physiological height vertigo. 1. Theoretical approach and psychophysics. Acta Otolaryngol (Stockh). 1980;89:513.
Alpers GW, Adolph D. Exposure to heights in a theme park: fear, dizziness, and body sway. J Anxiety Disord. 2008;22(4):591.
Brown JJ, Baloh RW. Persistent mal de debarquement syndrome: a motion induced subjective disorder. Am J Otolaryngol. 1987;8:219.
Cha YH, Brodsky J, Ishiyama G, Sabatti C, Baloh RW. Clinical features and associated syndromes of mal de debarquement. J Neurol. 2008;255(7):1038.
This page intentionally left blank
![]()
Bedside Examination of the Vestibular
Fistula Test
TESTS OF VESTIBULOSPINAL REFLEXES
Pastpointing Static Posture Walking Tests
TESTS OF VESTIBULO-OCULAR REFLEXES
Doll’s Eye Test (Oculocephalic Response) Head-Thrust Test
Dynamic Visual Acuity Cold Caloric Test Rotational Testing
TESTS FOR PATHOLOGIC NYSTAGMUS
Methods of Examination
TYPES OF PATHOLOGIC NYSTAGMUS
Spontaneous Nystagmus Gaze-Evoked Nystagmus Positional Nystagmus Vibration-Induced Nystagmus Head-Shaking Nystagmus
Hyperventilation-Induced Nystagmus
OTHER OCULAR OSCILLATIONS
Dissociated Spontaneous Nystagmus Voluntary Ocular Oscillations (Voluntary
Nystagmus)
Convergence Retraction Nystagmus Saccadic Intrusions
Ocular Bobbing
Palato-Ocular Myoclonus
OCULAR TILT REACTION
As noted earlier, the vestibular system works in conjunction with the visual and somatosensory systems to achieve ocular and postural stability. To examine the vestibular system adequately, one must isolate it from these other sensory systems. Historically this has been a difficult task at the bedside, which is why most general textbooks only describe ways to assess the “audio” component of the audio-vestibular sys- tem or eighth cranial nerve. The contribution of the visual system can be removed with eye closure, but then the eye movements gener- ated by the vestibular system cannot be observed. There is no simple way to eliminate somatosensation.
Unlike the visual system, in which the optic nerve can be directly visualized and acuity accurately measured, the inner ear is located
deep within the temporal bone, and the subjec- tive sensation to vestibular stimulation is ill defined. One can visualize the tympanic mem- brane and some structures within the middle ear, but this generally provides no information about the status of the inner ear.
The neurotologist should be familiar with the normal anatomy of the external canal and tym- panic membrane (see Chapter 2), be capable of removing cerumen that interferes with visu- alization of the tympanic membrane, and be able to recognize certain common disorders on inspection (Fig. 6–1). Otoscopy is performed
149
Figure 6–1. Appearance of the tympanic membrane in a normal subject (A) and in patients with a superior marginal perforation and cholesteatoma (B), a tympanic glomus body tumor (C), and a step deformity caused by a longitudinal temporal bone fracture (D).
with the largest speculum that fits comfortably into the external canal: the pinna is gently pulled posterior and superior to straighten the canal. The tympanic membrane is normally translucent; changes in color indicate middle ear disease (e.g., an amber color with middle ear effusions). Tympanosclerosis, the conse- quence of a resolved otitis media or trauma, appears as a semicircular crescent or horse- shoe-shaped white plaque within the tympanic membrane. It is rarely associated with hearing loss but is an important clue to past otitic infections. The pars flaccida region, the area superior to the lateral process of the malleus, should be carefully inspected for evidence of a retraction pocket or attic cholesteatoma. The ossicles and the color of the underlying mucous membrane of the middle ear can often be assessed through a normal translucent tym- panic membrane. Pneumatoscopy allows one to determine the mobility of the tympanic membrane. Lack of mobility may indicate an unsuspected perforation (usually under an anterior overhang), fluid in the middle ear, or severe scarring of the tympanic membrane or middle ear.
Fistula Test
A fistula test is performed by transiently increasing and decreasing the pressure in the external canal with a pneumatoscope.1 A posi- tive fistula sign (a transient burst of nystagmus and vertigo) occurs in patients with a perfo- rated tympanic membrane and erosion of the bony labyrinth from conditions such as chronic infection, surgery, or trauma. The change in pressure is transmitted directly to the peri- lymph, compressing the membranous labyrinth and stimulating the semicircular canal cristae. The resulting nystagmus usually lasts from 10 to 20 sec. The direction of the nystagmus may be toward or away from the involved ear and is often the same for both positive and negative pressure changes.
Lesser ocular and subjective responses may occur in patients with an intact tympanic mem- brane. Hennebert 2 first described this sign in patients with congenital syphilis, but it can occur in patients with a wide range of labyrin- thine disorders. The response is a slow ocular deviation followed by a few beats of nystagmus even with sustained pressure. The abnormal
eye movements are in the plane of the affected semicircular canal. With semicircular canal dehiscence syndrome the pressure changes are transmitted to the abnormal canal due to the presence of a “third window” in the bony laby- rinth (see Fig. 16–3 in Chapter 16). In the case of endolymphatic hydrops (idiopathic or sec- ondary to infection), fibrous adhesions between the medial surface of the stapedial footplate and the membranous labyrinth could result in displacement of endolymph when the footplate moves.3 This sign can occasionally be elicited during routine pneumatoscopy in normal sub- jects. In these cases it is usually present bilater- ally. The mechanism by which pressure changes in the external auditory canal result in a pres- sure gradient across a normal vestibular recep- tor organs is unclear.
Fistulas of the labyrinth can also be tested for by inducing pressure changes by pressing and releasing the tragus (induces less pressure changes in the external canal than pneumatos- copy), Valsalva maneuver against closed glottis (increases intracranial pressure which can be transmitted to the inner ear by an intracranial fistula such as superior canal dehiscence), and Valsalva maneuver against pinched nostrils (increases pressure transmitted to the middle ear via the Eustachian tube).4
TESTS OF VESTIBULOSPINAL REFLEXES
As discussed in Chapter 3, the labyrinths influ- ence spinal cord motoneurons through the lat- eral vestibulospinal tract, the reticulospinal tract, and the descending medial longitudinal fasciculus (MLF). Labyrinthine stimulation of the spinal cord increases extensor tone and decreases flexor tone, resulting in a facilitation of the antigravity muscles. Both otolith and semicircular canal signals influence spinal cord anterior horn cells, but the former are more important in maintaining posture.
Pastpointing
Pastpointing refers to a reactive deviation of the extremities caused by an imbalance in the vestibular system (Fig. 6–2). The test is per- formed by having patients place their extended index finger on that of the examiner’s, close their eyes, raise the extended arm and index finger to a vertical position, and attempt to return the index finger to the examiner’s. Consistent deviation to one side is pastpointing (See Video 6–1).

Past Pointing
2 Test
3 1
Romberg Test


Tandem Walking
Figure 6–2. Bedside tests of vestibulospinal function.
Pastpointing tests represent one of the earli- est attempts to clinically assess vestibular func- tion. In 1910, Bárány5 published a review of pointing deviation and emphasized the impor- tance of having patients sit with their eyes closed to avoid confusion with other orienting information. Bárány showed that caloric stimu- lation consistently induced pastpointing in the direction of the slow component of induced nystagmus. Cold caloric irrigation (inhibiting the horizontal ampullary nerves’ spontaneous firing rate) resulted in pastpointing toward the irrigated ear, and warm caloric irrigation induced the opposite effect. As expected, patients with acute unilateral loss of vestibular function past-pointed toward the damaged side. Bárány and numerous others emphasized that repeated testing shows a large variability, occasionally with drift in the wrong direction. Subsequent investigators tried to improve test accuracy by eliminating tactile feedback and using small finger lamps that could be photo- graphed, but the large variability among nor- mal subjects and patients remained.
It is apparent that results from a single point- ing test can be misleading and should not be considered clinically relevant when in isolation. Extralabyrinthine influences should be elimi- nated as much as possible by having the patient seated with eyes covered and arms and index fingers extended throughout the test. The standard finger-to-nose test will not identify pastpointing, inasmuch as joint and muscle proprioceptive signals permit accurate localiza- tion even when vestibular tone is asymmetric. Although patients with acute peripheral ves- tibular damage usually past-point toward the side of loss, compensation rapidly corrects the pastpointing and can even produce a drift to the other side. The cortical and subcortical pathways to the spinal anterior horn cells, illus- trated in Figure 1–14 in Chapter 1, apparently account for the compensation.
Static Posture
Patients with damage to the vestibular system often suffer instability of the trunk and lower limbs so that they sway back and forth or even fall to one side. In 1846, Romberg6 noted that patients with proprioceptive loss from tabes dorsalis were unable to stand with feet together and eyes closed. Bárány7 first emphasized the
importance of vestibular influences in main- taining the Romberg position (Fig. 6–2). As with pastpointing, he noted that patients with acute unilateral labyrinthine lesions swayed and fell toward the diseased side, that is, in the direction of the slow component of nystagmus. However, like the pastpointing test, the Romberg test was found to be rather insensi- tive for detecting chronic unilateral vestibular impairment, and sometimes the patient would fall toward the intact ear. The so-called sharp- ened Romberg test is a more sensitive indica- tor of vestibular impairment. For this test the patient stands with feet aligned in the tandem heel-to-toe position with eyes closed and arms folded against the chest. Most normal subjects under the age of 70 can stand in this position for 30 sec: older normal subjects and patients with unilateral or bilateral vestibular impair- ment usually cannot sustain the position. Abnormalities on this test, however, are not specific to the vestibular system.
Although lower mammals consistently develop ipsilateral hypotonia of extensor mus- cles after labyrinthectomy, one rarely finds this in human patients. Occasionally, slight asym- metry in posture is found with the ipsilateral upper extremity slightly flexed and abducted compared to the contralateral upper extremity. The clinically elicited deep tendon reflexes are also unaffected by vestibular lesions. Apparently, other supraspinal influences on the anterior horn cells rapidly compensate for the loss of tonic vestibular signals.
Walking Tests
Unterberger8 was the first to systematically study the tendency of vestibular stimulation or unilateral vestibular lesions to induce blind- folded subjects to turn in the earth’s vertical axis when walking. The direction of turning coincided with the direction of pastpointing and falling (in the direction of the slow compo- nent of nystagmus). Fukuda9 obtained similar results by having subjects take 50 to 100 steps on the same spot and recording the angle of rotation as well as forward and backward move- ments. Both of these tests were performed with arms extended parallel and horizontal in front of the subject, so upper extremity deviation (pastpointing) may have added to the tendency to rotate in a given direction. Tandem gait tests
(Fig. 6–2) are widely used as part of the routine neurologic examination, and most clinicians recognize normal and abnormal performances. When performed with eyes open, tandem walk- ing is primarily a test of cerebellar function, because vision compensates for chronic vestibular and proprioceptive deficits. Acute vestibular lesions, however, typically impair tandem walking, even with the eyes open.
Recently, there has been an emphasis on timed walking tests to provide a semiquantita- tive measure of balance and risk of falling in older patients. These tests are not specific for vestibular function but rather provide an over- all measure of gait and balance. Gait speed (over a set path), timed tandem stance and walking, and maximum step length (ability to maximally step out and return to the initial position) have all been shown to predict the likelihood of falls in older people.10,11
TESTS OF VESTIBULO-OCULAR REFLEXES
Doll’s Eye Test (Oculocephalic Response)
The doll’s eye test involves slowly moving the head back and forth in the horizontal plan to induce reflex eye movements. In an alert human these eye movements result from com- bined visual and vestibular stimulation. Therefore, a patient with complete loss of ves- tibular function will have compensatory eye movements on the test if the visuomotor sys- tem is intact. On the other hand, an alert human can also use the visuomotor system (i.e., the smooth pursuit system) to overcome or sup- press the vestibular eye movement in this test that uses slow passive movements of the head. In a comatose patient, however, the doll’s eye test is a useful bedside test of the vestibulo- ocular reflex since the pursuit system is not functioning.12 Conjugate compensatory eye movements indicate normally functioning vestibulo-ocular pathways (those shown in Fig. 3–8 in Chapter 3). This test is a standard component of the coma exam and also brain death exam because the reflex eye movements indicate intact function not only of the inner ear but also the brain stem. Absence of reflex
eye movements in a comatose patient is usually an ominous sign, indicating massive brain stem damage if acute drug intoxication or metabolic disorders can be ruled out.13,14
The doll’s eye test can also provide highly localizing information. For example, if the test results in disconjugate eye movements, then one could localize the lesion to MLF, the ocul- omotor neurons, or the ocular muscles (depend- ing on the abnormal pattern).
Head-Thrust Test
The head-thrust test is a simple way to identify a complete unilateral or bilateral loss of ves- tibular function at the bedside.15 The discovery of this test was a major breakthrough in the ability to examine the vestibular system at the bedside. Prior to the head-thrust test, the bed- side assessment was mostly limited to search- ing for surrogate signs of an acute vestibular imbalance (i.e., nystagmus). Tests such as past- pointing, the Unterberger test, and the doll’s eye test could be used, but these had major limitations as noted previously.
The head-thrust test is used to directly assess for vestibular de-afferentation and thus is anal- ogous to the test for an afferent pupillary defect (i.e., the swinging light test). Though the affer- ent pupillary defect was first reported around 1900,16 the head-thrust test was not reported until 1988.15 Despite the consensus opinion among neurotologists regarding the substantial value of the head-thrust test, it remains a test that is underappreciated and underutilized in the general medical community.
The head-thrust test is performed by grasp- ing the patient’s head and applying brief, small- amplitude, high-acceleration head thrusts, first to one side and then to the other (Fig. 6–3, See Video 6–2 and Video 6–3). Before the move- ment, the patient is instructed to fixate on the examiner’s nose. During and after the quick movements, the examiner closely watches the patient’s eye position looking specifically for “catch-up” saccades, which are the sign of an inappropriate compensatory vestibular system response. If the vestibular system is intact on both sides, then movement to either side trig- gers the eye movements (the eye movements are triggered by a reflex, the vestibulo-ocular reflex) that keep the patient’s eyes fixated on the examiner’s nose. On the other hand, if a


(a) 20°


Fixed target
![]()



(b) 20°
![]()



![]()
Figure 6–3. The head thrust test. The head thrust test is a test of vestibular function that can be easily done during the bedside examination. This maneuver tests the vestibulo-ocular reflex (VOR). The patient sits in front of the examiner and the examiner holds the patient’s head steady in the midline. The patient is instructed to maintain gaze on the nose of the examiner. The examiner then quickly turns the patients head about 10-15 degrees to one side and observes the ability of the patient to keep the eyes locked on the examiner’s nose. If the patient’s eyes stay locked on the examiner’s nose (i.e., no corrective saccade) (picture a), then the peripheral vestibular system is assumed to be intact. If, however, the patient’s eyes move with the head (picture b) and then the patient makes a voluntary eye movement back to the examiner’s nose (i.e., corrective saccade), then this indicates a lesion of the peripheral vestibular system and not the central nervous system. Thus, when a patient presents with the acute vestibular syndrome, the test result shown in picture A would suggest a CNS lesion (because the VOR is intact), whereas the test result in picture B would suggest a peripheral vestibular lesion (because the VOR is not intact).
With permission from: Edlow JA, Newman-Toker DE, Savitz SI. Lancet Neurology. 2008; 7:951–964.
The passive head movement is fast enough (high enough acceleration) that the smooth pursuit system cannot be used to keep the patient’s eyes fixated on the examiner’s nose (recall how the smooth pursuit system can cover up for vestibular impairment with the low- velocity and low-frequency movements of the doll’s eye test). In addition, the patient also can- not initiate a voluntary saccade fast enough to keep the eye’s on the nose and for this reason a “catch-up” saccade is required to move the eye back to the nose. Of course, if a patient can cor- rectly predict the direction of the head move- ment and then time the initiation of a saccade accurately, then a voluntary saccade can come very close to covering up for a vestibular lesion. In the same way that the afferent pupillary defect is mostly a test of the optic nerve func- tion, the head-thrust test is mostly a test of the vestibular nerve function. Intact end-organ (e.g., labyrinthine) function is of course required to generate the vestibulo-ocular reflex but a disorder of the end-organ may not result in a positive test unless it is severe or complete. Similarly, retinal lesions, unless severe, do not typically result in an afferent pupillary defect. For this reason, the head-thrust test is typically not positive in patients with Meniere’s disease unless a procedural lesion has been per- formed.17 In addition, the accuracy of the head- thrust test is also influenced by the severity of the nerve lesion. For example, the head-thrust test may not be positive if the vestibular nerve lesion is only mild to moderate in severity (i.e., resulting in approximately less than a 50% paresis) but is nearly always positive with lesions that are moderate to severe.18,19 It is not completely fair to gauge the value of the quali- tative positive versus negative results of the head-thrust test to the quantitative results of the caloric test. The reason is because the head thrust is a test of the vestibular system at high acceleration, whereas the caloric test is a test of
the vestibular system at a very low acceleration. Furthermore, the caloric test, though generally regarded as the gold standard test for a unilat- eral vestibular lesion, is known to have con- cerning levels of false-positive results based on the limitations of the test and thus is not an optimal gold standard test.20
The head-thrust test is nearly always positive in patients with acute vestibular neuritis because the lesion is of the vestibular nerve and is typically at least a moderate to severe lesion.21,22 For this reason, the head thrust is particularly valuable in the presentation of the acute vestibular syndrome because a positive head-thrust test is a very strong indicator of a lesion of the vestibular nerve (and thus the most common cause, vestibular neuritis). A negative head-thrust test in the acute vestibu- lar syndrome suggests the vestibular nerve is intact and thus the likelihood of a a brainstem or cerebellar lesion increases.21,22
The head-thrust test is also particularly help- ful for identifying a bilateral vestibulopathy. A bilateral vestibulopathy can be difficult to rec- ognize based on the symptom report and gen- eral examination. As a result, many patients likely go undiagnosed. However, a positive bilateral head-thrust test can clinch the diagno- sis at the bedside.
Dynamic Visual Acuity
The dynamic visual acuity test is performed by having the patient shake the head rapidly back and forth in the horizontal plane at approxi- mately 2 Hz while reading a Snellen visual acu- ity chart at the standard distance.23,24 Inasmuch as the smooth pursuit system functions best below 1 Hz and almost not at all at 2 Hz, this is primarily a test of the horizontal vestibulo- ocular reflex. A drop in acuity of more than one line on the Snellen chart suggests an abnormal vestibulo-ocular reflex. The test is most useful for identifying bilateral vestibular loss but can also be abnormal with unilateral vestibular loss and with cerebellar lesions.25,26
Cold Caloric Test
Because of its ready availability, ice water (approximately 0°C) is commonly used for bedside caloric testing.27 Tap water can be
brought to 4°C in about 10 minutes after add- ing ice cubes. To bring the horizontal canal into the vertical plane, the patient lies in the supine position with the head tilted 30 degrees forward or in the sitting position with the head tilted 60 degrees backward (see Fig. 1–4C in Chapter 1). Infusion of ice water induces a burst of nystagmus with slow phase toward the side of infusion and the fast phase in the oppo- site direction, usually lasting from 1 to 3 min. The volume of ice water recommended for this test varies from 0.5 to 2 ml. Regardless of the volume used, however, it is critical that the stimulus reach the eardrum (i.e., not be injected into the canal wall or in a canal blocked by cerumen). Direct visualization of the eardrum is mandatory. We suggest using 1 or 2 ml of ice water infused directly against the tympanic membrane through a small rubber hose. The ear being infused is turned upward for approx- imately 30 sec after the infusion to be certain that the water stays against the drum. In an alert subject, a burst of nystagmus will develop within 30 sec to 1 min after infusion and last from 1 to 3 min. In a comatose patient, only a slow tonic deviation toward the side of stimula- tion is observed because the brain does not generate the fast phases. In normal subjects, duration and speed of induced nystagmus var- ies greatly, but >20% asymmetry in nystagmus duration suggests the possibility of a lesion on the side of the decreased response, though this test is less valid than standard bithermal caloric testing with nystagmography (see Bithermal Caloric Test in Chapter 7).
In terms of the bedside vestibular evaluation in an alert subject, the use of the cold caloric test has largely been replaced by the head- thrust test.
Rotational Testing
Qualitative rotational testing of the horizontal vestibulo-ocular reflex can be performed at the bedside by using a swivel chair. Bárány7 intro- duced a rotatory test in which the chair on which the patient was seated was manually rotated 10 times in 20 sec and then suddenly stopped with the patient facing the observer. The duration of postrotatory nystagmus in each direction was then measured. In normal subjects an average of 22 sec was required for cessation of postrota-
was large. Much of this variability could be traced to the difficulty in manually maintaining constant velocity and then a uniform sudden deceleration. Furthermore, the vestibular response to the initial acceleration was often not completed before deceleration began, resulting in interaction between the two responses. As with the ice water caloric testing, this type of qualitative testing can provide only gross information about the presence and sym- metry of vestibular function, and thus it is not felt to be as valuable as the head-thrust test. One aspect of rotational testing that is useful at the bedside is the fixation-suppression test. With this test the subject extends the arm rig- idly and attempts to fixate on the extended thumb while the entire body is rotated back and forth en bloc. Normal subjects can completely suppress their vestibulo-ocular reflex, keeping their eyes fixed in the center of the orbits (as shown in Fig. 1–9 in Chapter 1). Abnormal fixation-suppression (nystagmus) indicates impairment of the smooth pursuit systems and thus is an indicator of a central lesion, often involving the cerebellum (see “Visual-Vestibular Interaction” in Chapter 7).
TESTS FOR PATHOLOGIC NYSTAGMUS
Nystagmus can be defined as a nonvoluntary rhythmic oscillation of the eyes. It usually has clearly defined fast and slow components alter- nating in opposite directions. By convention, the direction of the fast component defines the direction of nystagmus (e.g., left-beating nys- tagmus indicates the fast phase is to the left). Physiologic nystagmus refers to nystagmus that occurs in normal subjects, whereas pathologic nystagmus implies an underlying abnormality (Table 6–1). Spontaneous nystagmus refers to nystagmus that occurs with the eyes in the pri- mary position, without external stimulation
Table 6–1 Types of Nystagmus
![]()
Physiologic Pathologic
![]()
Rotational-induced Spontaneous
Caloric induced Gaze-evoked
Optokinetic Positional
End-point
tory nystagmus, but intersubject variability
such as movement of the head or surroundings. Gaze-evoked nystagmus is induced by changes in gaze position. Nystagmus that is not present in the sitting position but is present in some other head and body position is called posi- tional nystagmus. This definition excludes nys- tagmus present in the sitting position that is modified by a change in position.
Methods of Examination
The clinical examination for pathologic nystag- mus should include a systematic study of changes in (1) fixation, (2) eye position, and (3) head position. Omission of any of these three maneuvers may lead to overlooking the pres- ence of nystagmus or misinterpreting its type. Sometimes pathologic nystagmus can be trig- gered by vibration, head shaking, or hyperven- tilation.
Spontaneous nystagmus may be present with fixation, or it may occur only when fixation is inhibited (See Video 6–4). There are several simple methods for achieving the latter at the bedside. Frenzel glasses consist of +30 lenses mounted in a frame that contains a light source on the inside so that the patient’s eyes are eas- ily visualized (Fig. 6–4). The light can be pow- ered by a battery, making the entire system portable. Frenzel glasses should be used only in a darkened room, because the patient can fixate (at least partially) through the lenses in a lighted room. An ophthalmoscope can also be used to block fixation and bring out a
spontaneous nystagmus. While the fundus of one eye is being visualized the patient is asked to lightly cover the other eye with one hand. Nystagmus appears as a slow drift of the retina in one direction interrupted by flicking move- ments in the opposite direction (the direction of the nystagmus is reversed, inasmuch as one is visualizing the back pole of the eye). Alternatively, one can simply shine a penlight in one eye while intermittently occluding the other eye.28 In some cases, simply holding a blank sheet of paper in front of the patient’s vision and asking the patient to stare through it is enough to bring out the spontaneous nystag- mus. When this is done, the examiner has to look at the patient’s eyes from the side. Occasionally, nystagmus can be seen even through closed lids. This can be misleading, however, because lid-twitch movements often mimic nystagmus.
The effect of change in eye position is evalu- ated by having the patient fixate on a target 30 degrees to the right, left, up, and down. Because horizontal eye deviation beyond 40 degrees may result in a low-amplitude, high-frequency torsional nystagmus in normal subjects (so- called end-point or end-gaze nystagmus), extreme eye positions should be avoided. Each eye position is held for at least 20 sec. First- degree nystagmus refers to nystagmus that is present only on gaze in the direction of the fast component. Second-degree nystagmus is pres- ent in the midposition (primary position) and on gaze in the direction of the fast component, and third-degree nystagmus is present even on

Figure 6–4. Frenzel glasses.
| ||
|
|
|
|
| ||
|
| |
– |
| ||
|
|
|
|
| ||
|
| |
– |
Peripheral Spontaneous Symmetric Gaze-evoked
| ||
|
|
|
|
– | ||
|
|
|
|
| ||
|
|
|
|
– | ||
|
|
|
|
Congenital Spontaneous Downbeat Spontaneous
Figure 6–5. Method for describing the effect of eye position on nystagmus amplitude and direction. Arrows indicate direc- tion of nystagmus (direction of fast component) in each eye position.
gaze away from the fast component. These terms are not applicable to all varieties of nys- tagmus and, therefore, can lead to confusion. A simple description can be rapidly summarized with a box diagram as illustrated in Figure 6–5. The size, shape, and direction of the arrows provide information about the amplitude and direction of the fast component of nystagmus in each eye position.
Routinely, two types of positional testing are used: slow and rapid. With the first, the patient slowly moves into the supine, right lateral, and left lateral positions. Positional nystagmus induced by slow positioning is persistent, low in frequency, and often present only when fixa- tion is inhibited. Paroxysmal positional nystag- mus, however, is induced by a rapid change from the erect sitting to the supine head- hanging left (left Dix-Hallpike test), center, or right position (right Dix-Hallpike test) or from the supine to head-right or head-left position (supine positional testing). It is initially high frequency but rapidly dissipates within 30 sec to 1 min (see Chapter 10).
For vibration-induced nystagmus, a hand- held vibrator (approximately 100 Hz) is placed on the mastoid and suboccipital bones on each side for 10 to 15 seconds. The test for head- shaking nystagmus is performed by having the
patient shake the head back and forth in the horizontal and vertical planes for approximately 10 cycles. For hyperventilation-induced nys- tagmus, the patient breathes rapidly in and out for about a minute and a half.
Spontaneous Nystagmus
MECHANISM
Spontaneous nystagmus results from an imbal- ance of tonic signals arriving at the oculomotor neurons. Because the vestibular system is the main source of oculomotor tonus, it is the driv- ing force of most types of spontaneous nystag- mus (tonic signals arising in the pursuit and optokinetic systems may also play a role, par- ticularly with congenital nystagmus).29 A ves- tibular imbalance results in a constant drift of the eyes in one direction (slow phase) inter- rupted by fast components in the opposite direction. If the imbalance results from a peripheral vestibular lesion, patients can use their pursuit system to cancel it. If it results from a central vestibular lesion, their pursuit
Table 6–2 Differentiation between Spontaneous Nystagmus of Peripheral and Central Origin
![]()
![]()
Appearance Fixation Gaze Mechanism Localization
Peripheral Combined
horizontal, torsional
Inhibited Unidirectional
(Alexander’s law)
Asymmetric loss of peripheral vestibular tone
Labyrinthine
or vestibular nerve
Central Often pure vertical, horizontal, or torsional
Usually little effect
May change direction
Imbalance in central oculomotor tone; usually central vestibular, may be pursuit or OKN
CNS, usually brain stem or cerebellum
![]()
CNS, central nervous system; OKN, optokinetic nystagmus
system usually cannot suppress it (because cen- tral vestibular and pursuit pathways are highly integrated; see “Visual–Vestibular Interaction” in Chapter 3). The features that separate peripheral from central varieties of spontane- ous nystagmus are summarized in Table 6–2.
PERIPHERAL SPONTANEOUS NYSTAGMUS
Lesions of the peripheral vestibular system (labyrinth or eighth nerve) typically interrupt tonic afferent signals originating from all of the receptors of one labyrinth so that the resulting nystagmus has combined torsional, horizontal, and vertical components. The hori- zontal component dominates, because the tonic activity from the intact vertical canals and otoliths partially cancels out. Gaze in the direction of the fast component increases the frequency and amplitude, whereas gaze in the opposite direction decreases the frequency and amplitude (Alexander’s law) (See Video 6–4, Video 6–5, and Video 6–6).30 Peripheral spon- taneous nystagmus is “unidirectional” meaning that the primary direction does not change. Thus, if the primary position nystagmus is right-beating, then it will not change to left- beating nystagmus even on left gaze. The slow phase is linear, resulting in a saw-toothed wave- form. As noted previously, peripheral sponta- neous nystagmus is strongly inhibited by fixation. Unless the patient is seen within a few days of the acute episode, spontaneous nystag- mus will not be present when fixation is permitted.
CENTRAL SPONTANEOUS NYSTAGMUS
Central spontaneous nystagmus is usually prominent with and without fixation. It may be purely vertical, horizontal, or torsional, or have some combination of torsional and linear components (See Video Video 6–7 and Video 6–8). As with peripheral spontaneous nystag- mus, gaze in the direction of the fast compo- nent usually increases nystagmus frequency and amplitude, but unlike peripheral sponta- neous nystagmus, gaze away from the direction of the fast component will often change the direction of the nystagmus (i.e., direction changing nystagmus). There is typically a null region several degrees off center in the direc- tion opposite that of the fast component where nystagmus is minimal or absent. Gaze beyond this null region results in reversal of nystagmus direction. The slow phase of central spontane- ous nystagmus may be linear, exponentially increasing, or exponentially decreasing.31
With spontaneous downbeat nystagmus the vertical amplitude increases with horizontal gaze deviation.32 Downward gaze also increases the amplitude in about two-thirds of cases, but in the other one-third it decreases it. Upward gaze may reverse the direction to upbeat. Downbeat nystagmus has been produced in monkeys after lesions of the uvula and floccu- lonodular lobes of the cerebellum.33 In the human it is localizing to the cervicomedullary junction (which includes the midline cerebel- lar regions). Common causes of downbeat nystagmus include cerebellar atrophy, verte- brobasilar ischemia, multiple sclerosis, and Arnold-Chiari malformation.34,35 The latter
produces downbeat nystagmus, presumably by pressure on the flocculonodular region of the cerebellum; in some cases it may be reversed by decompression of the foramen magnum region.36 Spontaneous upbeat nystagmus usu- ally results from lesions of the dorsal central medulla in the region of the medial vestibular and prepositus hypoglossi nuclei.35,37 Common causes include infarction, infiltrating tumors, and multiple sclerosis. Pure torsional sponta- neous nystagmus is frequently associated with syringomyelia and syringobulbia. A high- frequency, small-amplitude pendular sponta- neous nystagmus is commonly seen in the late stages of multiple sclerosis.38 This pendular nystagmus converts to a sawtooth pattern on lateral gaze to either side. Lesions involving the vestibular nuclear region can produce a horizontal torsional nystagmus similar to that seen with peripheral lesions, but, unlike the latter, the direction of nystagmus does not reliably indicate the side of lesion and the nystagmus persists with fixation because of damage of visual–vestibular interaction pathways.39
Central spontaneous nystagmus has gener- ally been attributed to an imbalance in either central vestibulo-ocular or smooth pursuit pathways.32 Horizontal and vertical pathways are usually separate so that lesions can com- monly lead to pure horizontal or pure vertical nystagmus. Often central spontaneous nystag- mus is altered by position change, suggesting that peripheral otolith input can alter the cen- tral imbalance. Marti and colleagues suggested that downbeat nystagmus results from damage to the inhibitory vertical gaze-velocity sensitive Purkinje cells in the cerebellar flocculus.40 These neurons have spontaneous activity and most exhibit downward on-directions. Loss of these vertical flocculus Purkinje cells would lead to disinhibition of their brainstem target neurons and spontaneous upward drift (i.e., downbeat nystagmus).
CONGENITAL NYSTAGMUS
Congenital spontaneous nystagmus is almost always highly dependent on fixation, disappear- ing or decreasing with loss of fixation.41 In some instances a slow nystagmus in the reverse direc- tion occurs when fixation is inhibited. One common variety, so-called latent congenital
nystagmus, occurs only when either eye is covered, permitting monocular fixation. The resulting nystagmus beats toward the fixating eye. Latent congenital nystagmus is usually associated with other congenital ocular defects such as concomitant squint and alternating hyperphoria.42
Several characteristic clinical features help distinguish congenital from acquired spontane- ous nystagmus. It is usually purely horizontal and may diminish or disappear with conver- gence. The waveform may be pendular to saw- tooth shaped, with many variations in between.43 Different waveforms occur in the same patient in different eye positions. Gaze in the direction of the fast component converts a pendular nys- tagmus to a jerk nystagmus; often there is a null region where the nystagmus is minimal. Several different waveforms may be seen in members of the same family with congenital nystagmus. The frequency of congenital nystagmus is usually >2 beats/sec and at times reaches 5 to 6 beats/sec. Nystagmus of this high frequency is unusual other than on a congenital basis. Of course, most patients are aware that the nystag- mus has been present since infancy.
The pathophysiologic mechanism of congen- ital nystagmus is only partially understood.31 Convincing evidence exists that the slow com- ponent causes the target to slip from the fovea, and the fast component brings the target back to the fovea. The slow component is not the result of, but the cause of, decreased vision. Maneuvers designed to decrease the target slippage (fitting glasses with prisms and extraoc- ular muscle surgery) improve visual acuity. Patients with congenital nystagmus can make normal-velocity saccades, indicating that the extraocular muscles and orbital mechanics are normal. The vestibular system also appears to be normal in most of these patients. Abnormalities in smooth pursuit and optoki- netic slow phases are uniformly present, but it is difficult to know whether these abnormalities are due to a superimposition of the spontane- ous nystagmus on attempted tracking eye movements or to an underlying abnormality.44 Recently mutations have been identified in two genes associated with X-linked congenital nys- tagmus: FRMD7 causing X-linked idiopathic congenital nystagmus and GPR143 causing X-linked ocular albinism and congenital nystag- mus.45,46 Characterizing these genes and other
PERIODIC ALTERNATING NYSTAGMUS
Periodic alternating nystagmus (PAN) is a spontaneous nystagmus that periodically changes direction without a change in eye or head position.48 Cycle length varies between 1 and 6 min, with null periods between each half cycle varying from 2 to 20 sec. The nystagmus slowly builds in intensity and reaches a peak slow component velocity near the center of each half cycle before slowly decreasing. This type of nystagmus has been reported in associ- ation with such varied conditions as encephali- tis, brainstem ischemia, demyelinating disease, syringobulbia, syphilis, and trauma and as a congenital disorder. Unlike patients with other forms of congenital nystagmus, patients with congenital PAN frequently complain of oscil- lopsia because they are unable to adapt to the constantly changing direction of nystagmus. It is usually present with fixation, although cases have been reported in which PAN occurs only with loss of fixation.
Necropsy studies from three patients with acquired PAN showed diffuse brainstem involvement, with a predilection for the caudal brain stem.49 Reported cases have been associ- ated with downbeat nystagmus, further sug- gesting caudal brainstem dysfunction. The pathophysiologic mechanism for production of PAN is unknown. The PAN cycles can be altered in both phase and magnitude by a ves- tibular stimulus (rotatory or caloric), suggest- ing that the PAN rhythm is not the result of an independent central nervous system (CNS) pacemaker but, rather, a response pattern of the central vestibulo-ocular reflex arc.50 It is
from the primary position (See Video 6–9, Video 6–10, and Video 6–11). The eyes drift back toward the center with an exponentially decreasing wave form; corrective saccades (fast components) constantly reset the desired gaze position. Gaze-evoked nystagmus is therefore always in the direction of gaze. The site of abnormality can be anywhere from the neuro- muscular junction to the multiple brain centers controlling conjugate gaze (Table 6–3). Dysfunction of the oculomotor integrator (see Chapter 3) may be a common mechanism for several types of gaze-evoked nystagmus.
SYMMETRIC
Symmetric gaze-evoked nystagmus (equal amplitude to the left and right) is most com- monly produced by ingestion of drugs such as phenobarbital, phenytoin, alcohol, and diaze- pam. With these agents, high-frequency, small- amplitude nystagmus (<2 degrees) is found in all directions of gaze. A rough correlation exists between nystagmus amplitude and blood drug level.52,53 The nystagmus initially appears at extreme horizontal gaze positions and moves toward the midposition with higher drug levels. In addition to its association with drug ingestion, symmetric gaze-evoked nystagmus commonly occurs in patients with myasthenia gravis, multiple sclerosis, and cere- bellar atrophy.
Table 6–3 Causes of Gaze-Evoked Nystagmus
![]()
Localization Common Causes
![]()
important to recognize this unusual form of spontaneous nystagmus because the acquired variety is markedly diminished by baclofen, a
Symmetric Nonlocalizing or
cerebellum
Asymmetric Unilateral brain stem
Drugs,
metabolic disorders
gamma-amino butyric acid (GABA) agonist.51
Tumor,
and/or cerebellum
Unfortunately, congenital PAN does not respond to baclofen.
Gaze-Evoked Nystagmus
MECHANISM
Patients with gaze-evoked nystagmus are unable to maintain stable conjugate eye deviation away
Rebound Cerebellum
Dissociated MLF, extraocular
nerve, or muscle
![]()
MLF, medial longitudinal fasciculus.
infarction Tumor,
infarction, atrophy
Multiple sclerosis, myasthenia gravis
Asymmetric horizontal gaze-evoked nystagmus always indicates a structural brain lesion. When it is caused by a focal lesion of the brain stem or cerebellum, the larger amplitude nystagmus is usually directed toward the side of the lesion. Large cerebellopontine angle tumors com- monly produce asymmetric gaze-evoked nys- tagmus from compression of the brain stem and cerebellum (Bruns’ nystagmus). Some patients with large acoustic neuromas develop a combination of asymmetric gaze-evoked nys- tagmus from brainstem compression and peripheral spontaneous nystagmus from eighth nerve damage.54,55 Asymmetric gaze-evoked nystagmus may be present during the recovery from gaze paralysis (either cortical or subcorti- cal in origin), in which case it is large in ampli- tude and low in frequency and present only in one direction of gaze (the direction of the previous gaze paralysis).
REBOUND
Rebound nystagmus is a type of gaze-evoked nystagmus that either disappears or reverses direction as the lateral gaze position is held. When the eyes return to the primary position, another burst of nystagmus occurs in the direc- tion of the return saccade. Thus, the patient may have a transient primary position nystag- mus in either direction. Rebound nystagmus occurs in patients with cerebellar atrophy and focal structural lesions of the cerebellum; it is the only variety of nystagmus thought to be specific for cerebellar involvement.56
DISSOCIATED
Dissociated, or disconjugate, gaze-evoked nys- tagmus commonly results from lesions of the medial longitudinal fasciculus (MLF), so-called internuclear ophthalmoplegia. With early MLF lesions the eyes appear to move conjugately, but the abducting eye on the side opposite the MLF lesion develops a regular small-amplitude, high-frequency nystagmus in the direction of gaze. With more extensive MLF lesions, the adducting eye lags behind and develops a low- amplitude nystagmus while the abducting eye overshoots the target and develops large- amplitude nystagmus that has a characteristic peaked waveform.57 A MLF nystagmus can be
bilateral or unilateral, depending on the extent of MLF involvement. Bilateral MLF nystag- mus is most commonly seen with demyelinat- ing disease, whereas unilateral MLF nystagmus most often accompanies vascular disease of the brain stem.58 Patients with myasthenia gravis develop dissociated gaze-evoked nystagmus similar to MLF nystagmus (pseudo-MLF nys- tagmus) because of unequal impairment of neuromuscular transmission in adducting and abducting muscles. Unlike MLF nystagmus, the dissociated nystagmus with myasthenia progressively increases in amplitude as the gaze position is maintained.59
Positional Nystagmus
MECHANISM
Beginning with Bárány, positional nystagmus was attributed to lesions of the otoliths and their connections in the vestibular nuclei and cerebellum, as these are the receptors that are sensitive to changes in the direction of grav- ity.5,60 Subsequently, other mechanisms for the production of positional nystagmus have been identified, forcing reexamination of these tra- ditional concepts. If a semicircular canal cupula is altered so that its specific gravity no longer equals that of the surrounding endolymph or if debris inappropriately enters a semicircular canal, the canal becomes sensitive to changes in the direction of gravity and can produce positional nystagmus (see Causes of Benign Positional Vertigo in Chapters 10 and Alcohol and Thiamine Defeciency in Chapter 17).
Traditional classifications of positional nys- tagmus are often confusing and can be difficult to apply in clinical practice. Some classifica- tions have been based on clinical observations obtained while the patient is fixating, whereas others have been based on electronystagmog- raphy (ENG) recordings with eyes closed or with eyes open in darkness. Some investigators use slow positioning maneuvers, but others employ only rapid positioning. These different methods make it difficult to compare classifica- tions. Nylen60 initially described three types of positional nystagmus based on visual inspec- tion of nystagmus direction and regularity. Type I, direction changing, and type II, direc- tion fixed, remained constant as long as the position was maintained. Type III was less clearly defined, comprising all paroxysmal
varieties of positional nystagmus and some per- sistent varieties that did not fit into types I and
II. Numerous modifications of Nylen’s original classification have subsequently been pro- posed, and the definition of each type has changed. Most investigators do agree that two broad categories of positional nystagmus can be identified: paroxysmal and persistent.
PAROXYSMAL POSITIONAL NYSTAGMUS (POSITIONING NYSTAGMUS)
The most common type of paroxysmal posi- tional nystagmus is induced by a rapid change from erect sitting to the supine head-hanging left or right position (Dix-Hallpike test) (Fig. 6–6). It is initially high in frequency but dissipates rapidly. There is a 3- to 10-sec latency before onset and the nystagmus rarely lasts longer than 30 sec.61,62 The nystagmus has

(a)
(b)
combined torsional (fast component toward the undermost ear) and vertical (fast compo- nent toward the forehead) components (See Video 6–12 and Video 6–13). Although infre- quent bilateral cases do occur, the nystagmus is usually prominent only in one head-hanging position, and a burst of nystagmus occurs in the reverse direction when the patient moves back to the sitting position. Another key feature is that the patient experiences severe vertigo with the initial positioning, but with repeated posi- tioning, vertigo and nystagmus rapidly disap- pear (fatigability). This type of paroxysmal positional nystagmus is specific for the posterior canal variant of canalithiasis (see Chapter 10). Horizontal canal variants also exist but are less common. They are induced by turning the patient’s head to the side while the patient lies supine.
Paroxysmal positional nystagmus can also result from brainstem and cerebellar lesions. The central type does not decrease in ampli- tude or duration with repeated positioning, does not have a clear latency, and usually lasts longer than 30 sec. The direction is unpre- dictable and may be different in each position. It is often purely vertical with fast phase directed downward (i.e., toward the cheeks). The presence or absence of associated vertigo is not a reliable differential feature. Central paroxysmal positional nystagmus can be the initial presenting sign of a posterior fossa tumor such as a medulloblastoma or cerebellar glioma.63,64 It is therefore critical to distinguish it from the benign peripheral variety (Table 6–4).
PERSISTENT POSITIONAL NYSTAGMUS
This type of positional nystagmus remains as long as the position is held, although it may fluctuate in frequency and amplitude. It may be in the same direction in all positions or change directions in different positions. Direction-changing and direction-fixed static positional nystagmus are most commonly asso- ciated with peripheral vestibular disorders,
although both occur with central lesions.65
Figure 6–6. Method for inducing paroxysmal positional nystagmus (Dix-Hallpike maneuver). Patient is taken rap- idly from sitting to head-hanging right (a) or head-hanging left (b) position.
One variety of persistent direction-changing positional nystagmus (apogeotropic) is thought to result from otolithic debris attached to the cupula or lodged in the ampulla of the horizontal semicircular canal (see Chapter 10).66 As with spontaneous nystagmus, lack of
Table 6–4 Differentiation between Peripheral and Central Paroxysmal Positional Nystagmus
![]()
![]()
Appearance Latency Duration Fatigability Mechanism Localization
Peripheral Torsional, upbeat, or
horizontal geotropic
Central Pure vertical, often
downbeat
Usual < 60 sec Usual Change in
cupula-specific gravity
Unusual > 60 sec Unusual Damage to
central otolith-ocular pathways
Labyrinth
Brain stem or cerebellum
![]()
suppression with fixation and signs of associ- ated brainstem dysfunction suggest a central lesion.
CENTRAL POSITIONAL NYSTAGMUS
Positional nystagmus can also stem from a cen- tral lesion. The most common pattern is a per- sistent downbeating nystagmus. In fact, a persistent downbeating positional nystagmus may be the most prominent examination find- ing in a patient with a central lesion causing dizziness symptoms. Typical causes include a genetic cerebellar ataxia syndrome or a struc- tural lesion (e.g., Chiari malformation or tumor). Central positional nystagmus does not mimic the key characteristics of BPV from the posterior semicircular canal but can mimic the positional nystagmus of BPV from the horizon- tal semicircular canal.
Vibration-Induced Nystagmus
Vibration applied to the mastoid can often induce a typical peripheral vestibular nystag- mus in patients with compensated unilateral vestibular lesions.67,68 With vestibular neuritis the slow phase is nearly always directed toward the lesion side but with Meniere’s syndrome it can be directed to either side. Vibration may activate hair cells in the inner ear although the exact mechanism for vibration induced nystag- mus is poorly understood. In some cases of anterior semicircular dehiscence syndrome vibration over the suboccipital cranium will induce nystagmus in the plane of the anterior canal.69 In this case vibration activates the ante- rior semicircular canal just as noise or pressure
changes activate the canal (see Fig. 16–3B and 3C in Chapter 16).
Head-Shaking Nystagmus
Patients with a compensated vestibular imbal- ance due to either peripheral or central lesions may develop a transient nystagmus after vigor- ous head shaking.70 With unilateral peripheral vestibular lesions, the abnormal side is in the direction of the slow phase. With central ves- tibular lesions, the direction of nystagmus is nonlocalizing. Sometimes vertical nystagmus is induced by horizontal head shaking.71 The results of vertical head shaking are more diffi- cult to interpret because some normal subjects will have transient vertical nystagmus after ver- tical head shaking.
Hyperventilation-Induced Nystagmus
Hyperventilation can induce a near-faint dizzi- ness in anyone, particularly in anxious patients, but hyperventilation-induced nystagmus is less common.72 Patients with compressive lesions of the vestibular nerve, such as with an acoustic neuroma or cholesteatoma, or with demyelination of the vestibular nerve root entry zone, such as with multiple sclerosis, may develop nystagmus after hyperventilation. Presumably metabolic changes associated with hyperventilation trigger the partially damaged nerve to fire inappropriately. Hyper- ventilation-induced nystagmus has also rarely been associated with labyrinthitis or perilymph fistula.
Dissociated Spontaneous Nystagmus
Several different lesions of the posterior fossa can result in a spontaneous nystagmus with tor- sional, horizontal, and vertical components varying in each eye. The nystagmus is usually synchronized, however, in that the fast compo- nent occurs at exactly the same time in both eyes. Tumors, vascular disease, and demyeli- nating disease of the brain stem produce this form of dissociated nystagmus.73 Frequently the eye on the side of the lesion shows the larg- est amplitude oscillation. Monocular nystag- mus results from similar posterior fossa lesions; this unusual form of dissociated nystagmus also has been reported with such varied entities as congenital syphilis, meningitis, optic nerve glioma, cerebral trauma, unilateral amblyopia, and high refractive error.74,75 As expected, these patients are typically bothered by severe oscillopsia.
Seesaw nystagmus is an unusual type of dis- sociated nystagmus in which one eye rhythmi- cally rises and intorts and the other eye falls and extorts. It may be congenital but most often is produced by acquired lesions near the optic chiasm, particularly those producing a bitemporal field defect and decreased central visual acuity.76 Lesions associated with seesaw nystagmus include craniopharyngiomas, syrin- gobulbia, brainstem infarction, and diffuse choroiditis; compression of the midbrain teg- mentum may be the common denominator.24
Voluntary Ocular Oscillations (Voluntary Nystagmus)
Some normal subjects are able to produce rapid oscillations of the eyes at will, apparently by producing rapid sequenced saccades back and forth.77 Some cases may overlap with con- genital nystagmus.78 The main significance of these ocular gymnastics is that they may be mistaken for pathologic nystagmus. High in frequency (90 to as high as 1380 cycles/min) and low in amplitude (2 to 5 degrees), these rapid horizontal movements cannot be main- tained for more than 20 to 30 sec before fatigue
sets in. Several siblings in the same family may have the ability to produce voluntary ocular oscillations. Keyes79 reported two generations of the same family who could produce the eye movements, suggesting a dominant mode of inheritance.
Convergence Retraction Nystagmus
This dramatic ocular motor disorder results from lesions involving the diencephalic midbrain junction. When the patient attempts to make voluntary upward saccades or when involuntary upward saccades (fast components) are induced by an optokinetic or vestibular stimulus, the patient develops co-contraction of all extraocu- lar muscles and the eyes rhythmically retract and converge.80 (See Video 6–14). In other cases convergence nystagmus occurs without retrac- tion, apparently because of asynchronous adducting saccades.81 Convergence retraction nystagmus is usually associated with other signs of midbrain dysfunction (impaired upward gaze, pupillary abnormalities, accommodative spasm, retraction of the lids, and skew deviation), con- stituting the dorsal midbrain syndrome. This syndrome is most frequently produced by dys- germinomas of the pineal region but is also asso- ciated with other tumors and vascular lesions involving the tectal or pretectal area.
Saccadic Intrusions
Included under this category are square wave jerks, macrosquare wave jerks, macrosaccadic oscillations, ocular flutter, and opsoclonus. The common feature is that unwanted saccades dis- rupt steady fixation. Square wave jerks refer to small-amplitude involuntary saccades that take the eyes off the target, followed after a normal intersaccadic interval (200 msec) by a correc- tive saccade bringing the eyes back to the tar- get. Infrequent small square wave jerks can be seen in normal subjects, especially the young and elderly.82 Persistent large-amplitude square wave jerks (1 to 5 degrees) are abnormal but nonlocalizing. They are prominent with cerebellar lesions and with progressive supra- nuclear palsy and have been reported with dif- fuse cerebral lesions, Huntington’s disease, and schizophrenia.83,84 Macrosquare wave jerks
(10 to 50 degrees) have been observed in mul- tiple sclerosis and olivopontocerebellar atro- phy. Macrosaccadic oscillations are typically seen in patients with saccade overshoot dysme- tria (i.e., those with cerebellar lesions). After refixation, patients make a series of hypermet- ric saccades, apparently because they are unable to make a small enough saccade to bring the target onto the fovea. Ocular flutter refers to a burst of to-and-fro horizontal saccades occurring either spontaneously or after a sac- cade to a target (See Video 6–15). This burst of saccades lacks the characteristic delay normally present between serial saccades. Ocular flutter is typically seen with diffuse involvement of brainstem-cerebellar pathways, being particu- larly prominent in such varied disorders as Friedreich’s ataxia, brainstem encephalitis, and paraneoplastic syndromes.
The most prominent saccadic oscillations are seen with opsoclonus.85 With this rare eye movement disorder, the eyes are constantly making random conjugate saccades of unequal amplitude in all directions. As with ocular flutter, there is typically no intersaccade inter- val. The phenomenon occurs with several different types of CNS disease and probably represents a mixed group of eye movement disorders. The inappropriate saccades are most prominent immediately before or after a refixation and are only slightly affected by loss of fixation. One variety of opsoclonus prob- ably represents a continuum with square wave jerks and ocular flutter. Other more dramatic varieties of opsoclonus have been reported in patients with brainstem encephalitis, as a remote effect of tumors (e.g., neuroblastoma), and in association with toxins (e.g., the pesti- cide kepone).24 These saccade disorders prob- ably represent a release of the brainstem sac- cade burst neurons from supranuclear control. Functional MRI showed activation of the deep cerebellar nuclei in two patients whose opsoclonus was markedly diminished with eye closure.86
Ocular Bobbing
Ocular bobbing consists of abrupt, nonrhyth- mic, conjugate, downward jerks of the eyes, followed by slow return to midposition.31 The abnormal movements are classically seen in comatose patients with intrinsic pontine lesions
that also produce absent reflex horizontal eye movements, but they have also been reported with posterior fossa lesions that compress the pons and with metabolic encephalopathy.87 Inverse ocular bobbing or ocular dipping refers to a slow downward movement of the eyes fol- lowed by a rapid return to midposition. Reverse bobbing consists of a rapid deviation of the eyes upward followed by a slow return to the pri- mary position. These latter phenomena may be variations of ocular bobbing because all can be seen in the same subject at different times.88 As with typical ocular bobbing, they are usually seen with metabolic disorders or structural lesions of the pons.
Palato-Ocular Myoclonus
This is a rhythmic oscillation of the eyes associ- ated with synchronous oscillation of the palate. An associated rhythmic oscillation of die phar- ynx, larynx, mouth, tongue, diaphragm, extrem- ities, and intercostal muscles may also occur.89 The eye movements are typically pendular oscillations that are often vertical but may have a horizontal or torsional component. The fre- quency varies from 1 to 3/sec, and the move- ments may continue with loss of fixation. Ocular myoclonus often disappears during sleep even though the palatal movements continue. Palato-ocular myoclonus is seen in association with lesions disrupting the connections between the cerebellar dentate nucleus, the red nucleus, and the inferior olivary nucleus (Guillain- MoDaret triangle). It most commonly accom- panies vascular lesions but also occurs with tumors and degenerative disease. When seen in association with vascular lesions, it often develops months after the brainstem or cere- bellar infarction. Intravenous scopolamine has been reported to abolish the ocular oscillations temporarily, but there is no good long-term treatment.90
If a subject is tilted in the frontal plane (about the nasal occipital axis), the head reflexively tilts and the eyes counter-roll and skew toward the opposite side. The functional role of this reflex in visual stabilization during natural body
pontomedullary
Right ponto- mesencephalic
Figure 6–7. Ocular tilt responses associated with lesions at different locations within the peripheral and central vestibular pathways. The ocular tilt reaction consists of a head tilt toward the side of the lower eye, a skew deviation with one eye higher than the other, and counterroll (torsion) of both eyes, with the top poles rolling toward the side of the lower eye. The ipsilateral eye is down with lesions of the labyrinth and pontomedtillary regions but it is up with lesions in the pontomes- encephalic region.
movements is minimal, however, as the magni- tude of the compensatory head tilt and ocular counter-rolling is only about 10% of angular displacement of the head. The ocular tilt reac- tion is principally a labyrinthine reflex; it is independent of the position of the head, rela- tive to the body (indicating that neck position is not important). The ocular tilt reaction can be elicited by electrical stimulation of the rostral midbrain tegmentum in the region of the inter- stitial nucleus of Cajal.91 Clinically, the ocular tilt reaction has been seen in patients with peripheral labyrinthine lesions (a complication of stapedectomy), lesions of the lateral medulla (particularly Wallenberg’s syndrome), and with
Lesions in these regions would, of course, result in an ocular tilt reaction in the opposite direction (Fig. 6–7). Paroxysmal tonic ocular tilt reactions have been reported in patients with multiple sclerosis and in a patient with a focal brainstem abscess.94,95 Such patients may respond to carbamazepine or baclofen.
REFERENCES
Hain TC, Ostrowski VB. Limits of normal for pres- sure sensitivity in the fistula test. Audiol Neurootol. 1997;2(6):384.
Hennebert C. A new syndrome in hereditary syphilis of
lesions of the rostral midbrain (Fig. 6–7).92–94 On the basis of animal and clinical data,
the labyrinth. Presse Med Belg Brnx. 1911;63:407.
Nadol JB. Positive Hennebert’s sign in Meniere’s ease. Arch Otolaryngol. 1977;103:524.
dis-
Halmagyi and colleagues93 proposed that the
excitatory ocular tilt reaction arises in the utri- cle of the dependent ear, passes through or synapses in the vestibular nuclei of the same side, and then projects to the opposite side of the upper brain stem. Therefore, stimulation of the utricular nerve or the region of the ves- tibular nuclei results in a contralateral ocular tilt reaction, whereas stimulation of the mid-
Baloh RW. Semicircular canal dehiscence syndrome: leaks and squeaks can make you dizzy. Neurology. 2004;62:684.
Bárány R. Neue Untersuchungsmethoden, die Beziehungen zwischen Vestibularapparat, Kleinhirn, Grosshirn and Ruckenmark betreffend [in German]. Wien Med Wochenschr. 1910;60:2033.
Romberg MH. Lehrbuch der Nervenkrankheiten des Menschen. Berlin, Germany: A Dunker; 1846.
Bárány R. Physiologie and Pathologie des Bogen
brain results in an ipsilateral ocular tilt reaction.
Gangsapparates beini Menschen. Vienna: Deuticke;
1907.
Unterberger S. Neue objectiv registrierbare Vestibularis—Korperdrehreaktion, erhalten dutch Freten auf der Stelle: Der “Tretversuch“[in German]. Arch Ohr Nas Kehlk Heilk. 1938;145:478.
Fukuda T. The stepping test: two phases of the labyrin- thine reflex. Acta Otolaryngol (Stockh). 1959;50:95.
Cho BL, Scarpace D, Alexander NB. Tests of step- ping as indicators of mobility, balance, and fall risk in balance-impaired older adults. J Am Geriatr Soc. 2004;52(7):1168.
Lark SD, Pasupuleti S. Validity of a functional dynamic walking test for the elderly. Arch Phys Med Rehabil. 2009;90(3):470.
Leigh RJ, Hanley DE, Munschauer FE, Lasker AG. Eye movements induced by head rotation in unre- sponsive patients. Ann Neurol. 1984;15:465.
Mueller-Jellsen A, Neunzig H-P, Emskotter TH. Outcome prediction in comatose patients: significance of reflex eye movement analysis. J Neurol Neurosurg Psychiatry. 1987;50:389.
Morrow SA, Young GB. Selective abolition of the vestibular-ocular reflex by sedative drugs. Neurocrit Care. 2007;6(1):45.
Halmagyi GM, Curthoys IS. A clinical sign of canal paresis. Arch Neurol. 1988;45:737.
Pearce J. The Marcus Gunn pupil. J Neurol Neurosurg Psychiatry. 1996;61:520.
Minor LB. Intratympanic gentamicin for control of vertigo in Meniere’s disease: vestibular signs that spec- ify completion of therapy. Am J Oto. 1999;20:209.
Perez N, Rama-Lopez J. Head-impulse and caloric tests in patients with dizziness. Otol Neurotol. 2003;24: 913.
Beynon GJ, Jani P, Baguley DM. A clinical evalu- ation of head impulse testing. Clin Otolaryngol. 1998;23:117.
Assessment: Electronystagmography: Report of the Therapeutics and Technology Assessment Committee. Neurology. 1996;46:1763.
Newman-Toker DE, Kattah JC, Alvernia JE, Wang DZ. Normal head impulse test differentiates acute cerebellar strokes from vestibular neuritis. Neurology. 2008;70:2378.
Kattah JC, Talkad AV, Wang DZ, Hsieh YH, Newman- Toker DE. HINTS to diagnose stroke in the acute vestibular syndrome. Three step bedside oculomotor examination more sensitive than early MRI diffuision weighted imaging. Stroke. 2009;40:3504.
Longridge NS, Mallinson AI. The dynamic illegible E (DIE) test: a simple technique for assessing the ves- tibulo-ocular reflex to overcome vestibular pathology. Can J Otolaryngol. 1987;16:97.
Burgio DL, Blakely BW, Myers SE The high frequncy oscillopsia test. J Vestib Res. 1992;2:221.
Kaeser PF, Borruat FX. Altered vision during motion: an unusual symptom of cerebellar dysfunction, quanti- fiable by a simple clinical test. Acta Ophthalmol. ePub ahead of print, Sep 2, 2009.
Dannenbaum E, Paquet N, Chilingaryan G, Fung J. Clinical evaluation of dynamic visual acuity in subjects with unilateral vestibular hypofunction. Otol Neurotol. 2009;30(3):368.
Schmäl F, Lübben B, Weiberg K, Stoll W. The minimal ice water caloric test compared with estab- lished vestibular caloric test procedures. J Vestib Res. 2005;15(4):215.
Newman-Toker DE, Sharma P, Chowdhury M, Clemons TM, Zee DS, Della Santina CC. Penlight- cover test: a new bedside method to unmask nystag- mus. J Neurol Neurosurg Psychiatry. 2009;80(8):900.
Rucker JC. An update on acquired nystagmus. Semin Ophthalmol. 2008;23(2):91.
Jacobson GP, McCaslin DL, Kaylie DM. Alexander’s law revisited. J Am Acad Audiol. 2008;19(8):630.
Leigh RJ, Zee DS. The Neurology of Eye Movements. 4th ed. New York: Oxford University Press; 2005.
Baloh RW, Spooner JW. Downbeat nystagmus: a type of central vestibular nystagmus. Neurology. 1981;31:304.
Zee DS, Yamazaki A, Batter PH, Gucer G. Effects of ablation of flocculus and parallocculus on eye move- ments in primates. J Neurophysiol. 1981;46:878.
Halmagyi GM, Rudge P, Gresty MA, Sanders MD. Downbeating nystagmus. Arch Neurol. 1983;40:777.
Baloh RW, Yee RD. Spontaneous vertical nystagmus.
Rev Neurol (Paris). 1989;145:527.
Spooner JW, Baloh RW. Arnold-Chiari malformation: improvement in eye movements after surgical treat- ment. Brain. 1981;104:51.
Fisher A, Gresty M, Chambers B, Rudge P Primary position upbeat nystagmus: a variety of central posi- tional nystagmus. Brain. 1983;106:949.
Aschoff JC, Conrad B, Kornhuber HH. Acquired pen- dular nystagmus with oscillopsia in multiple sclerosis: a sign of cerebellar nuclei disease. J Neurol Neurosurg Psychiatry. 1974;37:570.
Baloh RW, Yee RD, Honrubia V. Eye movements in patients with Wallenberg’s syndrome. Ann NY Acad Sci. 1981;374:600.
Marti S, Straumann D, Büttner U, Glasauer S. A mod- el-based theory on the origin of downbeat nystagmus. Exp Brain Res. 2008;188(4):613.
Cogan DG. Congenital nystagmus. Can J Ophthalmol. 1967;2:4.
Dell’Osso LF, Schmidt D, Darolf RB. Latent, manifest latent, and congenital nystagmus. Arch Ophthalmol. 1979;97:1877.
Yee RD, Wong EK, Baloh RW, Honrubia V. A study of congenital nystagmus: waveforms. Neurology. 1976;26:326.
Yee RD, Baloh RW, Honrubia V Kim YS. A study of congenital nystagmus: vestibular nystagmus. J Otolaryngol. 1981;10:89.
He X, Gu F, Wang Z, et al. A novel frameshift muta- tion in FRMD7 causing X-linked idiopathic congenital nystagmus. Genet Test. 2008;12(4):607.
Fang S, Guo X, Jia X, Xiao X, Li S, Zhang Q. Novel GPR143 mutations and clinical characteristics in six Chinese families with X-linked ocular albinism. Mol Vis. 2008;14:1974.
Self J, Lotery A. A review of the molecular genetics of congenital idiopathic nystagmus (CIN). Ophthalmic Genet. 2007;28(4):187.
Baloh RW, Honrubia V, Konrad HR. Periodic alter- nating nystagmus. Brain. 1976;99:11.
Keane JR. Periodic alternating nystagmus with down- ward beating nystagmus. A clinico-anatomic case study of multple sclerosis. Arch Neurol. 1974;30:399.
Leigh RJ, Robinson DA, Zee DS. A hypothetical expla- nation for periodic alternating nystagmus: Instability in the optokinetic vestibular system. Ann NY Acad Sci. 1981;374:619.
Halmagyi GM, Rudge P, Gresty AM, et al. Treatment of periodic alternating nystagmus. Ann Neurol. 1980;8:609.
Gallagher BB, Baumel IP, Mattson RH, Wood-burg SG. Primidone, dipenylhydantoin and Phenobarbital. Aspects of acute and chronic toxicity. Neurology. 1973;23:145.
Hogan RE, Collins SD, Reed RC, Remler BF. Neuro-ophthalmological signs during rapid intrave- nous administration of phenytoin. J Clin Neurosci. 1999;6(6):494.
Baloh RW, Konrad HR, Dims D, Honrubia V. Cerebellar-pontine angle tumors. Results of quan- titative vestibulo-ocular testing. Arch Neurol. 1976;33:507.
Lloyd SK, Baguley DM, Butler K, Donnelly N, Moffat DA. Bruns’ nystagmus in patients with vestibular schwannoma. Otol Neurotol. 2009;30(5):625.
Hood JD. Further observations on the phenomenon of rebound nystagmus. Ann NY Acad Sci. 1981;374:352.
Baloh RW, Yee RD, Honrubia V. Internuclear oph- thalmoplegia. I. Saccades and dissociated nystagmus. Arch Neurol. 1978;35:484.
Cogan DG, Kubik SC, Smith WL. Unilateral inter- nuclear ophthalmoplegia: report of eight clinical cases and one post-mortem study. Arch Ophthalmol. 1950;44:783.
Spooner JW, Baloh RW. Eye movement fatigue in myasthenia gravis. Neurology. 1979;29:29.
Nylen CO. Positional nystagmus. A review and future prospects. J Laryngol Otol. 1950;64:295.
Baloh RW, Honrubia V, Jacobson K. Benign positional vertigo: clinical and oculographic features in 240 cases. Neurology. 1987;37:371.
Aw ST, Todd MJ, Aw GE, McGarvie LA, Halmagyi M. Benign positional vertigo: a study of its three dimen- sional spatio-temporal characteristics. Neurology. 2005;64:1897.
Grand W. Positional nystagmus: an early sign of medulloblastoma. Neurology. 1971;21:1157.
Gregorius FK, Crandall PH, Baloh RW. Positional vertigo in cerebellar astrocytoma. Report of two cases. Surg Neurol. 1976;6:283.
Lin J, Elidan J, Baloh RW, Honrubia V. Direction changing positional nystagmus: incidence and mean- ing. Am J Otolaryngol. 1986;7:306.
Baloh RW, Yue Q, Jacobson KM, Honrubia V. Persistent direction-changing positional nystagmus. Another variant of benign positional nystagmus? Neurology. 1995;45:1297.
Park HJ, Shin JE, Lim YC, Shin HA. Clinical sig- nificance of vibration-induced nystagmus. Audiol Neurootol. 2008;13(3):182.
Dumas G, Perrin P, Schmerber S. Nystagmus induced by high frequency vibrations of the skull in total uni- lateral peripheral vestibular lesions. Acta Otolaryngol. 2008;128(3):255.
White JA, Hughes GB, Ruggieri PN. Vibration- induced nystagmus as an office procedure for the diag- nosis of superior semicircular canal dehiscence. Otol Neurotol. 2007;28(7):911.
Hain TC, Spinder J. Head-shaking nystagmus. In: Sharpe JA, Barber HO, eds. The Vestibulo-ocular Reflex and Vertigo. New York: Raven Press; 1993.
Strupp M. Perverted head-shaking nystagmus: two possible mechanisms. J Neurol. 2002;249(1): 118.
Bance ML, O’Driscoll M, Patel N, Ramsden RT. Vestibular disease unmasked by hyperventilation. Laryngoscope. 1998;108:610.
Cogan DG. Dissociated nystagmus with lesions in the posterior fossa. Arch Ophthalmol. 1963;70:121.
Nathanson M, Bergman PS, Berker MB. Monocular nystagmus. An J Ophthalmol. 1955;40:685.
Donin JF. Acquired monocular nystagmus in children.
Can J Ophthalmol. 1967;2:212.
Rambold H, Helmchen C, Straube A, Büttner U. Seesaw nystagmus associated with involuntary tor- sional head oscillations. Neurology. 1998;51:831.
Shults WT, Stark L, Hoyt WF, Ochs AL. Normal saccadic structure of voluntary nystagmus. Arch Ophthalmol. 1977;95:1399.
Lewis RF, Traish AS, Lessell S. Atypical voluntary nys- tagmus. Neurology. 2009;72(5):467.
Keyes MJ. Voluntary nystagmus in two generations.
Arch Neurol. 1973:29:63.
Gay AJ, Brodkey J, Miller JE. Convergence retrac- tion nystagmus: an electromyographic study. Arch Ophthalmol. 1963:70:456.
Ochs AL, Stark L, Hoyt WF, D’Amico D. Opposed adducting saccades in convergence retraction nystag- mus. Brain. 1979;102:497.
Herishanu YO, Sharpe JA. Normal square wave jerks.
Invest Ophthalmol Vis Sci. 1981;20:268.
Fukazawa T, Tashiro K, Hamada T, Kase M. Multisystem degeneration: drugs and square wave jerks. Neurology. 1986;36:1230.
Troost BT, Daroff RB. The ocular motor defects in pro- gressive supranuclear palsy. Ann Neurol. 1977:2:397.
Wong A. An update on opsoclonus. Curr Opin Neurol. 2007;20:25.
Helmchen C, Rambold H, Sprenger A, Erdmann C, Binkofski F. Cerebellar activation in opsoclonus. An fMRI study. Neurology. 2003;61:412.
Bosch EP, Kennedy SS, Aschenbrener CA. Ocular bobbing: the myth of its localizing value. Neurology. 1975;25:949.
Stark SR, Masucci EF, Kurlzke JF. Ocular dipping.
Neurology. 1984;34:391.
Tahmoush AJ, Brooks JE, Keltner JL. Palatal myo- clonus associated with abnormal ocular and extremity movements. Arch Neurol. 1972:27:431.
Gresty MA, Ell JJ, Findley LJ. Acquired pendular nystagmus: its characteristics, localizing value and pathophysiology. J Neurol Neurosurg Psychiatry. 1982;45:431.
Westheimer G, Blair M. The ocular tilt reaction – a brain stem oculomotor routine. Invest Ophthalmol. 1975;14:833.
Brandt T, Dieterich M. Pathological eye head coordi- nation in roll: tonic ocular lilt reaction in mesenceph- alic and medullary lesions. Brain. 1987;1(10):649.
Halmagyi GM, Gresty MA, Gibson WPR. Ocular tilt reaction with peripheral vestibular lesions. Ann Neurol. 1979;6:80.
Hedges TR, Hoyt WF. Ocular tilt reaction due to an upper brain stem lesion: paroxysmal skew deviation, torsion, and oscillation of the eyes with head tilt. Ann Neurol. 1982;11:537.
Oh SY, Choi KD, Shin BS, Seo MW, Kim YH, Kim JS. Paroxysmal ocular tilt reactions after mesodien- cephalic lesions: report of two cases and review of the literature. J Neurol Sci. 2009;277(1-2):98.
This page intentionally left blank
![]()
Laboratory Examination of the Vestibular
Methods of Recording Eye Movements Interpreting the Recording
Recording Pathologic Nystagmus Bithermal Caloric Test
Tests of Visual–Ocular Control
ROTATIONAL TESTING OF VESTIBULO-OCULAR REFLEXES
Relationship between Stimulus and Response Results in Normal Subjects
Results in Patients
VISUAL–VESTIBULAR INTERACTION
Methodology
Results in Normal Subjects Results in Patients
TESTS OF OTOLITH–OCULAR REFLEXES
Ocular Counterrolling Eccentric Rotation
Off-Vertical Rotation Linear Acceleration
VESTIBULOSPINAL TESTING
Static-Force Platforms
Moving-Platform Posturography
VESTIBULAR-EVOKED POTENTIALS
Brain Stem and Cortical
Vestibular Evoked Myogenic Potentials (VEMPs)
Nystagmography is a method for recording nystagmus, although it can be used for record- ing any type of eye movement. One can quan- tify the slow-component velocity, frequency, and amplitude of spontaneous or induced nys- tagmus, and the changes in these measure- ments brought about by loss of fixation (with eyes open in darkness).1 In addition, visually controlled eye movements (saccades, smooth pursuit, and optokinetic nystagmus) can be recorded and quantitatively assessed.
Methods of Recording Eye Movements
Electrooculography (or electronystagmogra- phy, ENG) is the simplest and still most readily
available system used for nystagmography (Fig. 7–1).2,3 The potential difference between the cornea and retina, known as the corneal– retinal potential, acts as an electric dipole, ori- ented in the direction of the long axis of the eye. In relation to a remote location, an electrode placed in the vicinity of the eye becomes more positive when the eye rotates toward it and less positive when it rotates away from it. Recordings are usually made with a three-electrode system, using differential amplifiers. Two of the (active) electrodes are placed on each side of the eye, and the reference (ground) electrode is placed somewhere remote from the eye. The two active electrodes measure a potential change of equal amplitude but opposite direction. The difference in potential between these elec- trodes is amplified and then transferred to a recording device for a graphical representation. Because the differential amplifiers monitor the
171
–
–
–
–
–
+
+
+
+
Electrodes + Amplifier
Pen Recorder
Figure 7–1. Electronystagmography. Electrode placement (top); Method for recording eye movements (bottom). See text for details.
difference in voltage between the two active electrodes, remote electrical signals (electro- cardiographic or electroencephalographic, for example) arrive at the electrodes with approxi- mately equal amplitude and phase and are canceled out.
Infrared video recording of eye movements (videonystagmography, VNG) is a newer, more flexible eye movement recording system that is gradually replacing ENG in many clinical labo- ratories (Fig. 7–2).4 The infrared cameras used are fitted into various types of goggles, which are then placed around the patient’s head. With some goggles, the cameras are placed directly in front of the eyes, which is fine for recording eye movements in dark conditions (i.e., caloric and positional tests). To allow patients to better track targets (e.g., during saccade and smooth pursuit testing), other goggles were designed with the cameras mounted on top (thus out of the patient’s line of vision) and a reflective glass is used so that the eyes can be recorded. The infrared video camera is interfaced with a digi- tal computer. At regular intervals, images are stored by the computer for subsequent data analysis. Specialized digital signal–processing
algorithms are then used to determine horizon- tal, vertical, and even torsional eye position.
Magnetic search coils are another method of recording eye movements. This technique uses a contact lens embedded with two coils of wire, one that senses horizontal and vertical move- ments, and the other that senses torsional movements. When the subject sits in a mag- netic field, voltages are induced in these search coils that can be used to measure eye position. The magnetic search coil technique is the gold standard technique for measuring eye move- ments because it allows measurement of eye rotations around all threes axes with high sensitivity and low noise.
Each eye movement recording system has advantages and disadvantages. ENG is rela- tively inexpensive, easily administered, nonin- vasive, does not interfere with vision, and does not require head restraint. Furthermore, it is reasonably accurate even for the large- amplitude horizontal eye movements that are encountered during routine ENG testing. The disadvantages of ENG include the inability to measure torsional eye movements and rela- tively inaccurate measurement of vertical eye
Video Cameras Video Cameras
Reflective lenses
Right Eye Left Eye
![]()

5 10 15
Figure 7–2. Videonystagmography. Two different examples of goggle systems, one with cameras mounted in front of the eyes and the other with cameras mounted on top of the goggles and out of the patient’s line of vision (top); example of appearance of video-recorded eyes (middle); example of videonystagmography caloric tracing and smooth pursuit tracing (bottom).
movements, interference of eye blink artifacts, poor signal-to-noise ratio, and dependence on lighting conditions in the test room.5,6 Infrared video recordings have the advantage of accu- rately recording horizontal, vertical, and torsional eye movements.4 In addition to providing a digitized paper recording, there is a video recording that can be reviewed when there are questions about the paper recording, and it can also be used for teaching purposes. With video recordings there is a much faster setup time, as the goggles containing the video cameras are
simply strapped on. Usually the computer finds and calibrates the eyes in less than a minute. With ENG, calibrations must be repeated fre- quently throughout the test battery, since the corneal–retinal potential fluctuates, particu- larly if the lights are turned on and off. In addition, with the video system there is no interference from extraneous electrical signals from in- or outside the body. Disadvantages of the video systems include possible obstruction of vision by the video cameras, difficulty wear- ing eyeglasses, difficulty in keeping the camera
system fixed on the forehead, and difficulty with the digital tracking system in some patients with poor contrast between the pupil and iris. Eye blinks can interfere with the video record- ing systems, but VNG can be used to monitor eyelid dynamic disorders.7 The main disadvan- tage of the magnetic search coil technique is that it requires the use of a thick contact lens, which can cause discomfort for the patient or rarely corneal abrasion. Accordingly the mag- netic search coil technique is generally only used in research laboratories.
With newer versions of VNG systems with high-frequency sampling rates, the degree of precision now rivals that of the magnetic search coil technique,8 so such systems are being incorporated into more research laboratories. Although increased precision of VNG and the magnetic search coil technique is a clear advan- tage over ENG in research studies,9 it remains uncertain whether these advantages over ENG enhance clinical diagnostic capabilities or can result in better patient outcomes.
Ultimately, the performance of any of these techniques to record eye movements depends substantially on practical factors like the skills of the technician performing the test, the coop- eration of the patient, and the expertise of the clinician interpreting the test. If the test is not properly set up or instructions are not effec- tively communicated to the patient, then the results will be largely questionable. For the caloric test, the ear canals often need to be cleaned, the irrigation tube must be appropri- ately positioned, and the temperature of the water monitored. These are not difficult tasks, but appropriate training, experience, and care are required. There are many patient factors (e.g., decreased alertness from sedating medi- cines, apathy, or severe nausea) that could result in an invalid test even when performed
by a top technician. Finally, the clinician inter- preting the test must be familiar with these and other nuances of the test.
Interpreting the Recording
By convention, for horizontal recordings, eye movements to the right are displayed so that they produce upward pen deflection and those to the left produce downward pen deflection. For vertical recordings, upward and downward eye movements produce upward and downward deflections, respectively. For torsional eye movements, clockwise eye movements produce upward pen deflections while counterclockwise eye movements produce downward pen deflec- tion. Figure 7–3 illustrates the relationship between components of a typical beat of hori- zontal nystagmus. Values chosen for each component are those commonly seen with spon- taneous vestibular nystagmus recorded in the dark. The fast component moves to the left, so by convention the nystagmus is to the left (i.e., “left-beating”). A 10-degree fast component would have an average velocity (a/fd; a = ampli- tude, fd = fast duration) of approximately 100 degrees/sec. The slow-component velocity (a/sd; a = amplitude, sd = slow duration) is usu- ally much slower—in this case, 10 degrees/sec. It is approximately the product of the amplitude times frequency as long as the fast duration is small compared with the slow duration. Although the magnitude of each nystagmus measurement shown in Figure 7–2 can be calculated directly from the paper recording, such a procedure is tedious and therefore subject to error. Digital computers are ideally suited for making such measurements. Using a programmed algorithm, computers can calculate the amplitude, dura- tion, and velocity of each of the slow and fast
![]()

R sc fc a
L
10°
![]() sd fd
sd fd
1 sec
f = sd 1 fd vsc = a
![]()
vsc a·f if sd >> fd
+ sd
Figure 7–3. Single beat of nystagmus recorded with electronystagmography. a, amplitude; f, frequency; fc, fast component; fd, fast duration; sc, slow component; sd, slow duration, vsc, velocity of slow component.
Nystagmography can be used to evaluate any type of eye movement disorder, and the testing procedure should be flexible enough to deal with any abnormality encountered. It is useful, however, to have a standard test battery that will at least screen all areas of potential abnor- mality (Table 7–1). In most clinical laborato- ries, the test battery includes (1) recording for pathologic nystagmus, (2) the bithermal caloric test, and (3) tests of visual–ocular control.
Recording Pathologic Nystagmus
The same systematic search for pathologic nys- tagmus outlined in the previous chapter should be conducted during the nystagmography examination. Recording with eyes open in darkness is more effective than using Frenzel glasses for identifying peripheral spontaneous and positional nystagmus. Approximately 20% of normal subjects have a low-velocity sponta- neous nystagmus (i.e., <4 degrees/sec) and as many as 75% have a low-velocity positional nystagmus when tested with eyes open in darkness.10,11 Apparently the vestibular system is unable to stabilize the position of the eyes when visual signals are removed.
SPONTANEOUS NYSTAGMUS
The effect of change in ocular position and fixation on peripheral spontaneous nystagmus
is illustrated with the ENG recordings in Figure 7–4. The patient was tested 3 days after and again 2 weeks after a left labyrinthectomy. On the initial recording, nystagmus was pres- ent even while fixating on a target, although it was more prominent without fixation. On the subsequent recording, nystagmus occurred only when the lights were turned off. This pattern is typical of an acute peripheral vestib- ulopathy of any cause. The nystagmus is exag- gerated with gaze in the direction of the fast component (Alexander’s law). As a general rule, nystagmus with fixation (nystagmus seen on routine neurologic examination) disappears within 1 to 2 weeks after the occurrence of an acute peripheral vestibular lesion. By contrast, spontaneous nystagmus can be recorded in the dark for as long as 5 to 10 years after an acute peripheral vestibular lesion. In some patients, the spontaneous nystagmus emerges only with mental alerting tasks (e.g., when performing serial 7 subtractions from 100).12 Changes in head position with respect to gravity may alter the direction and magnitude of peripheral spontaneous nystagmus.
Eye movement recordings can help differ- entiate congenital and central varieties of spon- taneous nystagmus from peripheral varieties. Spontaneous downbeating nystagmus is a com- mon central nystagmus pattern. The downbeat nystagmus shown in Figure 7–5a increases in frequency and amplitude with downward gaze, but it reversed direction with upward gaze. Loss of fixation did not change the nystagmus frequency or amplitude (Fig. 7–5b). The spon- taneous vertical nystagmus was superimposed on attempted vertical pursuit (Fig. 7–5c).
Table 7–1 Standard Nystagmography Test Battery
![]()
Recording for Pathologic Nystagmus
Fixation at midposition
Fixation inhibited with eyes open in darkness (constant mental alerting) Gaze held 30° right, left, up, and down
Rapid and slow positional changes
Bithermal Caloric Test
30°C and 44°C water infused into each ear, eyes open in darkness, constant mental alerting, allow at least 5 min between each infusion
Visual Tracking Tests
Saccades: 5°–40°, target can be series of dots or lights Smooth pursuit: target velocity 20°–40°/sec Optokinetic nystagmus: stripe velocity 20°–40°/sec
Optokinetic after-nystagmus: lights turned off after 1 min constant velocity optokinetic nystagmus in each direction
![]()
14 Days Post Labyrinthectomy
![]()
30° right 30° right
![]()
10°
![]()
15° right 5 sec
![]()
Center
![]()
15° left
![]()
30° left
![]()
Lights out
![]()
Center
Center
![]()
30° left
![]()
Lights out
30° right
![]()
Center
![]()
30° left
![]()
Figure 7–4. Peripheral spontaneous nystagmus (bitemporal horizontal ENG recording) taken 3 days and 14 days after the patient underwent a left labyrinthectomy. By 14 days, the nystagmus disappeared when in the light with fixation but was still prominent in the dark when fixation was inhibited.
The waveform of the congenital nystagmus illustrated in Figure 7–6a changed in different horizontal gaze positions (from pendular to near sawtooth). When fixation was inhibited by darkness, the nystagmus almost disappeared (Fig. 7–6b). Horizontal smooth pursuit was markedly impaired in both directions (Fig. 7–6c). By comparison, peripheral spontaneous nystagmus does not change direction with change in gaze position, does increase with loss of fixation, and usually does not impair smooth pursuit. The slow component with acquired spontaneous nystagmus is typically
linear, producing a sawtooth pattern, whereas the slow component with congenital nystagmus usually increases exponentially.13
GAZE-EVOKED NYSTAGMUS
The most common type of gaze-evoked nystagmus (drug-induced) has approximately equal ampli- tude in all directions of gaze. A large-amplitude, asymmetric gaze-evoked nystagmus is often seen with lesions of the cerebellar-pontine (CP) angle (so-called Bruns’ nystagmus).14 The lesion is usu- ally on the side of the large-amplitude nystagmus.
(a)
30° up
(b)
![]()
Lights off
![]()
15° up
![]()
![]()
Center
![]()
15° down
5 sec
![]()
![]()
Pursuit down
10°
Pursuit up
Figure 7–5. Spontaneous downbeat nystagmus (monocular vertical ENG recordings). (a) Nystagmus increased on down- ward gaze and changed direction on upward gaze. (b) Loss of fixation had little effect. (c) Nystagmus was superimposed on attempted vertical pursuit.
![]()
![]()
30° right
![]()
15° right 5 sec Center
(b)
Lights off Lights on
10°
15° left
(c)
Pursuit right
Pursuit left
30° left
![]()
Figure 7–6. Congenital nystagmus (bitemporal horizontal ENG recordings), (a) Waveform changed in different gaze posi- tions, (b) Nystagmus decreased with loss of fixation, (c) Nystagmus was superimposed on attempted horizontal pursuit.
Rebound gaze-evoked nystagmus decays as the lateral gaze position is held (Fig. 7–7, upper trace) and recurs transiently after returning to the primary position, with fast components occurring in the direction of the return saccade (Fig. 7–7, lower trace).15
POSITIONAL NYSTAGMUS
Although benign paroxysmal positional nystag- mus can be readily identified on routine physi- cal examination, recording the nystagmus documents its stereotyped profile. For this rea- son, we include the Dix-Hallpike positioning test as part of our nystagmography examina- tion; the technician also observes the nystag- mus while it is being recorded. Dix-Hallpike positioning can also be used to identify central positional nystagmus, which is typically down- beating. VNG is ideal for documenting vertical and torsional positional nystagmus patterns since the video recording can be reviewed. (Fig. 7–8).16 If ENG is used with only horizon- tal electrodes, then vertical or torsional nystagmus could be missed.17
As suggested earlier, persistent low velocity positional nystagmus is a common finding on nystagmography even in control subjects when recordings are made with eyes open in dark- ness. An average slow-phase velocity that exceeds 4 degrees/sec is considered abnormal, but nonlocalizing. Both direction-fixed and direction-changing static positional nystagmus occur with peripheral and central vestibular lesions.18 Lack of suppression of the nystagmus with fixation indicates a central lesion.
Bithermal Caloric Test
MECHANISM OF STIMULATION
Robert Barany received the Nobel Prize for proposing the mechanism of caloric stimula- tion of the vestibular system. The test uses a nonphysiologic stimulus (water or air) to induce endolymphatic flow in the semicircular canals by creating a temperature gradient from one side of the canal to the other (Fig. 7–9).19,20 Irrigation of the external auditory canal with
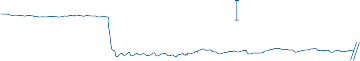
Center 15°
Left 30°
![]()
Center
5 sec
Figure 7–7. Rebound nystagmus (horizontal bitemporal ENG recording). Nystagmus decays as the lateral gaze position is held and recurs on return to the primary position. The reverse occurred with gaze to the right.
10
0
–10
Eye Position (deg)
Eye Position (deg)
–20
20
10
0
–10
–20
![]()
Horizontal | ||
Horizontal | ||
20
![]()
10
0
–10
![]()
–20
Torsional
Vertical
5 10
![]()
![]()
15 20 25
Time (seconds)
Figure 7–8. Three-dimensional recording of benign paroxysmal positional nystagmus. Scleral search coil in right eye. Positive values indicate clockwise (as seen from the subject), up and right. Vertical lines indicate start and end of positioning maneuver. (Courtesy Michael Fetter, Tübingen, Germany).
water or air that is below or above body tem- perature transfers a temperature gradient from the external auditory canal to the inner ear by conduction. The horizontal semicircular canal develops the largest temperature gradient because it lies closest to the source of tempera- ture change. Because the vertical canals are relatively remote from the external ear, caloric stimulation of the vertical canals is unreliable. The endolymph circulates because of the dif- ference in its specific gravity on the two sides of the canal when the semicircular canal being investigated is in the vertical plane. Caloric testing of horizontal semicircular canal func- tion is usually performed with the patient in
same test is repeated with the patient lying on the abdomen, so that the horizontal canal is reversed in the vertical plane (i.e., the direction of the gravity vector with relation to the head is reversed), the direction of nystagmus induced by warm and cold stimulation is reversed.
Monkeys that have had their horizontal semicircular canals blocked with paraffin still have caloric-induced nystagmus, although
Ampullopetal deviation of cupula
Ampulla
Middle ear
the supine position, with head tilted 30 degrees up (placing the horizontal semicircular canals in the vertical plane; see Fig. 1–4c). With the warm caloric stimulus illustrated in Figure 7–9, the column of endolymph nearest the middle ear rises because of its decreased density. This
Crista 37°C
Utricle
c
44°C Water
Temp.
Temp.
gradient
gradient
a External
b canal
causes the cupula to deviate toward the utricle (ampullopetal flow) and produces horizontal nystagmus with the fast component directed toward the stimulated ear. A cold stimulus pro- duces the opposite effect on the endolymph column, causing ampullofugal endolymph flow and nystagmus directed away from the
Horizontal semicircular canal
c
Gravity vector
b
44°C
![]()
![]()
Temp. 37°C
a
stimulated ear. These induced eye movements can be remembered with the mneumontic COWS—cold opposite, warm same. If the
Distance from external canal
Figure 7–9. Mechanism of caloric stimulation of the horizontal semicircular canal (see text for details).
lesser in magnitude than prior to surgery.21 Presumably, the spontaneous afferent nerve activity increases and decreases because of heating and cooling of the afferent nerve, respectively. Consistent with this interpreta- tion, the caloric-induced nystagmus in canal- blocked animals does not reverse direction when the gravity vector is reversed.21 This mechanism could also explain the unexpected finding of caloric-induced nystagmus in space, outside of earth’s gravity.22 Other mechanisms, including differential pressure effects from the temperature gradient and central otolith–canal interactions, have also been proposed to account for caloric responses in space.23 From a clinical point of view, however, gravity is the main driving force for the caloric response. The response can be effectively shut off (in instances in which the patient becomes extremely uncomfortable) simply by having the patient sit up and tilt the head about 30 degrees downward so that the horizontal canals are in the horizontal plane.24
The caloric test is the most widely used clin- ical test of the vestibulo-ocular reflex (VOR) for two major reasons: (1) each labyrinth can be stimulated individually and (2) the stimulus is easy to apply without requiring complex equipment. Several limitations of the test must be appreciated if one is to assess the results properly, however. The slow-component veloc- ity and duration of caloric-induced nystagmus are dependent not only on the relationship between the temperature gradient vector and the gravity vector but also on the blood flow to the skin, presence of fluid in the middle ear chamber, the physical distance between the tympanic membrane and the horizontal canal (longer distance leads to less temperature transfer), and heat conductivity of the temporal bone.25,26 If local blood flow to the skin is decreased (from vasoconstriction due to pain or to anxiety), the maximum slow-component velocity of the response decreases (from decreased heat conductivity through skin), but its duration is prolonged (from delayed heat transfer). Patients with infection or fluid in the middle ear and mastoid air cells may have an increased caloric response (increased maxi- mum slow component velocity) because the fluid can increase heat conductivity from the external ear to the inner ear. Similarly, patients who have undergone mastoid surgery and reconstruction of their middle ear may have
increased responses due to a shortening of the conduction pathway. A thickened temporal bone, by contrast, would produce the opposite effect, because of decreased bone heat con- ductivity. Some of these factors no doubt underlie the large variability of caloric responses measured in normal subjects and explain the occasional unexpected increase or decrease in caloric response found in patients with tempo- ral bone disease. The caloric response can also be attenuated by small or narrow external audi- tory canals, or cerumen build-up within the external canal.
TEST METHODOLOGY
With the bithermal caloric test introduced by Fitzgerald and Hallpike,27 each ear is irrigated for a fixed duration (30 to 40 sec) with a con- stant flow rate of water that is 7° below body temperature (30°C) and 7° above body tem- perature (44°C). One must wait a minimum of 5 min from the end of one response to the next stimulus to avoid additive effects. The major advantages of this test methodology are
(1) both ampullopetal and ampullofugal endo- lymph flow are serially induced in each horizontal semicircular canal, (2) the caloric stimulus is highly reproducible from patient to patient, and (3) the test is tolerated by most patients. The major limitation is the need for constant temperature baths and plumbing to maintain continuous circulation of water through the infusion hose. Patients with migraine may have an increased chance of hav- ing a migraine headache after caloric testing.28 The magnitude of caloric-induced nystag- mus is highly dependent on the degree of fixa- tion permitted during the test procedure. Four different fixation conditions have been used for caloric testing: (1) eyes open, fixating; (2) eyes open, Frenzel glasses; (3) eyes open, total dark- ness; and (4) eyes closed. Without eye move- ment recording devices, obviously only the first two conditions can be used. Comparison of these four conditions in normal subjects shows a consistently lower coefficient of variation (standard deviation/mean) for response mea- surements when the test is performed with eyes open, either behind Frenzel glasses or in
total darkness.29
When caloric testing is performed with fixation (as initially described by Fitzgerald and Hallpike), two separate systems are being
evaluated: the VOR and the smooth pursuit system (see Visual–Vestibular Interaction, Chapter 3). Some normal subjects are very good at suppressing caloric-induced nystagmus with fixation29; others are not. Patients with impaired smooth pursuit (such as patients with cerebellar atrophy) may show no difference in caloric-induced nystagmus with or without fixation.30 When measured with fixation the responses in these patients will appear hyper- active when compared with those of subjects with a normal smooth pursuit system. Eye clo- sure and the associated upward deviation of the eyes can lead to suppression of both spontane- ous and induced nystagmus.29 It can also alter the nystagmus waveform, making it more diffi- cult to quantify with nystagmography. Patients with central nervous system (CNS) lesions often have a horizontal deviation of the eyes on closure, which can also change the waveform of induced nystagmus.31 To avoid these uncontrol- lable variables, caloric testing should be per- formed with eyes open (either in the dark or behind opaque goggles). For a brief period during the test, fixation can be permitted to evaluate the nystagmus suppressing functional status of the smooth pursuit system.32
NORMATIVE DATA
The response to caloric stimulation can be assessed in several ways. The simplest method is to measure the duration of nystagmus after
each infusion, using a stopwatch. Prior to the development of eye movement recordings, this was the only way to quantify the bithermal caloric test. Using nystagmography, however, it is possible to record multiple response mea- surements accurately. Figure 7–10 illustrates an ENG recording of a normal caloric response. The subject was supine, the head elevated 30 degrees, and the eyes open behind Frenzel glasses in a darkened room. Two hundred fifty milliliters of 44°C water was infused into the left ear during the 40 sec marked on the figure, resulting in ampullopetal endolymph flow in the left horizontal semicircular canal, producing left-beating horizontal nystagmus. The nystag- mus began just before the end of stimulation, reached a peak approximately 60 sec post stim- ulus, and then slowly decayed over the next minute. Next to the ENG tracing, nystagmus slow-component velocity, slow-component amplitude, and frequency are plotted versus time. Each measurement demonstrates beat- to-beat variability, but the velocity of the slow components shows the least variability. Furthermore, a decrease in slow-component velocity is the most sensitive indicator of vestibular damage.
As suggested earlier, the absolute magnitude of caloric response depends on several physical factors unique to each subject that are unre- lated to actual semicircular canal function (e.g., blood flow to the skin, fluid in the middle ear, the physical distance between the tympanic
(a)
44° Caloric infusion

5 sec
15°
(b)

Slow Comp. Vel. (deg/sec)
Slow Comp. Vel. (deg/sec)
30
20
10
0

Slow Comp. Ampl. (deg)
Slow Comp. Ampl. (deg)
20 (c)
10
0
0 30 60 90 120
8 (d)

Frequency (beats/sec)
Frequency (beats/sec)
6
4
2
0
0 30
60 90 120
Seconds
Figure 7–10. Caloric response produced by infusion of 250 ml of 44°C water into the left ear of a normal subject.
(a) Bitemporal electronystagmographic recording. Horizontal bar indicates duration of infusion. Plots are of slow- component velocity (b), slow-component amplitude (c), and frequency (d) versus time, generated by a digital computer.
membrane and the horizontal canal, and heat
( ( ) 00
[2]
![]()
conductivity of the temporal bone). The maxi- mum slow-component velocity (MSCV) after a
R L44 R L30
caloric stimulus can be as low as 5 degrees/sec and as high as 75 degrees/sec and still be within the 95% confidence interval for normal subjects.33 Because of this large intersubject variability, intrasubject measurements are more useful clinically (Fig. 7–11). The vestibu- lar paresis formula
compares the MSCV of nystagmus to the right with that of nystagmus to the left in the same subject. Dividing by the total response normal- izes the measurements to remove the large variability in absolute magnitude of normal caloric responses.
A caloric fixation suppression index is
![]()
( ( )
100
[1]
obtained by having the patient fixate on a target
R R44 L L44
during the middle of the response. Because the slow-component velocity of caloric-induced
compares the MSCV of right-sided (R) responses with that of left-sided (L) responses, and the directional preponderance formula
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
H O °C Nystagmus to R
nystagmus is constantly changing, it is impor- tant that the fixation period occur near the time of maximum response to obtain the best esti- mate of fixation suppression. The fixation suppression index is defined as (MSCV with fixation ÷ MSCV without fixation) × 100. With each of these formulas, the result is reported as a percentage of the total response.
In our laboratory, a vestibular paresis is defined as >25% asymmetry between left-and
Normal
Left vestibular paresis
Right directional preponderance
2
30°
44°
30°
44°
30°
44°
Ear L R
L
R L R
L
R
L R
L
R
Nystagmus to L
right-sided responses, and a directional pre- ponderance as >30% asymmetry between left- and right-beating nystagmus, and a fixation suppression index >70% is abnormal. These values are comparable to those reported by other investigators (many use >30% asymmetry to define a vestibular paresis), but it must be emphasized that each laboratory should estab- lish its own normal range because of the many methodologic variables discussed earlier.
Slow component velocity
Slow component velocity
RESULTS IN PATIENTS
Table 7–2 summarizes the abnormalities found in caloric testing, their meaning in terms of location of lesion, and the mechanism by which each abnormality is produced.
Peripheral Lesions
Assuming acceptable test conditions and per-
(40 sec) 1 2 3
![]() Time (min)
Time (min)
Duration
of irrigation
formance (e.g., experienced technician, coop- erating patient, clean and symmetric external auditory canals), the finding of a significant ves- tibular paresis with bithermal caloric stimula-
Figure 7–11. Normal and two common patterns of abnor- mal response to bithermal caloric testing. With a vestibu- lar paresis, the responses to cold (30°C) and warm (44°C) water are decreased on the same side. With a directional preponderance, the responses to warm water on one side and cold water on the opposite side are decreased.
tion suggests damage to the vestibular system that can be located anywhere from the end organ to the vestibular nerve root entry zone in the brain stem. By far, the most common cause of a caloric vestibular paresis is unilateral
Table 7–2 Interpreting the Results of Bithermal Caloric Testing
![]()
Result Location of Lesion Mechanism
![]()
Vestibular paresis Labyrinth, VIII nerve Decreased peripheral sensitivity Directional preponderance Not localizing Tonic bias in vestibular system
Hyperactive responses Cerebellum Loss of inhibitory influence on vestibular nuclei Dysrhythmia Cerebellum Loss of inhibitory influence on pontine nuclei Impaired fixation suppression CNS pursuit pathways Interruption of visual signals on way to
oculomotor neurons
Perverted nystagmus Fourth ventricular region Disruption of vestibular commissural fibers
![]()
peripheral vestibular disease. For the central vestibular system to be the source, the lesion would need to involve the eighth nerve root entry zone. In animal studies focal lesions in different vestibular nuclei did not produce a vestibular paresis. This is not to say that a cen- tral lesion cannot cause a vestibular paresis, but to simply say that a central lesion is much less likely. A recent study reported that 43% (10 out of 23) of patients with stroke (defined as a relevant lesion with restricted diffusion on magnetic resonance imaging [MRI]) causing acute vertigo who underwent caloric testing had a vestibular paresis of >25%.34 In 22% (5 out of 23) of these stroke patients the ves- tibular paresis was severe (>75%). Because peripheral causes of vestibular paresis are much more common than central causes, the likelihood of a central cause is low particularly when there are no other central ocular motor abnormalities (e.g., bi-directional gaze evoked nystagmus) or other brainstem signs.
A directional preponderance on caloric testing occurs with peripheral end-organ and eighth nerve lesions and with CNS lesions (from brain stem to cortex).30 It indicates an imbalance in the vestibular system and is usually associated with spontaneous nystagmus: the velocity of the slow components of the spontaneous nystagmus adds to that of the caloric-induced nystagmus in the same direction and subtracts from that of the caloric-induced nystagmus in the opposite direc- tion.35 Occasionally, a directional preponderance will occur in patients without spontaneous nys- tagmus; most of these patients have peripheral lesions, although about 5% have central lesions.36 The need to distinguish between end-organ and eighth nerve lesions is a common clinical problem. Partial lesions of the eighth nerve should not, in theory, affect the duration of induced nystagmus, as it is related to the time course of cupular deflection and not to the ability
of the nerve fibers to transmit action potentials. However, end-organ lesions involving the cup- ula and hair cells should affect both the MSCV and duration of the responses. Unfortunately, this turns out not to be a reliable way of differ- entiating end-organ from eighth nerve lesions. Lesions involving the eighth nerve can reduce the duration of nystagmus, whereas end-organ lesions (particularly in the early stages) frequently result only in decreased MSCV (the duration of response remains unaffected). The magnitude of loss is of some help in differentiat- ing nerve from end-organ lesions. A complete or nearly complete unilateral paralysis is more commonly associated with nerve lesions than with end-organ labyrinthine lesions.
The vestibular paresis and directional pre- ponderance formulas are of little use in evaluat- ing patients with bilateral peripheral vestibular lesions, because caloric responses are symmet- rically depressed. Because of the wide range of normal values for MSCV, the patient’s value may decrease severalfold before falling below the normal range. For example, a patient may have a MSCV of 7 degrees/sec for each side, values that are typically within the normal range. However, if that patient has other features indi- cating a bilateral vestibulopathy, then it can be assumed that the MSCV would have been sub- stantially higher if it was tested prior to the pre- sentation. Baseline and serial measurements in the same patient are needed if one hopes to identify early bilateral vestibular impairment, such as that produced by ototoxic drugs.
Central Lesions
As suggested earlier, patients with CNS lesions may exhibit a vestibular paresis on caloric test- ing if the lesion involves the root entry zone of the vestibular nerve.34 The most common neu- rologic disorders associated with this finding
Lesions of the cerebellum can lead to increased caloric responses, possibly because of loss of the normal inhibitory influence of the cerebellum on the vestibular nuclei. Because of the wide range of normal caloric responses, however, it is unusual for any of the responses to exceed the upper normal range. Patients with the cerebellar atrophy syndromes demon- strate a wide range of caloric responses.37 Those with Friedreich’s ataxia often have bilaterally decreased responses because of associated atrophy of the vestibular nerve and ganglia, whereas those with a dominantly inherited spi- nocerebellar ataxia (SCA) syndrome could have decreased, normal, or even increased responses, depending on which areas of the medulla and pons are involved. Increased caloric responses, when they do occur, are usually found in patients with clinically pure cerebellar atrophy (e.g., SCA-6, see Chapter 18). Bilaterally reduced caloric responses are common in SCA-1 and SCA-3.38–40
An abnormal fixation suppression index on caloric testing typically occurs with lesions involving the smooth pursuit system (from the parietal-occipital cortex to the pons and cere- bellum). Lesions of the midline cerebellum produce the most profound impairment of fixa- tion suppression. When asymmetric, pursuit deficits in one direction correlate with suppres- sion deficits in the opposite direction.
Dysrhythmia refers to a marked beat-to-beat variability in caloric-induced nystagmus ampli- tude without any change in the slow-component velocity profile. The cerebellum is important for controlling the amplitude of nystagmus fast components, and loss of this control with cere- bellar lesions may lead to a disorganized nystagmus pattern. Unfortunately, from a diag- nostic point of view, caloric dysrhythmia also occurs in normal subjects when they are tired and inattentive. As will be shown in the next
section, rotatory stimuli are better suited than caloric stimuli for examining the pattern of induced nystagmus.
Vertical or oblique nystagmus produced by caloric stimulation of the horizontal semicircu- lar canals is called perverted nystagmus. Normal subjects commonly exhibit a small vertical component on nystagmography recordings of caloricinduced nystagmus, but vertical compo- nents larger than the horizontal components are clearly abnormal.41 Perverted nystagmus with caloric stimulation has been reported with both peripheral and central lesions, the latter usually being in the region of the floor of the fourth ventricle (near the vestibular nuclei).42 Perverted caloric nystagmus occurs in rhesus monkeys after unilateral focal lesions occur in the rostral medial vestibular nucleus. Warm caloric stimulation on the intact side produces downward nystagmus, and cold stimulation produces upward diagonal nystagmus. These findings are probably due to a disturbance of the commissural fibers between the vestibular nuclei.
Tests of Visual–Ocular Control
The central vestibulo-ocular connections are highly integrated with the visual-ocular stabi- lizing pathways, and both systems share the final common pathway of the oculomotor neu- rons (see Comparison of Vestibular- and Visual- Induced Eye Movements in Chapter 3). If the efferent limb of the VOR arc is damaged, visu- ally controlled eye movements are also abnor- mal; but if the afferent limb of the reflex is damaged, visually controlled eye movements are usually normal. Because eye movement– recording techniques used for quantifying the VOR can also be used to quantify visually con- trolled eye movements, an important “bonus” of information is obtained with little increased effort. Table 7–3 summarizes the types of sac- cade, smooth pursuit, and optokinetic abnor- malities commonly associated with focal lesions of the nervous system.
SACCADIC EYE MOVEMENTS
Methods of Testing and Results in Normal Subjects
One can induce saccadic eye movements with a series of dots or lights separated by known
Table 7–3 Summary of Visual Ocular Control Abnormalities Produced by Focal Neurologic Lesions
![]()
Location of Lesion Saccades Smooth Pursuit and OKN Slow Phase
![]()
Cerebellopontine angle Ipsilateral dysmetriaa Progressive ipsilateral impairment
Diffuse cerebellar Bilateral dysmetria Bilateral impairment
Intrinsic brain stem Marked slowing, increased delay time Ipsilateral or contralateral impairment
Basal ganglia Mild slowing, hypometria,b increased delay time
Bilateral impairment
Frontoparietal cortex Difficulty inhibiting reflex saccades Ipsilateral impairment
![]()
aUnder- and overshoots.
bUndershoots only.
OKN, optokinetic nystagmus.
angular degrees, or with a dot of light generated on a screen and moved through a series of step- wise jumps of different amplitudes.43 The ENG recording in Figure 7–12A illustrates the high speed and accuracy of saccadic eye movements induced in a normal subject by a target moving in steps of random amplitude. Normal subjects consistently undershoot the target for jumps larger than 20 degrees, requiring a small cor- rective saccade to achieve the final position. Overshoots of the target are rare. A character- istic delay time of approximately 200 msec occurs between each target jump and induced saccade.
Computer algorithms have been developed to rapidly quantify these saccade parameters.43 Saccades are easily identified on the basis of their characteristic velocity profile. The rela- tionship between peak velocity and amplitude (the so-called main sequence) is nonlinear, with decreasing peak velocities occurring at higher amplitudes (Fig. 7–12B). For example, the average peak velocity for a 15 degrees sac- cade is 400 degrees/sec, whereas that for a 30 degrees saccade is 550 degrees/sec. Saccade accuracy is defined as the ratio of the saccade amplitude divided by the target displacement amplitude times 100. The mean saccade accuracy
![]()
Right eye
![]()

Left eye
Target
15°
R
5 sec
L
800
deg/sec
deg/sec
600
400
200

0
Left eye
Right eye

40 20 0
20 40 40 20 0
Amplitude (deg)
20 40
Figure 7–12. Saccadic eye movements induced in a normal subject by a target moving in steps of random amplitude (3°–36°) and changing intervals between jumps (0.5–2.5 sec). (top) Monocular horizontal electronystagmographic record- ings. (bottom) Computer-generated plots of peak velocity versus amplitude for entire sequence (dotted lines represent normal mean ± standard deviation).
Results in Patients
Slowing of saccadic eye movements can occur with lesions anywhere in the diffuse central pathways involved in generating saccades. The most pronounced slowing occurs with lesions of the pretectal and paramedian pontine gaze centers, the oculomotor neurons, and the extraocular muscles. Lesions involving these pathways impair both voluntary and involun- tary saccades. Damage to the oculomotor neurons, oculomotor nerves, and extraocular muscles causes a slowing of saccades when the paretic muscle is the agonist required to gener- ate the sudden force necessary to move the globe rapidly. Saccade slowing identified on eye movement recording can occur before clinical examination reveals the presence of strabismus.45,46 Recordings have been particu- larly helpful for identifying early lesions of the medial longitudinal fasciculus (MLF), mani- fested by slowing of adducting saccades made by the medial rectus on the side of the lesion (Fig. 7–13).47,48 A characteristic saccade abnor- mality is seen with myasthenia gravis. Saccades begin with normal velocity, but within a short time the transmitters at the myoneural junc- tion are depleted, and the remainder of the saccade is markedly slow.49 In some patients with severe oculomotor dysfunction, only brief bursts of oculomotor firing are possible before a complete block occurs. This results in the unusual situation in which a patient with almost complete absence of sustained eye movements can have small-amplitude, high-velocity sacca- des followed by a quick return to the primary position (so-called ocular quiver). These sac- cade abnormalities are usually rapidly reversed with intravenous Tensilon.50
Reversible saccade slowing is produced by fatigue and by ingestion of alcohol or tranquil- izers.51–53 This results from impaired synaptic transmission through the multineuronal net- works needed to generate the high-frequency firing for horizontal and vertical saccades. Patients with Huntington’s disease and pro- gressive supranuclear palsy develop slowing of saccades, apparently due to diffuse degenera- tion of supranuclear pathways.54,55 Focal disease
of the pretectum or paramedian pontine retic- ular formation produces selective slowing of vertical and horizontal saccades, respec- tively.56,57 Lesions of one paramedian pontine center produce ipsilateral saccade slowing. The pretectal centers for upward and downward saccades are separate (downward ventral dor- sal to the upward center) but are so close together that lesions usually involve both. Destruction of the pretectal and pontine supra- nuclear saccade centers results in complete absence of saccadic eye movements (voluntary and involuntary). Patients with such a dysfunc- tion produce only a slow tonic deviation of the eyes with vestibular or optokinetic stimuli because of the absence of fast components (see Fig. 7–29c).57
Impaired saccade accuracy is most com- monly seen with cerebellar disorders.58,59 Overshooting of the target (saccade overshoot dysmetria) is most apparent, as overshoots rarely occur in normal subjects (See Video 7–1). The velocity of these inaccurate saccades is normal unless the brain stem is also involved. Of the cerebellar atrophy syndromes, saccade dysmetria is most prominent with Friedreich’s ataxia.60,61 Monocular overshoots in the abduct- ing eye are characteristic of MLF lesions (see Fig. 7–13). Disorders of the cortical and sub- cortical supranuclear centers also affect the accuracy of saccades.62,63 Patients with Parkinson’s disease exhibit delayed saccade reaction time and hypometria of voluntary sac- cades. Complete removal of one hemisphere or the presence of a large frontal parietal lesion results in hypometria of horizontal saccades made in the contralateral direction.64 Vertical saccades are unaffected. Animals with lesions of the frontal eye fields may have normal- appearing saccade metrics but have difficulty inhibiting reflex saccades.65 Patients with lesions of the frontal cortex and basal ganglia have sim- ilar difficulties.66,67 This can be demonstrated with the antisaccade test, in which a fixation tar- get is illuminated in the periphery and the patient is instructed to make a saccade in the exact opposite direction. Normal subjects can reliably perform this task, but patients with lesions involving cortical and subcortical presac- cade structures often make unwanted saccades to the fixation target before refixating in the desired location.
Patients with acquired and congenital oculo- motor apraxia68 and ataxia telangiectasia69
![]()
![]()
![]()
Left eye
Target
15°
R
5 sec L
800
deg/sec
deg/sec
600
400
200
0
Left eye

40 20 0
Right eye

20 40 40 20 0 20 40
Amplitude (deg)
Figure 7–13. Saccadic eye movements in a patient with bilateral medial longitudinal fasciculus lesion caused by multiple sclerosis. Recordings are as in Figure 7–12. Adducting saccades are markedly slow; abducting saccades have normal velocity but overshoot the target.
exhibit prolonged reaction time for the initia- tion of voluntary saccades and use a series of hypometric saccades to produce refixations. Nystagmus fast components (involuntary sac- cades) are also abnormal, such that the eyes deviate in the direction of the slow component rather than in the direction of the fast compo- nent. To compensate for impaired voluntary saccades, these patients often use head thrusts to perform refixation. Because their VOR is intact, the head thrusts produce con- troversive deviation of the eyes, necessitating an overshoot of the head thrusts to obtain fixa- tion. Fixation is then maintained as the head is slowly returned on line with the target. The site of the anatomic defect that produces these abnormalities in voluntary saccades is unknown.
SMOOTH PURSUIT
Methods of Testing and Results in Normal Subjects
Examining physicians can test smooth pursuit eye movements by slowly moving their finger or a pencil back and forth and asking the patient to follow it as well as possible. The target should be moved as smoothly as possible and the movement should not be too fast (about 1/2 cycle/sec is an ideal rate). A more exact rela- tionship between the velocity of the target and
the eye is determined by using precise targets and eye movement recording. A pendulum hanging from the ceiling or a metronome pro- vides an inexpensive reproducible sinusoidally moving target. Precise control of the target can be achieved over a series of velocities by pro- jecting a dot onto a screen with a motor-con- trolled device. Figure 7–14b illustrates an ENG recording of horizontal smooth pursuit in a normal subject as he follows a sinusoidally moving dot on a white screen (0.3 Hz, maxi- mum amplitude 18 degrees). The accuracy of smooth pursuit can be quantified by repeatedly sampling eye and target velocity and plotting the two velocities against each other (see Fig. 7–14d). A computer algorithm makes the comparison between eye and target velocity after saccade waveforms have been removed.70 The slope of this eye target velocity relation- ship (in this case, 0.95) represents the gain of the smooth pursuit system. The mean gain determined from similar plots in 25 normal young subjects was 0.95 ± 0.07. Older normal subjects (>70 years of age) show marked vari- ability in pursuit ability, and therefore pursuit testing must be interpreted with caution in older patients.71 Also, smooth pursuit gain decreases with both increasing frequency and increasing velocity of the target. Each labora- tory must establish normative data for its stan- dard test protocol.


15°
5 sec R
(b)
L
15°
![]()
(c)
15°
(d)


(e)
48
32
16 Target
16 32 48 velocity
(deg/sec
Target
16 32 48 velocity
16 (deg/sec
32
48
Eye velocity (deg/sec)
Figure 7–14. Smooth pursuit of a target (a) moving with a sinusoidal waveform in a normal subject (b) and a patient with cerebellar atrophy (c). Bitemporal horizontal recording. Eye velocity is plotted against target velocity (both sampled 10 times/sec) after saccades have been removed for the normal subject (d) and patient (e).
Results in Patients
Patients with impaired smooth pursuit require frequent corrective saccades to keep up with the target, producing so-called cogwheel, or sacca- dic, pursuit (see Fig. 7–14c). As expected, the gain (given by the slope of the eye velocity–target velocity plot) of the smooth pursuit system is markedly decreased in such patients (see Fig. 7–14e). It must be emphasized, however, that normal subjects may intermix saccades with smooth pursuit movements, particularly if they are inattentive or fatigued, or if the target velocity exceeds the limit of their smooth pursuit system.72 Therefore, quantitative analysis of intersaccadic eye velocity is a more reliable way of assessing the accuracy of smooth pursuit than simply observ- ing the frequency of superimposed saccades.
Abnormalities of smooth pursuit are of lim- ited localizing value, as they occur with disor- ders throughout the CNS. Acute lesions of the peripheral labyrinth or vestibular nerve tran- siently impair smooth pursuit contralateral to the lesion when the eyes are moving against the slow component of spontaneous nystagmus.73 This asymmetry in smooth pursuit disappears within a few weeks despite the continued pres- ence of spontaneous nystagmus in darkness. Just as tranquilizing drugs, alcohol, and fatigue affect saccadic eye movements, they also impair smooth pursuit eye movements.74 Barbiturates may impair smooth pursuit before affecting sac- cadic eye movements, which suggests an increased sensitivity of the smooth pursuit system. Patients with diffuse cortical disease75 (degenerative or vascular), basal ganglia
disease54,76(Parkinson’sdiseaseandHuntington’s disease), or diffuse cerebellar disease37,58 consis- tently have bilaterally impaired smooth pursuit eye movements. Focal disease of one cerebellar hemisphere or one side of the brain stem usu- ally produces ipsilateral impairment of smooth pursuit, although large cerebellar pontine angle tumors are frequently associated with bilater- ally impaired smooth pursuit.14 Focal cortical lesions in the parietooccipital region impair ipsilateral smooth pursuit (Fig. 7–15a).77,78
OPTOKINETIC NYSTAGMUS
Methods of Testing and Results in Normal Subjects
The simplest optokinetic stimulus is a striped cloth that can be moved across the patient’s visual field in each direction. While the patient stares at the cloth, the amplitude of induced nystagmus in each direction is compared. This type of test permits identification of absent or markedly asymmetric optokinetic nystagmus (OKN). The test sensitivity is improved by using an optokinetic stimulus of known velocity and recording the induced nystagmus. Figure 7–16 shows such a recording of OKN induced by a striped drum completely surrounding a subject and moving at a constant velocity of 30 degrees/sec. At the arrow, the lights were turned off and optokinetic after-nystagmus (OKAN) was recorded. A plot of slow- component velocity is provided beneath the tracing. Typically, the OKN slow-component velocity approaches that of the drum velocity as
![]()
Target position
![]()
![]()
15º
Patient 1
![]()
Patient 2
1 sec
15º
![]()
L
![]()
(b)
![]()
Drum velocity
60º/sec
0º/sec
15º
60º/sec
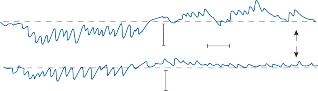
Patient 1
Patient 2
15º
15º
1 sec R
L
Figure 7–15. Right monocular recordings of horizontal smooth pursuit (a) and optokinetic nystagmus (OKN) (b) from two patients with left parietal lobe lesions.77 In A the patients tracked a laser dot moving in a sinusoidal fashion (left: 0.2 Hz, peak velocity 22.5°/sec; right: 0.4 Hz, peak velocity 45°/sec). In B a surrounding optokinetic drum moved in a sinusoidal pattern (0.05 Hz, peak velocity 60°/sec). Pursuit and OKN slow phases to the left were markedly impaired.
long as the drum velocity does not exceed 30 to 40 degrees/sec. As with smooth pursuit gain, the gain of OKN (slow-component velocity/ drum velocity) drops off with increasing fre- quency and drum velocities in normal subjects (Fig. 7–17).79 The OKAN velocity is more vari- able than OKN velocity even in young normal subjects. There is a rapid exponential dropoff followed by a gradual decay, as shown in Figure 7–16. The mean OKAN slow-component
velocity (after the initial rapid dropoff) and the mean OKAN duration in 20 normal subjects after 1 min of 30 degrees/sec optokinetic stim- ulation was 6.3 degrees/sec ± 4.5 degrees/sec and 23.75 sec ± 23.1 sec, respectively.79
Results in Patients
As a general rule, abnormalities of optokinetic slow components parallel abnormalities in
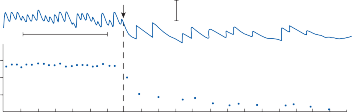
a
SCV (deg/sec)
SCV (deg/sec)
b 5 sec
30
20
10
Lights out
15º
0
55 60 0 5 10
Time (sec)
Figure 7–16. Optokinetic nystagmus (OKN) induced by a surrounding striped drum moving at a constant velocity of 30°/sec. At the arrow the lights were turned off, and optokinetic after-nystagmus (OKAN) was recorded. a Bitemporal electronystagmographic recording. b Plot of slow-phase velocity (SCV) versus time.
![]()
Slow Phase Eye Velocity (deg/sec)
Slow Phase Eye Velocity (deg/sec)
60
50
40
30
20
10
Optokinetic Drum Velocity (deg/sec)
![]()
Figure 7–17. Normal mean ± 1 standard deviation for horizontal optokinetic nystagmus slow-phase velocity at different drum velocities. , ramp acceleration from 0° to 70°/sec in 1 min; , constant velocity for 30 sec; , sinusoidal (0.05 Hz).
smooth pursuit, and abnormalities of fast components correlate with abnormalities of voluntary saccades.79 Symmetrically decreased slow-component velocity is produced by dif- fuse disease of the cortex, diencephalon, brain stem, and cerebellum.37,54,58,62,75 As with smooth pursuit, focal lateralized disease of the parietal occipital region, brain stem, and cerebellum result in impaired OKN when the stimulus moves toward the damaged side (see Fig. 7–15b).77,78 Lesions of the occipital lobe, although associated with a hemianoptic visual field defect, are not associated with impaired smooth pursuit or OKN, presumably because each parietal lobe receives oculomotor signals from both occipital lobes. Some patients with severely impaired smooth pursuit exhibit a gradual buildup in OKN slow-component velocity.80 This is a feature of OKN normally seen in afoveate animals that have only a sub- cortical OKN system (see Chapter 3). Presumably, in normal humans the cortical pursuit system dominates the subcortical OKN system, so normal OKN exhibits features of normal pursuit. When the cortical pursuit sys- tem is lesioned, however, the remaining OKN may exhibit features of the subcortical system. Patients who are unable to produce saccadic eye movements produce only a slow tonic devi- ation of the eyes in the direction of an optoki- netic stimulus. Although patients with slow
saccades produce OKN, the waveform is rounded, and the amplitude and slow- component velocity are decreased. The delayed ending of the impaired fast component sub- tracts from the initial part of the slow compo- nent in the opposite direction. The many causes of saccade slowing were outlined in detail in the previous section.
Abnormalities of OKAN are typically seen with peripheral vestibular lesions.81 Unilateral lesions result in asymmetric OKAN (present only in the direction of the spontaneous nystagmus), whereas bilateral lesions (e.g., due to ototoxic drugs) result in diminished or absent OKAN in both directions.79
ROTATIONAL TESTING OF VESTIBULO-OCULAR REFLEXES
The determination of which vestibular subor- gans are stimulated by rotation of the head requires knowledge of three factors: (1) the axis of rotation, (2) the orientation of the skull (and thus the labyrinth) with respect to the rotation, and (3) the orientation of the rotation with respect to gravity (Fig. 7–18). Currently, rotational tests of the vestibulo-ocular reflexes concentrate on the horizontal semicircular canal ocular reflex (yaw, z-axis rotation) because
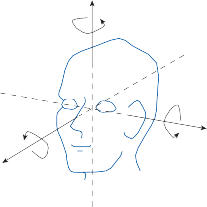
Roll
Pitch
y-axis
x-axis
Yaw

Off-center rotation Off-vertical rotation
Figure 7–18. Definitions of different axes and orientations for rotational testing.
it is the easiest reflex to stimulate and record. Rotational tests of the vertical semicircular canals and otoliths are still in the developmen- tal stage and are not generally available in most clinical settings (Table 7–4).
Rotational testing of the horizontal semicir- cular canal offers several advantages over caloric testing. Multiple graded stimuli can be applied in a relatively short period of time, and the testing is usually well tolerated by patients.
Table 7–4 Vestibular Tests and the Suborgans They Stimulate
Test | Horizontal Canals | Vertical Canals | Otoliths |
Conventional rotational chair | + | ||
Upright pitch rotation | + | + | |
Onside pitch rotation | + | ||
Static ocular counterrolling | + | ||
Dynamic ocular counterrolling | + | + | |
Eccentric rotation | + | + | |
Off-vertical rotation | + | + | |
Linear track | + | ||
Parallel swing | + |
According to the pendulum model intro- duced in Chapter 2, the slow-component veloc- ity of rotational-induced nystagmus should be proportional to the deviation of the cupula, which, in turn, is proportional to the intensity of stimulation. As will be demonstrated in the following sections, this model’s applicability to different forms of rotational stimulation pro- vides a rational approach to the evaluation of clinical rotational testing.
Relationship between Stimulus and Response
PASSIVE WHOLE-BODY YAW ROTATION
With standard rotational chairs, the angular acceleration in the z-axis can be precisely con- trolled and multiple response measurements
can be accurately monitored. Figure 7–19 illus- trates the nystagmus responses of a normal subject to three common types of angular acceleration used in clinical laboratories. The subject was rotated in the z-axis with the eyes open in complete darkness while he performed continuous mental arithmetic to maintain alert- ness. Each stimulus produced a peak angular chair velocity of 120 degrees/sec.
As with caloric testing, maximum slow- component velocity is the response measure- ment most useful for quantifying testing. The coefficient of variation (standard deviation divided by the mean) for maximum slow- component velocity after a rotational stimulus is about one-half the coefficient of variation after a caloric.82 Even with this increased preci- sion, however, there is still large variation in the rotational responses of normal subjects. Factors such as stress, fatigue, level of mental alertness, and habituation all contribute to the variability (see Chapter 3). Complete darkness is needed so that the patient cannot fixate on a target.
The slow-component velocity profiles (Fig. 7–19d–f) for each stimulus can be pre- dicted by the pendulum model discussed in Chapter 2. Note the similarity between these profiles and the time course of cupula devia- tion illustrated in Figure 2–15. An important
![]()
![]()
Chair velocity
![]()
EOG
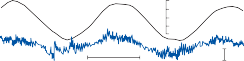
Chair velocity
EOG
Chair velocity
10 sec
10 sec
120
0
120
15º
15º
120
60
0
120
60
0
d
![]()
60
40
Slow component velocity (deg/sec)
Slow component velocity (deg/sec)
20
0
10
![]()
0 60
5

10 | 20 | 30 | 40 | 50 |
10 | 20 | 30 | 40 | 50 |
80 e
40
0
40
80
![]()
30 f
20
10
20 30 40 50
80 100
EOG
10 sec
15º
0
0 10 20 30 40 50
Seconds
Figure 7–19. Nystagmus recording (a–c) and slow-component velocity profile (d–f) with three types of angular accel- eration, each resulting in a maximum velocity of 120°/sec. With the impulse stimulus (a) the change in velocity occurs in < 1 sec, with an acceleration of 140°/sec2. The sinusoidal stimulus (b) has a frequency of 0.05 Hz (20 sec/cycle) and a maximum acceleration of 38°/sec2. The constant acceleration stimulus (c) is 4°/sec2 for 30 sec (horizontal bitemporal electrooculorgraphic [EOG] recordings).
Two types of measurements are typically used to quantify the response to these tradi- tional rotational stimuli: a magnitude (gain) and a timing (time constant or phase shift) measurement. The gain is defined as the peak slow-phase eye velocity divided by the peak stimulus (chair) velocity. The time constant of the step response is defined as the time required for the response to decay to 1/e or to 37% of the maximum value. For a sinusoidal test, the phase is typically measured by comparing the time of the maximum head velocity (measured by chair velocity) with the time of the maxi- mum slow-phase eye velocity. Consistent with models of the canal-ocular reflex, the maximum slow-phase eye velocity leads the maximum head velocity at low frequencies of sinusoidal rotation in normal subjects. The time constant (TCOR) of the canal-ocular reflex measured after
a step change in angular velocity is inversely
related to the phase lead () at low frequencies of sinusoidal rotation by84
in all three axes—yaw, pitch, and roll. Although expensive rotational devices are available for passively rotating the whole body at high accel- erations in all three planes, the tests can also be performed with passive head-on-body rotation by manually moving the head with small- amplitude thrusts in all three planes (as is done with the bedside head-thrust test).87 The eye movement recording system must be precise enough to measure the small-amplitude, high- velocity eye movements that are induced. The scleral search coil technique is a sensitive eye movement recording system that has been used to record these types of eye movements in research laboratories. With this system, the sub- ject is seated within a set of external reference magnetic field generator coils and wears a con- tact lens that contains tiny “receiving” coils to record horizontal, vertical, and torsional eye posi- tion.88 Similar coils can be attached to the head to accurately measure head position. VNG systems that are tightly coupled to the head to prevent slippage are just becoming available.89 ENG is not sensitive enough and, of course, it can only accurately record horizontal eye movements.
The unique advantage of the high-acceleration test is that one can measure gain in the time domain early after the onset of the stimulus. Early gain measurements (<50 msec after the stimulus) reflect an almost pure vestibulo- ocular response, since visual and other modula- tory central influences occur later in the response. Another unique advantage of the high-acceleration test is that it is more sensitive than traditional low-acceleration testing for
![]()
![]()
TCOR q
[3]
identifying the difference between ampullopetal and ampullofugal responses when there is a uni- lateral loss of function (see later discussion).
where = 2F.
HIGH-ACCELERATION
SMALL-AMPLITUDE ROTATION
Brief high-acceleration impulses provide a unique assessment of the VOR not available with traditional stimuli.85,86 Accelerations >2000 degrees/sec2 are typically applied for <200 msec, leading to a head position change of 10 to 15 degrees. By contrast, most commercially avail- able motorized chairs have a weight-dependent peak acceleration <150 degrees /sec2. Because only a small amplitude of head displacement is produced, the head accelerations can be applied
ACTIVE HEAD ROTATION (AUTOROTATION)
Active head-only rotational testing is a method for assessing the VOR that can potentially be performed at the bedside or in the clinic with inexpensive, transportable equipment.90–92 Patients are taught to move their head back and forth in a sinusoidal pattern, typically in the range of 0.5 to 3 or 4 Hz. The test can theo- retically be performed in all three axes, although subjects find it easiest to rotate their head in the yaw axis. To date, eye movements have mostly been recorded with ENG, but with this system only horizontal eye movements can be
accurately recorded. Auditory cueing is typically used to instruct the patient to perform the head rotations but somatosensory cues may provide an even better training stimulus.93 The test can be performed either at discrete sinusoidal fre- quencies or by a sweep of frequencies, although the latter stimulus may be more difficult to train. Gain and phase measurements can be made after frequency analysis of the data; this analysis is similar to that performed with pas- sive whole-body rotation. Of course, at these higher frequencies, no nystagmus is recorded and the sensitivity of the eye movement record- ing system becomes more important. A major advantage of the autorotation test is that it pro- vides a measurement of gain for the VOR in the frequency range (>1 Hz) in which it nor- mally functions.94 Disadvantages include the following: patients may have difficulty perform- ing the movements,92 responses may vary with practice,92 poor test-retest inter-individual repeatability,95 and extravestibular influences such as preprogrammed compensatory eye movements and neck-ocular responses can help compensate for vestibular loss.96,97
Results in Normal Subjects
PASSIVE WHOLE-BODY YAW ROTATION
Step Changes in Angular Velocity
The main advantage of the step stimulus is that it provides a rapid assessment of the gain and the time constant of the canal-ocular reflex in each direction. Because the stimulus is brief, however, if subjects are not maximally alert or if they attempt to suppress the response, the ini- tial peak will be blunted and the estimate of gain, inaccurate. For this reason, several mea- surements should be averaged in each direc- tion. The results of a typical response to a step change in velocity in a normal subject are shown in Figure 7–20a. Slow-component velocity (log- arithmic scale) is plotted versus time; each dot represents the average slow-component veloc- ity over a 25-msec interval. Fast components have been removed. The gain (peak slow-com- ponent velocity/peak chair velocity) can be read directly from these plots. The time constant (TCOR) represents the time required for the
slow-component velocity to fall to 37% of its
peak value given by the slope of a regression
line fitted to the data. The normal mean gain
and TCOR values calculated from similar plots in 20 normal subjects were 0.63 ± 0.18 and
12.2 ± 3.6 sec, respectively.84 These values are relatively stable over a wide range of step changes in velocity (0 to 250 degrees/sec), but both show a gradual decrease with larger- magnitude impulses (>120 degrees/sec).82,83
The variance associated with measurements comparing clockwise and counterclockwise responses in the same subject is less than the variance in response between subjects. A nor- malized difference formula, [(clockwise − counterclockwise) ÷ (clock wise + counter- clockwise)] × 100, is analogous to the directional preponderance formula used with caloric test- ing. Greater than 20% asymmetry on this nor- malized difference formula is considered abnormal in our laboratory.
Sinusoidal Changes in Angular Velocity
With sinusoidal rotational testing, the gain of the canal-ocular reflex can be measured at mul- tiple discrete frequencies after the subject has attained a steady-state response. It usually pro- vides a more accurate assessment of gain than is obtained with the step test. The main disadvan- tage is the more time that it takes to test a broad frequency range. Also, unlike the step test, sinusoidal testing measures only a single time constant (the low-frequency phase lead reflects the average time constant in both directions).
Two standard computer plots generated dur- ing sinusoidal rotational testing in a normal sub- ject are shown in Figure 7–21a. The subject was rotated at 0.05 Hz (peak velocity 60 degrees/ sec) with eyes open in the dark while perform- ing continuous mental alerting tasks. As with the step data shown in Figure 7–20, each dot repre- sents the average slow-component velocity over a 25-msec interval. The gain (peak slow-compo- nent velocity/peak chair velocity) in each direc- tion can be read directly from these plots, or an average gain can be calculated by performing a frequency analysis (Fourier analysis) on the data. From this analysis, one obtains the gain, dc bias, and phase relationship between the funda- mental of the slow-component velocity and the chair velocity.70 If the slow-component velocity data are symmetrical (as in normal subjects), the phase can be read directly from these plots by comparing the time of the maximum or the zero eye velocity with that of the chair velocity.
130
90
SCV
SCV
50
CW
Gain = 0.78
TCOR = 12.0
CCW
Gain = 0.75
TCOR = 14.1
10
0 10 20 30
0 10 20 30
(b)
130
90
SCV
SCV
50
10
CW CCW
Gain = 0.50 Gain = 0.95
TCOR = 5.2 TCOR = 11.2
0 10 20 30 0 10 20 30
Time (seconds)
Figure 7–20. Plots of nystagmus slow-component velocity (SCV) (log scale) versus time after a step change in angular velocity (100°/sec acceleration 140°/sec2) in a normal subject (a) and a patient with an acute right peripheral vestibular lesion (b). Gain is peak SCV divided by change in chair velocity. TCOR is the slope of a regression line best fit to the data. CCW, counter clockwise; CW, clockwise, (see text for details).
However, if the responses are asymmetric (as in Fig. 7–21b), an accurate assessment of phase can be obtained with a Fourier analysis of the data. The plot of slow-component velocity ver- sus stimulus velocity (Fig. 7–21, right) provides
a rapid visual assessment of dc bias and facilitates measurement of an average gain in each direction (i.e., the slope of the line in each direction). As with step-rotational stimuli, >20% asymmetry on the standard

80
40
Slow Component Velocity (deg/sec)
Slow Component Velocity (deg/sec)
0
–40
(a)
Gain = 0.58
DCBias = 1.2 | |
DCBias = 1.2 | |
Phase = 8
Gain R = 0.64
L = 0.54
DCBias = 0
Normal
–80
80
40
0
–40
(b)
Gain = 0.54
DCBias = 12.8 | |
DCBias = 12.8 | |
Phase = 25
Gain R = 0.72
L = 0.37
DCBias = 9
Patient
–80
0 20
Time (sec)
40 80 40 0 –40 –80
Stimulus Velocity (deg/sec)
Figure 7–21. Plots of nystagmus slow-component velocity versus time (left) and versus chair velocity (right) during sinu- soidal angular rotation (0.05 Hz, 60°/sec peak velocity) in a normal subject (a) and a patient with an acute right peripheral vestibular lesion (b) (same patient as in Fig. 6–20). The gain, phase (lead) and dc bias (+ rightward bias) were determined from frequency analysis (Fourier analysis) of the data.
The gain and phase of the canal-ocular reflex vary with frequency in normal subjects (Fig. 7–22),84 which is consistent with the pen- dulum model. Normal subjects exhibit an approximate 45-degree phase lead of eye veloc- ity relative to chair velocity at 0.01 Hz, but this phase lead is near zero by 0.2 Hz.
HIGH-ACCELERATION,
LOW-AMPLITUDE ROTATION
Typical responses to a high-acceleration impulse in all three planes in a normal subject are shown
in Figure 7–23.87 The impulses are passive, unpredictable, low-amplitude (10–20 degrees), high-acceleration 3000 to 4000 degrees/s2) head rotations (head thrusts) in yaw, pitch, and roll with the subject sitting upright. Head and eye position and velocity are almost equal and opposite (gain near 1.0) for yaw and pitch impulses, so there is relatively little change in gaze position. By contrast, the compensatory eye movement in the roll plane has a gain of about 0.8, so there is a gaze instability in this plane. This difference in normal gain in the three planes of rotation can be more readily seen in plots of eye velocity versus head veloc- ity shown in Figure 7–24 (top traces). In the yaw and pitch planes, the slope is near 1.0
![]()
![]()
![]()
![]()
![]()
![]()
![]()
1.0
![]()
Gain
Gain
0.1
0.05 ![]()
80
70
60
50
Phase
Phase
40
30
20
10
0
–10
0.01
0.1 1.0
Frequency (Hz)
Figure 7–22. Plots of the gain and phase (mean ± 1 standard deviation) of the horizontal canal-ocular reflex as a function
![]()
of frequency in 10 normal subjects (), 20 patients with compensated unilateral vestibular lesions ( ), and 22 patients with bilateral peripheral vestibular lesions (). All subjects were tested with mental alerting in the dark. The unilateral patients
had absent caloric response on one side; the bilateral patients had symmetrically decreased response to caloric stimulation.85 The peak velocities at different frequencies were 0.0125 Hz, 100°/sec; 0.05 and 0.2 Hz, 60°/sec; 0.4 and 0.8 Hz 30°/sec.
Position (RV)
Position (RV)
0.0
–0.1
–0.2
| 5º
Onset
Yaw Pitch
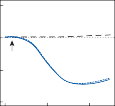
Roll
Head Eye
Gaze
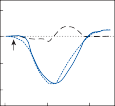
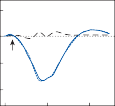
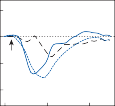
125
Velocity (º/sec)
Velocity (º/sec)
0
–125
–250
Right
0 100
200
Up
0 100 200
Time (ms)
CCW
0 100 200
Figure 7–23. Head, gaze and eye positions and velocities in normal subjects during a yaw-right, pitch-up, and a roll- counterclockwise (GCW) head thrust. The eye signals are inverted for illustration. The arrows indicate the onset of head movement. Note the gaze instability with roll thrusts. (From Au ST et al. Head impulses reveal loss of individual semicircu- lar canal function. J Vestib Res. 1999;9:173–180, with permission.)
(gain near 1.0), whereas the slope is about 0.8 in the roll planes.
ACTIVE HEAD ROTATION
Most normal subjects can produce near-perfect compensatory eye movements in the frequency range of 1 to 4 Hz with active sinusoidal head movements (Fig. 7–25).96 When the scleral search coil technique is used to record eye movements in the horizontal and vertical planes, the gain measurements are close to 1.0 and phase changes are near zero over this wide frequency range (Fig. 7–26). Interestingly, the VOR gain from active head rotations is consistently higher than the gain from passive head rotations in this frequency range in normal subjects (Fig. 7–26). Normative data from studies using ENG to record eye move- ments have been more variable. Some investi- gators have found gains in excess of 1.1 in the high-frequency range and others have reported poor test–retest reliability of the gain measurements.95,97,98 Vertical gain and phase measurements are particularly unreliable with ENG, with a wide range of normative val- ues being reported. In our experience this test is overused and overinterpreted.
Results in Patients
UNILATERAL PERIPHERAL LESIONS
Patients who suddenly lose vestibular function on one side have asymmetric responses to rota- tional stimuli because of (1) a dc bias resulting from spontaneous nystagmus and (2) the differ- ence in response to ampullopetal and ampull- ofugal stimulation of the remaining intact labyrinth.84 These features are readily seen in passive yaw rotation data shown in Figures 7–20b and 7–21b. The patient was tested shortly after the acute onset of vertigo due to a right peripheral vestibular lesion (probable viral neu- rolabyrinthitis). At the time of testing, he exhib- ited a spontaneous left-beating nystagmus (eyes open in the dark) with an average slow-phase velocity of 10 degrees/sec. This spontaneous nystagmus added to rotational-induced nystag- mus in the same direction and subtracted from that in the opposite direction. The effects of this dc bias and of the asymmetry in response to ampullopetal and ampullofugal stimulation of the intact labyrinth are best illustrated in the plot of eye velocity versus stimulus velocity from sinusoidal rotation (see Fig. 7–21b, right side). The dc bias (the eye velocity at the point of Y-intercept) is equivalent to the average
100
0
–100
YAW
PITCH Y

Y
ROLL X
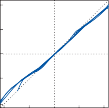
X
–200
200
100
0
–100
–200
Eye Velocity (deg/sec)
Eye Velocity (deg/sec)
200
100
0
–100
–200
200
100
0
–100
–200
200
100
0
–100
–200
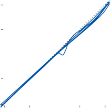
Z Normal | |
Z |
Z Normal | |
Z |
LuPCOZ Y X

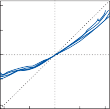
Z Y X


LuSD Z Y X
Z Y X


LuVD Z Y X
Z Y X



bVD Z Y X
Z Y X
Right Left Up Down CCW CW
–200 –100 0 100 200 –200 –100 0 100 200 –200 –100 0 100 200
Head Velocity (deg/sec)
Figure 7–24. Eye velocity as a function of head velocity during roll, pitch, and yaw head thrusts in a normal subject and in four patients with the following conditions: after left unilateral posterior canal occlusion (LuPCO); after left unilateral deafferentation of the superior branch of the vestibular nerve (LuSD); after left unilateral vestibular deafferentation (LuVD); and after bilateral vestibular deafferentation (bVD). The ten trials in each direction are displayed. CCW, counter- clockwise; CW, clockwise. (From Au ST et al. Head impulses reveal loss of individual semicircular canal function. J Vestib Res. 1999;9:173–180, with permission.)
slow-phase velocity of the spontaneous nystag- mus. The gain (slope) of the response with ampullopetal stimulation of the intact labyrinth is twice that with ampullofugal stimulation.
With compensation, the dc bias gradually disappears and the gain asymmetry between ampullopetal and ampullofugal stimula- tion decreases but does not disappear.99,100
Eye Velocity (deg/sec)
Eye Velocity (deg/sec)
100

50
0
–50
–100
(b)
Head Velocity (deg/sec)
Head Velocity (deg/sec)
100
50
0
–50
–100
0 1.0 2.0 3.0 4.0 5.0 6.0
(c) 100
80
Eye Velocity (deg/sec)
Eye Velocity (deg/sec)
60
40
20
0
–20
–40
–60
–80
–100

G = 1.04
0 1.0 2.0 3.0 4.0 5.0 6.0
–100
–50 0
50 100
Time (sec) Head Velocity (deg/sec)
Figure 7–25. Eye movements recorded with a scleral search coil during self-generated (active) head oscillations in the pitch plane. (a) head velocity, (b) Eye velocity data are fitted to sinusoids (solid curves) for those cycles meeting the statisti- cal criterion for acceptance, while cycles rejected due to artifacts are indicated by horizontal bars. (c) The graph plots phase- corrected eye velocity against head velocity; the slope corresponds to gain (G). The two flanking lines indicate boundaries of the region in which data points were accepted for analysis. (From Demer JL et al. Visual–vestibular interaction in humans during active and passive, vertical head movement. J Vestib Res. 1993;3:101, with permission.)
It remains most pronounced after high-intensity stimuli. These dynamic asymmetries in the canal-ocular reflexes can best be determined in the laboratory by using high-acceleration, small-amplitude impulses or high-frequency, high-acceleration sinusoidal rotations.86,87,101,102 Brief high-acceleration head thrusts in normal subjects result in little gaze instability, whereas in patients with a unilateral vestibular loss, accelerations toward the side of the lesion produce a prominent gaze shift in that direction because of the lack of a compensatory VOR response (Fig. 7–27a). This same gaze deviation phenomenon can be seen with high-frequency sinusoidal rotation; the deviation is most prominent at the highest fre- quencies when the head rotates toward the lesion side (Fig. 7–27b). Interestingly, this gaze deviation during ipsilesional rapid head movements is greater with passive rotation than with active head rotations, suggesting that with active rotations patients are able to compensate partially for the lack of a compen- satory VOR response. It follows that passive head rotations at high accelerations are best suited for identifying the dynamic
asymmetries associated with unilateral vestibu- lar lesions.101,102
By performing high-acceleration, small- amplitude head thrusts in all three planes, one can assess the function of the three pairs of semicircular canals (Fig. 7–24).86,87 For exam- ple, a patient who underwent blockage of the left posterior semicircular canal for treatment of benign paroxysmal positional vertigo showed a prominent decrease in VOR gain during upward pitch and counterclockwise roll head thrusts (Fig. 7–24, LuPCO). A patient with involvement of all three semicircular canals on the left side showed a prominent decrease in VOR gain with horizontal head thrusts to the left and approximate symmetrical decrease in gain with up-and-down pitch movements and a prominent decrease in gain with counterclock- wise roll (Fig. 7–24, LuVD). Thus, directional deficits in response to ipsilesional head thrusts allows one to identify the individual semicircu- lar canal lesions. The abnormalities are best identified by measuring VOR gain close to peak head velocity, where the disinhibition input from the intact side approaches saturation values. Recall that inhibition is produced by
1.50
VOR Gain
VOR Gain
1.25
1.00
0.75
0.50
0.0 1.0 2.0 3.0 4.0
Active VOR Passive VOR
Active VOR Passive VOR
5
4
3
2
Phase - deg
Phase - deg
1
0
–1
–2
–3
–4
–5
0.0 1.0 2.0
Frequency - Hz
3.0 4.0
Figure 7–26. Vestibulo-ocular reflex (VOR) gain and phase from nine normal subjects tested at multiple single frequen- cies during passive and active head rotations in the pitch plane. Gain was greater during active head rotations than during passive rotations in the mid-frequency range. Phase was near zero for both types of rotation.
ampullofugal stimulation of the horizontal semicircular canals and ampullopetal stimula- tion of the vertical canals. In addition to the dynamic asymmetries in VOR responses in the planes of the three sets of semicircular canals, patients with unilateral peripheral vestibular lesions develop anomalous (out of the plane of rotation) eye movements that contribute to gaze instability and to complaints of oscillopsia.102 For example, vertical head thrusts in the pitch plane result in anomalous torsional eye move- ments due to loss of the normally balanced ver- tical canal inputs (Fig. 7–28). Normally the ver- tical canals on each side generate a torsional component that is perfectly balanced with pitch rotation so that no torsional eye movements
are observed. However, with a unilateral loss of vestibular function, aberrant torsional eye movements are generated during up-and-down head thrusts. These aberrant torsional eye movements could contribute to sensations of tilt often reported by patients with unilateral vestibular lesions.102
Patients with compensated unilateral periph- eral vestibular lesions show a characteristic pattern of decreased gain and increased phase lead at low frequencies of sinusoidal z-axis rota- tion (see Fig. 7–22).103 These changes appear to be fixed in that they can be observed as long as 10 years after an acute unilateral peripheral vestibular loss.100 Their functional implications are minimal, however, as the visuomotor system
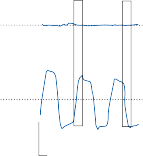
R L
R L
10
250 ms
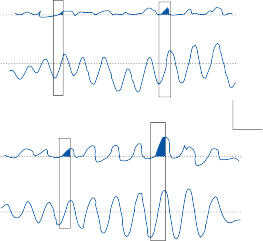
b R
L
R L
Control
Patient
10 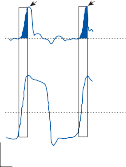
250 ms
Gaze
Head
Gaze
Head
Active
R L
5
500 ms
Gaze
R
L Head
Passive
Figure 7–27. Gaze stability during horizontal head rotations in the light in a normal subject and in a patient with right peripheral vestibular loss (after a nerve section). a The head is passively moved in quick steps (thrusts) to the right and left. Gaze is stable in the control whereas in the patient there are prominent gaze shifts to the right with head thrusts to the right. b Active and passive sinusoidal rotations of the patient’s head show the same gaze instability with movements to the right, but the gaze instability is much greater with passive than with active (self-generated) movements. (From Foster CA et al. Defects of gaze stability in multiple axes following unilateral vestibular lesions. Exp Brain Res. 1997;116:501, with permission).
can compensate for the loss of vestibular func- tion in the low-frequency range.
BILATERAL PERIPHERAL LESIONS
Rotational stimuli are ideally suited for testing patients with bilateral peripheral vestibular lesions because both labyrinths are stimulated simultaneously and the degree of remaining function is accurately quantified.84,86,104,105 Because the variance associated with normal rotational responses is less than that associated with caloric responses, diminished function is
identified earlier. Furthermore, artifactually decreased caloric responses occasionally occur in patients with angular, narrow external canals or with thickened temporal bones. Because the intensity of rotational stimuli is unrelated to these physical features, rotational-induced nys- tagmus is normal in such patients. Frequently patients with absent response to bithermal caloric stimulation have decreased but record- able rotational-induced nystagmus, particularly at higher frequencies of sinusoidal rotation (see later discussion). The ability to identify remaining vestibular function—even if
moving
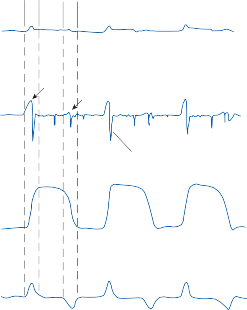
up
Head moving down
| 10º
CW CCW
Torsional gaze position
| 100º/s
Torsional gaze velocity
Torsional fast phase
| 10º
up down
Vertical head position
| 400º/s
Vertical head velocity
![]()
1 sec
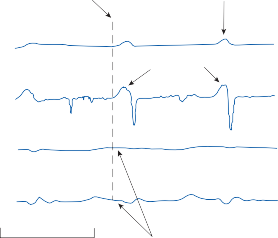
Head movement upward begins
Involuntary torsional movement
| 10º
CW
| 50º/s CCW
Slow phase
Torsional gaze position
Torsional gaze velocity
| 10º
| 50º/s CW
CCW
Torsional head position
Torsional head velocity
1 sec
No torsional head movement has occured
Figure 7–28. Aberrant torsional eye movements induced by self-generated, vertical head thrusts in a patient with a left-sided peripheral vestibular loss. a Clockwise (CW) torsional eye movements are more prominent during upward (first arrow) than downward (second arrow) head movement. Gaze deviations were corrected with torsional quick phases (asterisks). b There are no torsional head movements during these vertical head thrusts to account for the torsional eye movement responses. CCW, counterclockwise. (From Foster CA et al. Defects of gaze stability in multiple axes following unilateral vestibular lesions. Exp Brain Res. 1997;116:501, with permission.)
Patients with bilateral peripheral vestibular loss show the same pattern of low-frequency gain and phase deficits on sinusoidal testing (only more pronounced) observed in patients with compensated unilateral peripheral vestibu- lar lesions (see Fig. 7–22).106 Rarely, patients may have no response to rotation at frequencies below 0.05 Hz and yet have normal responses at higher frequencies.84 These findings have important clinical implications with regard to testing patients with suspected bilateral periph- eral vestibular disease. Given that the results of the bithermal caloric test reflect the results of low-frequency sinusoidal stimulation, the absence of caloric response does not indicate an absence of vestibular function. In fact, a patient could have absent caloric response yet normal response to traditional rotational testing at higher frequencies (up to 1 Hz). However, high- acceleration, small-amplitude head thrusts can be very sensitive for identifying bilateral periph- eral vestibular loss, particularly if one focuses on the first 100 msec after the impulse.107 Central compensation mechanisms cannot make up for the loss of peripheral vestibular function in this time domain. Interestingly, patients who show minimal or no early VOR response after high- acceleration head thrusts can have reasonably good responses during sinusoidal head rotation at higher frequencies.107 These nonvestibular oculomotor responses are presumably gener- ated by using whatever residual vestibular func- tion remains and other sensory clues that are available. These nonvestibular compensatory eye movements are most pronounced with active head rotations, presumably because the patient can use information generated by the volitional head movements to improve the com- pensatory eye movements. This must be kept in mind, however, when using autorotational tests to screen for bilateral vestibular loss.
CENTRAL VESTIBULAR LESIONS
As with lesions of the peripheral vestibular structures, lesions of the central VOR pathways can lead to a decrease or an asymmetry in the
gain of rotational-induced nystagmus. Lesions involving the nerve root entry zone and vestib- ular nuclei may produce responses indistin- guishable from those produced by peripheral vestibular lesions. The spectrum of abnormali- ties associated with central lesions, however, is much more diverse than a simple decrease in the gain. The gain may be increased in some patients with cerebellar lesions.108 The highly organized pattern of the nystagmus produced in normal subjects may be disorganized, result- ing in so-called dysrhythmic nystagmus. If the production of fast components is impaired, the nystagmus waveform is distorted or there may be only a slow tonic deviation of eyes from side to side. In this case, high-acceleration impulses or high-frequency sinusoidal rotation are the only stimuli that can be used to measure VOR gain. Finally, central lesions often interfere with the integration of visual and vestibular sig- nals, producing abnormalities on tests of visual– vestibular interaction (see later discussion).
Low-frequency sinusoidal rotational stimuli are ideally suited for studying the pattern of induced nystagmus. Figure 7–29 illustrates the responses to sinusoidal rotation (eyes open in darkness) in (a) a normal subject, (b) a patient with cerebellar atrophy, (c) a patient with a left pontine lesion (astrocytoma), and (d) a patient with a bilateral lesion of the medial longitudi- nal fasciculus (MLF). In the normal subject the eyes alternately deviate in the direction of the fast component for each half-cycle of induced nystagmus. As discussed in Chapter 3, the eye position in the orbit for initiation of fast components is near the midline. Fast compo- nents (saccades) are generated in the parame- dian pontine reticular formation, and the cerebellum controls the amplitude of both vol- untary and involuntary saccades. In the patient with cerebellar atrophy (Fig. 7–29b), the nys- tagmus pattern is disorganized with fast com- ponents occurring in random fashion, causing marked beat-to-beat variability in amplitude. This type of abnormality has been termed dys- rhythmia and is commonly found in patients with all varieties of cerebellar lesions. Patients with dysrhythmic vestibular nystagmus also demonstrate dysmetria of voluntary saccades.
The patient with a left pontine lesion (see Fig. 7–29c) could not produce voluntary or involuntary saccades (fast components) to the left, so during the half-cycle that normally produces left-beating nystagmus, the eyes

Right eye
60º/sec
0º/sec
60º/sec

15º
Right eye
Right eye
Right eye
Left eye
5 sec
15º
15º
15º
![]()
![]() L
L
15º
Figure 7–29. Electronystagmographic recordings of nystagmus response to sinusoidal rotation at 0.05 Hz, peak velocity 60°/sec in a normal subject (a) and in patients with cerebellar atrophy (b), left pontine glioma (c), and bilateral medial longitudinal fasciculus lesions caused by multiple sclerosis (d).
tonically deviated to the right. In patients with bilateral pontine lesions, the eyes tonically deviate to the right and left with each half-cycle of rotation because of the complete absence of fast components.57 One might mistakenly inter- pret this as a decreased or absent vestibular response.
In the patient with a bilateral MLF lesion (Fig. 7–29d), there is a dissociation in fast compo- nents between the two eyes. When either paretic adducting eye is required to make a fast compo- nent, the nystagmus beats are rounded because of a decrease in the frequency of action potentials arriving at the medial rectus motor neurons via the damaged MLF. Abducting fast components,
however, are normal because the abducting mus- cles (abducens nuclei) receive their innervation for fast components directly from the parame- dian pontine reticular formation with no involve- ment of the MLF. Frequently, the abducting fast components are actually too large. The oculomo- tor control centers attempt to overcome the block at the MLF by increasing the innervation sent from the paramedian pontine region to the ocul- omotor neurons.109,110 Because (according to Herring’s law) this increased innervation is sent equally to both medial and lateral rectus oculo- motor neurons, the difference in amplitude between adducting and abducting fast compo- nents is further magnified.
VISUAL–VESTIBULAR INTERACTION
The model introduced in Chapter 3 (Fig. 3–21) represented two general types of visual–vestibular interaction: one mediated via the “direct” (pur- suit) pathway and the other via the indirect (velocity storage) pathway. Because the direct pathway is dominant in humans, clinical tests have focused on pursuit–VOR interaction. In rare patients with selective lesions of the direct pathway, it is possible to demonstrate visual– vestibular interaction mediated via the indirect velocity storage pathway (see later discussion).
Methodology
Since the visual tracking systems function best at low frequencies and low velocities, visual– vestibular interaction is most prominent at low frequencies and low velocities. In the laboratory,
visual–vestibular interaction is typically tested by rotating the subject either sinusoidally or with a step change in velocity while (1) the surround- ing optokinetic drum is stationary (visualvestib- ulo-ocular reflex [VisVOR], a synergistic interaction of the visual and vestibular systems) or (2) the drum and chair are coupled so that they move together (fixation suppression of the vestibulo-ocular reflex [VOR-FIX], an antago- nistic interaction between the visual and vestibular systems).111 Fixation suppression can also be tested by rotating the subject in the dark with a single fixation light attached to the chair.
Results in Normal Subjects
Typical responses of a normal subject to low- frequency sinusoidal (0.05 Hz) optokinetic (OKN), vestibular (VOR), and visual-vestibular (VisVOR and VOR-FIX) stimulation are shown in Figure 7–30 (left). In each case, the peak stimulus velocity is 60 degrees/sec. At this low
40
0
–40
Slow Phase Velocity (deg/sec)
Slow Phase Velocity (deg/sec)
40
0
–40
40
0
–40
40
0
Normal Subject Right Peripheral Bilateral Peripheral Right

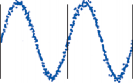


Left
OKN

VOR

VisVOR
VOR-FIX
–40
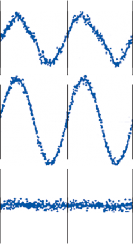


0 10 20 30 0 10 20 30
Time (sec)
0 10 20 30
Figure 7–30. Plots of slow-phase velocity versus time from a low-frequency visual–vestibular test battery (see text for details) in a normal subject (left), a patient who underwent a right labyrinthectomy (center), and a patient with bilateral vestibulopathy secondary to ototoxic drugs (right) (0.05 Hz, peak velocity 60°/sec). OKN, optokinetic nystagmus; VisVOR, visual-vestibulo-ocular reflex; VOR, vestibulo-ocular reflex; VOR-FIX, fixation suppression of VOR. (From Baloh RW, et al. Quantitative vestibular testing. Otolaryngol Head Neck Surg. 1984;92:1,45, with permission.)
FIX 0.03 ± 0.02. At high frequencies (>1 Hz) and velocities (>50 degrees/sec), the OKN (and pursuit) gain decreases (e.g., see Fig. 7–17). Above 2 Hz, the VisVOR and VOR-FIX gain are approximately the same as the VOR gain (near 1.0).
Results in Patients
Patients with peripheral vestibular lesions have decreased and/or asymmetric VOR gain, but visual–vestibular responses are usually normal at low stimulus frequencies and velocities
(see Fig. 7–30, center and right). Even with a complete loss of vestibular function, the visuo- motor system can provide good ocular stability. At high frequencies and velocities, however, the VisVOR gain decreases if the VOR gain decreases.112
Three abnormal patterns of visual–vestibular interaction seen on low-frequency sinusoidal testing in patients with central lesions are shown in Figure 7–31.111 Patients with lesions involving the vestibular nucleus region (e.g., Wallenberg’s syndrome) exhibit prominent ocul- omotor abnormalities (see Stroke Syndromes, Chapter 14). With eyes open in the sitting posi- tion, there is a tonic pulling of the eyes toward the side of the lesion, resulting in spontaneous nystagmus with the fast phase toward the intact side. With eyes closed or with eyes open in darkness, the spontaneous nystagmus may change direction. The responses illustrated in Figure 7–31 (left) are from a 32-year-old man who had the acute onset of vertigo, nausea, vomiting, dysphagia, and falling to the left.
40
0
–40
Slow Phase Velocity (deg/sec)
Slow Phase Velocity (deg/sec)
40
0
–40
40
0
–40

Left Lateral Medullary

Bilateral Vestibulocerebellum
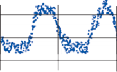
Left Parietal Lobe
OKN
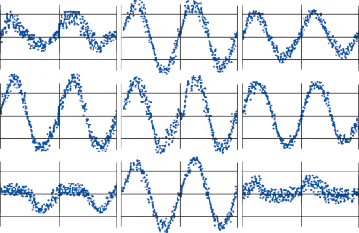
VOR
VisVOR
40
0
–40
0 10 20 30 0 10 20 30 0 10 20 30
Time (sec)
VOR-FIX
Figure 7–31. Plots of slow-phase velocity versus time from a low-frequency visual–vestibular test battery in a patient with infarction of the left lateral medullary region (left), a patient with midline cerebellar atrophy (center), and a patient with a glioma in the deep parietal lobe on the left side (right) (0.05 Hz, peak velocity 60°/sec). OKN, optokinetic nystagmus; VisVOR, visual-vestibulo-ocular reflex; VOR, vestibulo-ocular reflex; VOR-FIX, fixation suppression of VOR. (From Baloh RW, et al. Quantitative vestibular testing. Otolaryngol Head Neck Surg. 1984;92:145, with permission.)
Patients with lesions involving the vestibulo- cerebellum are unable to modify vestibular responses with vision.114 This is illustrated by the patient data shown in Figure 7–31 (center),
As noted earlier, in patients with minimal or no sinusoidal VOR, VisVOR, and VOR-FIX, responses are almost identical (e.g., Fig. 7–31, center). These patients may show evidence of visual–vestibular interaction with impulse stim- uli, however. The patient with cerebellar atro- phy, whose data are shown in Figure 7–32, had absent pursuit and sinusoidal OKN but exhib- ited a gradual buildup in OKN after a step onset in drum velocity. The gain (initial peak eye veloc- ity/peak stimulus velocity) of the step responses was the same regardless of whether the patient received (1) a VisVOR stimulus (i.e., a step from 0 to 60 degrees/sec in the light with a
100
40
20
in which the VOR, VisVOR, and VOR-FIX gains are approximately the same (nearly 1.0) and the OKN gain is markedly decreased in both directions. This patient was a 31-year-old woman who complained only of unsteadiness and oscillopsia. The results of neurologic exam- ination were normal except for spontaneous downbeat nystagmus and truncal ataxia. Computerized tomography (CT) and magnetic resonance (MR) scanning documented atrophy of the midline cerebellum.
Lesions of the visuomotor pathways from the parieto-occipital cortex to the pons (i.e., those shown in Fig. 3–23) lead to impaired smooth pursuit and optokinetic slow phases toward the side of the lesion.77 The abnormal visual-ocular control does not impair VOR responses but does alter visual–vestibular inter- action. Typical responses to the four sinusoidal rotational test conditions in a patient with a deep parietal lobe lesion are shown in Figure 7–31 (right). This 21-year-old man developed bitemporal headaches and slowly progressive
10
Slow Phase Velocity (deg/sec)
Slow Phase Velocity (deg/sec)
0
100
40
20
10
0
100
40
20
10
VOR
VOR-FIX
VisVOR
right facial and upper-extremity weakness. An angiogram identified a tumor blush in the left parietal region. A left parietal brain biopsy revealed a grade II astrocytoma. The OKN gain was normal to the right and markedly decreased to the left. The VOR gain was normal in both directions, but the patient was unable to inhibit VOR slow phases to the right with fixation (i.e., the VOR-FIX gain was increased to the right). The VisVOR gain was slightly asymmetric, with lower gain to the left than to the right.
0
0 20 40
Time (sec)
Figure 7–32. Vestibulo-ocular reflex (VOR), fixation sup- pression of VOR (VOR-FIX), and visual-VOR (VisVOR) responses to step rotational stimuli (0° to 60°/sec, 140°/sec2 acceleration) in a patient with cerebellar atrophy. The log of slow-phase eye velocity is plotted against time. For the VOR the chair was stopped in the dark, for the VOR-FIX the chair was stopped in the light, and for the VisVOR the chair was maintained at a constant velocity in the light with a stationary surround. (From Baloh RW, et al. Late cortical cerebellar atrophy. Brain. 1986;109:159, with permission.)
fixed surround), (2) a VOR-FIX stimulus (i.e., stopping the rotating chair in the light with a fixed surround), or (3) a VOR stimulus (i.e., start- ing or stopping the chair in darkness). However, the rate of decay in slow-phase velocity (i.e., the time constant) was prolonged after a VisVOR stimulus and shortened after a VOR-FIX stimu- lus, compared with the VOR stimulus. Thus, one type of visual–vestibular interaction (that medi- ated via the velocity storage pathway) was pre- served in a patient with absent smooth pursuit.
In summary, in addition to tests of the VOR, rotational testing includes tests of visual– vestibular interaction. Lesions of the periph- eral vestibular system typically impair only the VOR, whereas lesions of the CNS impair OKN and visual–vestibular interaction. The pattern of abnormal response can help localize lesions within the central pathways.
TESTS OF OTOLITH–OCULAR REFLEXES
Ocular Counterrolling
The otolith-ocular reflex produces torsional eye movements during static head tilts. Rotating the head toward the right shoulder causes the eyes to counterrotate to the left (see Chapter 3). Such rotation of the head in the coronal plane is called roll, and the counterrotation of the eyes is called ocular counterrolling.115,116 Dynamic roll movements also stimulate the vertical semicir- cular canals because of the angular acceleration of the movement, so when using roll stimulation a distinction should be made between static and dynamic ocular counterrolling.
The clinical use of ocular counterrolling has been hampered by difficulties both in stimulus delivery and in the measurement of response. In order to rotate someone in the coronal plane, the subject must be securely fastened to a cum- bersome device. In addition, the amount of torsional eye movement produced by a static tilt in the coronal plane is relatively small. For example, if the head is tilted 45 degrees, the eyes counterroll only about 7 degrees. Electrooculography is insensitive to this type of movement, so video recording or the magnetic scleral search coil must be employed.
Unilateral peripheral vestibular lesions can pro- duce asymmetries in static ocular counterrolling;
roll to the side of the lesion results in less coun- terrolling than roll away from the side of the lesion.117,118 With some types of central lesions one can see a roll rather than a counterroll response (i.e., the eyes rotate in the direction of head tilt).119,120 However, the responses are too variable to be a reliable test for identifying the side of a unilateral lesion.
Eccentric Rotation
Eccentric (off-center) rotation is delivered by seating a subject upright in a conventional rota- tional chair such that the head is away from the axis of rotation as if the head were placed at the end of the arm of a centrifuge (see Fig. 7–18). During angular acceleration with the head eccentric, the labyrinth is exposed to both rota- tional and linear (tangential and centrifugal) acceleration, and thus both the otolith organs and the horizontal semicircular canals are stim- ulated. Once a constant angular velocity is achieved, however, only the otoliths are stimu- lated. The net linear acceleration delivered to the subject is the vector summation of the lin- ear acceleration produced by the movement itself and the linear acceleration produced by gravity (See Fig. 1-3b). The advantages of eccentric rotation are that conventional rota- tional chairs (with minor modifications) and EOG methods can be used for this test.
With sinusoidal angular acceleration the dif- ference between eye movements induced with the head at the center of rotation and those with the head eccentric is the contribution of the oto- lith organs.121–123 An even simpler test of otolith function is to have the subject estimate the sub- jective vertical (with a vertical light bar) during constant velocity eccentric rotation.124,125 Unlike other tests of subjective vestibular sensation, the sensation of tilt experienced during eccen- tric rotation appears to be highly reproducible. Patients who have undergone a unilateral ves- tibular neurectomy experience less of a sensa- tion of tilt when the lesioned ear is outermost.124 The deficit is maximum in the first postopera- tive week but persists for at least 24 weeks.126
Off-Vertical Rotation
Off-vertical rotation is performed by seating the subject in a conventional rotational chair
and then tilting the entire apparatus, including the chair and subject (see Fig. 7–18).127,128 In this way, as the subject rotates, the head is con- tinually changing its orientation with respect to gravity. In the extreme case, in which the chair is tipped completely on its side (earth- horizontal axis, or so-called barbecue rotation), the subject is rotated from supine to lateral to prone to lateral, and so on.129,130 Once a con- stant velocity is achieved, only the otolith organs are stimulated (because the canals respond only to angular acceleration).
A major advantage of this type of otolith test is that a conventional rotatory chair can be used if the angle of inclination is kept small. Subjects can be placed into or moved from the appara- tus easily, and conventional EOG can record the eye movements because they are largely horizontal. A disadvantage is that the stimulus often produces nausea. Off-vertical constant velocity rotation in normal subjects induces two horizontal eye movement components: a bias and a modulation component.129,130 In patients with unilateral peripheral vestibular lesions, the bias component is diminished when the patient rotates toward the involved ear while the modulation component remains unchanged.131
Linear Acceleration
Another technique that has been used to study the otolith ocular reflex in the research labora- tory is to deliver a linear acceleration on a lin- ear track or a parallel swing.132–135 As with eccentric rotation, the otolith organs sense the net linear acceleration—that is, the vector summation of the linear acceleration induced by the device itself and that due to gravity. For the relatively simple case in which the subject is placed on the device facing the side as if looking out the side window of an automobile moving forward, a consistent horizontal eye movement (the linear VOR) can be recorded. For other head orientations vertical or torsional eye movements are induced, requiring eye movement recording techniques such as a magnetic scleral search coil or video system. Although patients with complete unilateral and bilateral vestibular loss consistently show a diminished linear VOR gain compared to controls,134,136,137 the test is not sensitive for identifying partial loss of vestibular function.
Furthermore, the asymmetry in the linear VOR gain after an acute unilateral vestibular lesion disappears within a few months.137
Current tests of vestibular function concen- trate on the vestibulo-ocular system: the vestibulospinal system has been relatively neglected. A major reason for this neglect is that it is difficult to assess the role of the ves- tibulospinal system in isolation of the other sensory systems.
Static-Force Platforms
The simplest method of recording human pos- tural sway employs a force plate. There are sev- eral devices of this type, each designed with the basic idea of recording the position of a subject’s center of mass during upright stance. In fact, these devices measure the position of the center of pressure (COP), which is a good estimate of the position of the center of mass if the body is moving slowly. The COP is mea- sured with force transducers in the force plate and then differentiated to give instantaneous sway velocity (Fig. 7–33).138 The major limita- tion of such devices relates to two factors:
the nervous system uses a combination of sensory modalities during the maintenance of upright stance, and (2) static force plates do not yield controlled stimulus–response mea- sures of vestibulospinal function and thus must rely on spontaneous movements of the body. This latter consideration is analogous to mak- ing assessments of the vestibulo-ocular system by simply monitoring eye position in the absence of vestibular stimulation. The mea- surement of postural sway might be useful as a screening test for imbalance, but the informa- tion it provides is nonspecific and probably not helpful for identifying vestibular lesions.138–140
Moving-Platform Posturography
Moving-force platforms have been designed to overcome the limitations of static-force plat- forms discussed above by (1) controlling the relative contributions of the visual, somatosensory,
![]()
![]()
![]()
![]()
COPAP (mm)
COPAP (mm)
0
–80
Eyes open
Eyes closed
80
![]()
0
–80
Eyes open
![]()
![]()
Eyes closed
SAP (mm/s)
SAP (mm/s)
400
0 ![]()
400
0 ![]()
![]()
–400
0 5 10 0 5 10
Time (sec)
Figure 7–33. Examples of posturography raw data from a static and dynamic test in an older subject with eyes open and closed. Upper traces, sway position (center of pressure [COP]) in the anterior–posterior (AP) directions; lower traces, sway velocity in the anterior–posterior ![]() ) directions.
) directions.
and vestibular inputs that are normally used to maintain upright posture; and (2) incorporat- ing stimulus–response measurements. By cou- pling the platform to the sway of the subject, it is possible to maintain the angle between the foot and the lower leg at a constant value, thereby reducing a major source of somatosen- sory input to the postural control system.141 A similar effect can be achieved by having the subject stand on a thick foam rubber pad. If the subject simultaneously closes the eyes or if the movement of the visual enclosure is cou- pled to body sway, the subject is also deprived of visual information about postural sway. In this way, the influence of the labyrinth on upright posture via the vestibulospinal system can be studied in a more or less isolated fash- ion.142 The disadvantage of this technique is that during postural sway many of the subor- gans of the vestibular labyrinth are simultane- ously stimulated, including the vertical semicir- cular canals and the otolith organs bilaterally. For this reason, moving-platform studies are incapable of providing an assessment of the individual suborgans of the vestibular labyrinth. In addition, these devices do not assess the subject’s strategy in moving other body parts and joints. Not surprisingly, patients with bilat- eral peripheral vestibular loss perform poorly on these tests when visual and somatosensory
signals have been effectively removed.140,143,144 However, preliminary reports that moving- platform posturography can identify sites of lesion or specific vestibular disorders have not been confirmed. Dynamic posturography is not a diagnostic test but rather a method to quan- tify balance dysfunction under different sen- sory conditions (Fig. 7–34).145,146 It may be helpful for identifying people at risk for falling, although it is not clear if it is better at this than a careful clinical assessment.147 Posturography may also be helpful in distinguishing between organic and function balance disorders.148
Brain Stem and Cortical
The ability to record a human brainstem vestib- ular-evoked potential has obvious merits, as it would provide an objective measure of periph- eral vestibular function that would be indepen- dent of either the oculomotor or postural control systems. Despite the fact that short latency sensory-evoked potentials using auditory, visual, and somatosensory inputs have been developed and are in routine clinical use, vestibu- lar-evoked potentials are not routinely available.
120
80
Sway Velocity (mm/sec)
Sway Velocity (mm/sec)
40
0
160
AP

Sway
ML
Sway
160
120
80
40
0
160
AP

Sway
ML
Sway
160
120
80
40
0

160
AP

Sway
ML
Sway
160
120
80
40
0
160
AP

Sway
ML
Sway
Eyes Open
120
80
40
0
Static
120
80
40
0
Foam
120
80
40
0
AP-Tilt
120
80
40
0

ML-Tilt
Eyes Closed
![]() Young controls Older controls Older patients
Young controls Older controls Older patients
![]() Young controls Older controls Older patients
Young controls Older controls Older patients
![]()
![]()
Figure 7–34. Mean sway velocity (vertical bar = 1 standard deviation) in young (black bars) and older (gray bars) controls and older patients with imbalance (striped bars) for the four standard posturography test conditions. AP, anterior–posterior; ML, medial–lateral. In the two graphs to left, the platform was still; in the two graphs to the right, the platform tilted up and down sinusidally about a central axis (0.10 Hz, 4° peak amplitude) (see Fig. 6–33).
One reason for this lack of development is related to the difficulty of delivering a vestibu- lar stimulus that is capable of triggering a coor- dinated volley of neural activity, a requirement for eliciting a measurable evoked potential.149 The vestibular equivalent of an auditory click, visual flash, or somatosensory prick is a brief, abrupt, high-intensity rotational impulse with an angular acceleration in the range of 7000 degrees/sec2.
Prior research regarding human vestibular- evoked potentials has focused upon recording long-latency cortical potentials rather than brainstem–evoked potentials.150,151 The results of these studies are conflicting. It is still unclear whether the recorded potentials are specific for the vestibular stimulus. Short-latency ves- tibular-evoked potentials have been induced in animals and in humans149 using brief, high- acceleration head displacements, but because of the complex methodology required and the potential discomfort to the patients, this type of testing will not likely become available in the
clinical vestibular laboratory. Pulsed galvanic stimulation over the mastoid can induce a syn- chronized volley within the vestibular-cochlear nerve, but this stimulus is also uncomfortable and could even lead to potential nerve damage.
Vestibular Evoked Myogenic Potentials (VEMPs)
MECHANISM OF STIMULATION
Vestibular evoked myogenic potentials were first recorded in the 1960s, but it wasn’t until the 1990s that the clinical application of this technique was appreciated.152–154 Animal stud- ies show that both air and bone conducted sound activates otolith afferents (utricular and saccular) but rarely semicircular canal afferents.155 Consistent with this observation it was shown that sound-evoked potentials recorded from electrodes over the sterno- cleidomastoid muscle in patients persisted
despite profound sensorineural hearing loss but disappeared after vestibular nerve sec- tion.156 The motor neurons of cervical flexor muscles receive inhibitory input from the sac- cule and intramuscular recordings in the ster- nocleidomastoid muscle show that the initial positivity of the VEMP is produced by an inhi- bition of the underlying motor units.157,158 Based on this data there is a general consensus that cervical VEMPs (cVEMPs) are saccular and inferior vestibular nerve dependent. More recently it has been shown that VEMPs can also be recorded from electrodes placed near the eyes (ocular VEMPs or oVEMPs).159 These potentials are not due to eye movement or electro-oculographic potentials. Since the vast majority of otolith ocular connections originate from the utricle, oVEMPs could be useful for assessing utricular function.160 Consistent with this premise patients with vestibular neuritis (which typically involves just the superior divi- sion of the vestibular nerve) were found to often have normal cVEMPs but absent oVEMPs.161
TEST METHODOLOGY
For recording cVEMPs electrodes are placed over the most prominent part of the sterno- cleidomastoid muscles and reference elec- trodes are placed on the clavicles. The record- ingsmustbemadewhenthesternocleidomastoid muscles are contracting. The simplest way to achieve this is to have the subject raise their head while lying supine. Many different stimuli have been used, including air- and bone-con- ducted sounds, skull taps, and galvanic current, but the most commonly used stimulus in our laboratory is an air-conducted high-intensity (100 to 130 dB SPL) low-frequency tone burst with a center frequency of about 500 Hz (2 msec rise/fall time, 2 msec plateau). Since the myogenic evoked potentials are large in amplitude, usually more than 100 microvolts) only a small number need be averaged. Typically three stimulation sequences (rate of four per second) are given, each consisting of 64 tone bursts. To correct for interindividual/ interside/intertest variations in tonic muscle contraction, a separate channel is used to obtain a numerical value for background muscle tension (rectified EMG).
Standard methodology has not been devel- oped for recording oVEMPs, but the most
consistent potentials appear to be obtained from electrodes placed beneath the eyes and on the cheeks while the subject is instructed to look upward.162 The stimuli (e.g., tone burst) can then be introduced and the response recorded. Since oVEMPs are smaller in ampli- tude than cVEMPs, more averaging may be needed.
NORMATIVE DATA
The typical cVEMP consists of a positive/ negative wave labeled p13 and n23 based on the approximate latency and polarity (Fig. 7–35).163 cVEMP amplitude is linearly related with back- ground muscular tension, so the p13-n23 amplitude should be divided by the mean value of the rectified EMG. The response is much larger in the ear ipsilateral to the stimulus and is larger for tone bursts than for clicks (Fig. 7–35). Since the responses depend on normal sound transmission through the middle ear, patients with middle ear disease such as otosclerosis may have absent responses. Of particular importance, the p13-n23 amplitude of cVEMPs decreases with age so the test is less useful in older patients.163 Each laboratory should establish normative data, but in our lab- oratory greater than 50% amplitude asymme- try and absolute amplitude values below 100 microvolts are considered abnormal. We do not routinely use latency measurements.
Normative data have not been established for oVEMPs, but when the recording elec- trodes are placed beneath the eyes, contralat- eral responses are much larger than ipsilateral responses (i.e., the reflex is crossed). This is consistent with the crossed utriculo-ocular pathway.
RESULTS IN PATIENTS
Since cVEMPs reflect activity originating in the saccule and carried in the inferior vestibular nerve, vestibular lesions that damage the saccule and/or inferior vestibular cause abnormal cVEMPs. About 50% of patients with Meniere’s syndrome have a decreased or absent cVEMP on the involved side when tested between attacks.164,165 Furthermore, cVEMPs can be used to monitor the effect of intratympanic gentamicin injection for treatment of Meniere’s syndrome.166 cVEMPs are abnormal in about 55%–80% of patients with vestibular schwannomas.165,167,168
500 Hz toneburst, 130 dB peSPL click, 100 dB HL

![]()
n23
p13
n23
p13
n23
p13
n23
p13
Right SCM
![]()
![]()
Left SCM
![]()
![]()
100 V 100 V
![]()
![]()
20 msec 20 msec
Figure 7–35. Vestibular evoked myogenic potentials (VEMPs) recorded from the right and the left sternocleidomas- toid (SCM) in response to sound stimulation to the right ear in a healthy individual. Sound stimuli were either 500 Hz tonebursts with an intensity of 130 dB peSPL or 100 dB HL clicks. The vertical dashed lines shows stimulus onset (the 20 millisecondsec prestimulus recordings were used for measuring background muscular tension). Although there are typical positive-negative VEMPs on the side ipsilateral to the sound stimulation, there is only a weak inverted response on the con- tralateral side. Note that VEMPs in response to tone bursts have longer latencies compared with those in response to clicks and amplitudes in response to tonebursts are larger than those in response to clicks. (From Brantberg K. Vestibular evoked myogenic potentials (VEMPs): usefulness in clinical neurotology. Semin Neurol. 2009;29:541 with permission.)
Stim Right (affected ear) Right SCM
Stim Left (healthy ear) Left SCM
130 SPL
p13
n23
5 ms 100 µv
p13
n23
100 SPL n23
p13
Figure 7–36. Vestibular evoked myogenic potentials (VEMPs) illustrate the typical hyperactive response and reduced threshold in a patient with superior semicircular canal dehiscence syndrome on the right side. Active electrodes were placed over the superior part of the sternocleidomastoid muscle (SCM), and the reference electrodes were placed near the mid-portion of the clavicle. High-intensity (100 to 130 dB sound pressure level [SPL]), low-frequency (500-Hz tone bursts, 2-ms rise/fall time, and 2-ms plateau) sounds were presented monaurally via TDH-49P headphones. Three stimulation sequences (rate of four per second) were given, each consisting of 64 tone bursts. The mean curves are shown below the three repetitions for each stimulus.
Schmid-Priscoveanu A, Allum JH. Infrared and video oculography – alternatives to electrooculography? HNO. 1999;47:472.
Barber HO, Wright G. Positional nystagmus in nor-
arises from, but initial studies did not find it to be useful for predicting tumor location prior to sur- gery.168,169 Since vestibular neuritis usually involves just the superior division of the vestibular nerve, the cVEMP should be spared in most cases. Initial studies found that only about one-third of patients with vestibular neuritis have a decreased or absent cVEMP.165,170,171 Some cases have normal caloric responses but abnormal cVEMPs suggesting selective involvement of the inferior division of the vestibular nerve. cVEMPs have been particu- larly helpful for confirming the diagnosis of ante- rior semicircular canal dehiscence syndrome (Fig. 7–36).172–174 Patients with this syndrome typically have very large cVEMP amplitude and low cVEMP threshold (present at much lower stimulus intensities compared to the normal side).
oVEMPs may have an additional advantage in
mals. Adv Otol Rhinol Laryngol. 1973;19:276.
Sunami K, Tochino R, Zushi T, et al. Positional and positioning nystagmus in healthy subjects under videonystagmoscopy. Acta Otolaryngol Suppl. 2004;(554):35.
McGovern TN, Fitzgerald JE. The effect of mental alerting on peripheral vestibular nystagmus during spontaneous, gaze (30 degrees left, 30 degrees right) and body positional (left & right lateral lying) testing using electronystagmography (ENG). Int J Audiol. 2008;47(10):601.
Dell’Osso LF, Daroff RB. Congenital nystagmus waveforms and foveation strategy. Doc Ophthalmol. 1975;19:155.
Baloh RW, Konrad HR, Dirks D, Honrubia V. Cerebellar-pontine angle tumors. Results of quan- titative vestibulo-ocular testing. Arch Neurol. 1976;33:507.
Hood JD, Kayan A, Leech J. Rebound nystagmus.
Brain. 1973;96:507.
Linthicum FH, Waldorf R, Luxford WM. Infrared/
screening for anterior canal dehiscence syn- drome, since they reflect an excitatory response that does not saturate with increasing intensity as the inhibitory cVEMP response does.
REFERENCES
Fife TD, Tusa RJ, Furman JM, et al. Assessment: vestibular testing techniques in adults and children: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2000;55(10):1431.
Aschan G, Bergstedt M, Stable J. Nystagmography: recording of nystagmus in clinical neuro-otological examinations. Acta Otolaryngol Suppl (Stockh). 1956;129:1.
Markley BA. Introduction to electronystagmography for END technologists. Am J Electroneurodiagnostic Technol. 2007;47(3):178.
Clarke AH, Teiwes W, Scherer H. Videooculography
– an alternative method for measurement of three dimensional eye movements. In: Schmidt R, Zambarieri A, eds. Oculomotor Control and Cognitive Processes. Amsterdam, Netherlands: Elsevier; 1991:431.
Gonshor A, Malcolm R. Effect of changes in illumina- tion level on electro-oculography (EOG). Aerospace Med. 1971;42:138.
Proctor L, Hansen D, Rentea R. Corneoretinal poten- tial variations. Arch Otolaryngol. 1980;106:262.
Casse G, Adenis JP, Sauvage JP, Robert PY. Videonystagmography to assess eyelid dynamic disorders. Orbit. 2009;28(1):20.
Houben MM, Goumans J, vab der Steen. Recording three-dimensional eye movements: scleral search coils versus video oculography. Invest Ophthalmol Vis Sci. 2006;47:179.
video recordings of rotatory nystagmus arising from the posterior semicircular canal via the singular nerve. Semin Hear. 1989;10:191.
Baloh RW, Sakala SM, Honrubia V. Benign paroxysmal positional nystagmus. Am J Otolaryngol. 1979;1:1.
Lim J, Elidan J, Baloh RW, Honrubia V. Direction- changing positional nystagmus: incidence and mean- ing. Am J Otolaryngol. 1986;7:306.
Schmaltz G. The physical phenomena occurring in the semicircular canals during rotatory and thermic stimu- lation. Proc R Soc Med. 1932;25:359.
O’Neill G. The caloric stimulus. Temperature gen- eration within the temporal bone. Acta Otolaryngol (Stockh). 1987;103:266.
Paige G. Caloric vestibular responses despite canal inactivation. Invest Ophthalmol Vis Sci. 1984;25(suppl):229.
Scherer H, Brandt U, Clarke AH, et al. European vestibular experiments on the spacelab-1 mission: 3. Caloric nystagmus in microgravity. Exp Brain Res. 1986;64:255.
Scherer H, Clarke AH. The caloric vestibular reac- tion in space. Physiological considerations. Acta Otolaryngol (Stockh). 1985;100:328.
Coats AC, Smith SY. Body position and the inten- sity of caloric nystagmus. Acta Otolaryngol (Stockh). 1967;63:515.
Baertschi AJ, Johnson RN, Hanna GR. A theoretical and experimental determination of vestibular dynam- ics in caloric stimulation. Biol Cybern. 1975;20:175.
Zangemeister WH, Bock O. The influence of pneu- matization of mastoid bone on caloric nystagmus response. Acta Otolaryngol (Stock). 1979;88:105.
Fitzgerald G, Hallpike CS. Studies in human vestibu- lar function: 1. Observations of the directional prepon- derance of caloric nystagmus resulting from cerebral lesions. Brain. 1942;65:115.
Murdin L, Davies RA, Bronstein AM. Vertigo as a migraine trigger. Neurology. 2009;73:638.
Baloh RW, Solingen L, Sills AW, Honrubia V. Caloric testing. 1. Effect of different conditions of ocular fixa- tion. Ann Otol Rhinol Laryngol. 1977;86(suppl 43):1.
Baloh RW, Sills AW, Honrubia V. Caloric testing. III. Patients with peripheral and central vestibular lesions. Ann Otol Rhinol Laryngol. 1977;86(suppl 43):24.
Cogan DG. Neurologic significance of lateral conju- gate deviation of the eyes on forced closure of the lids. Arch Ophthalmol. 1948;39:37.
Lightfoot GR. The origin of order effects in the results of the bi-thermal caloric test. Int J Audiol. 2004;43(5):276.
Sills AW, Baloh RW, Honrubia V. Caloric testing. II. Results in normal subjects. Ann Otol Rhinol Laryngol. 1977;86(suppl 43):7.
Newman Toker DE, Kattah JC, Alvernia JE, Wang DZ. Normal head impulse test differentiates acute cerebellar strokes from vestibular neuritis. Neurology. 2008;70:2378.
Coats AC. Directional preponderance and spontaneous nystagmus. Ann Otol Rhinol Laryngol. 1966;75:1135.
Halmagyi GM, Cremer PD, Anderson J, Murofushi T, Curthoys IS. Isolated directional preponderance of caloric nystagmus: I. Clinical significance. Am J Otol. 2000;21(4):559.
Baloh RW, Konrad HR, Honrubia V. Vestibulo- ocular function in patients with cerebellar atrophy. Neurology. 1975;25:160.
Yoshizawa T, Nakamagoe K, Ueno T, Furusho K, Shoji S. Early vestibular dysfunction in Machado Joseph disease detected by caloric test. J Neurol Sci. 2004;221:109.
Gordon CR, Joffe V, Vainstein G, Gadoth N. Vestibulo- ocular arreflexia in families with spinocerebellar ataxia type 3 (Machado Joseph disease). J Neurol Neurosurg Pyschiatry. 2003;74:1403.
Burk K, Fetter M, Abele M, et al. Autosomal domi- nant cerebellar ataxia type I: ocularmotor abnormali- ties in families with SCA1, SCA2, and SCA2. J Neurol. 1999;246:789.
Elidan J, Gay I, Lev S. On the vertical caloric nystag- mus. J Otolaryngol. 1985;14:287.
Fredrickson JM, Fernandez C. Vestibular disor- ders in fourth ventricle lesions. Arch Otolaryngol. 1964;80:521.
Baloh RW, Sills A, Konrad HR, Honrubia V. The sac- cade velocity test. Neurology. 1975;25:1071.
Baloh RW, Honrubia V. Reaction time and accuracy of the saccadic eye movements of normal subjects in a moving-target task. Aviat Space Environ Med. 1976;47:1165.
Metz HS, Scott AB, O’Meara D, Stewart HL. Ocular saccades in lateral rectus palsy. Arch Ophthalmol. 1970;84:453.
Solingen LD, Baloh RW, Myers L, Ellison G. Subclinical eye movement disorders in patients with multiple sclerosis. Neurology. 1977;27:614.
Crane TB, Yee RD, Baloh RW, Hepler RS. Analysis of characteristic eye movement abnormalities in internuclear ophthalmoplegia. Arch Ophthalmol. 1983;101:206.
Meienberg O, Muri R, Rabineau PA. Clinical and oculographic examinations of saccadic eye movements in the diagnosis of multiple sclerosis. Arch Neurol. 1986;43:438.
Yee RD, Cogan DG, Zee DS, et al. Rapid eye move- ments in myasthenia gravis. II. Electro-ocular analysis. Arch Ophthalmol. 1976;94:1465.
Baloh RW, Keesey JC. Saccade fatigue and response to edrophonium for the diagnosis of myasthenia gravis. Ann NY Acad Sci. 1976;274:631.
Baloh RW, Sharma S, Moskowitz H, Griffith R. The effect of alcohol and marijuana on eye movements. Aviat Space Environ Med. 1979;50:18.
Gentles W, Llewellyn-Thomas E. Effect of benzodi- azepines upon saccadic eye movements in man. Clin Pharmacol Ther. 1971;12:56.3.
Wilkinson IMS, Kime R, Purnell M. Alcohol and human eye movement. Brain. 1974;97:785.
Leigh RJ, Newman SA, Folstein SE, et al. Abnormal ocular motor control in Huntington’s chorea. Neurology. 1983;33:1268.
Troost BT, Daroff RB. The ocular motor defects in progressive supranuclear palsy. Ann Neurol. 1977;2:397.
Baloh RW, Furman J, Yee RD. Dorsal midbrain syn- drome: clinical and oculographic findings. Neurology. 1985;35:54.
Baloh RW, Furman J, Yee RD. Eye movements in patients with absent voluntary horizontal gaze. Ann Neurol. 1985;17:283.
Zee DS, Yee RD, Cogan DG, et al. Ocular motor abnormalities in hereditary cerebellar ataxia. Brain. 1976;99:207.
Buttner N, Geschwind D, Jen JC, et al. Oculomotor phenotypes in autosomal dominant ataxias. Arch Neurol. 1998;55:1353.
Furman JM, Perlman S, Baloh RW. Eye movements in Friedreich’s ataxia. Arch Neurol. 1983;40:343.
Fahey MC, Cremer PD, Aw ST, et al. Vestibular, sac- cadic and fixation abnormalities in genetically con- firmed Friedreich ataxia. Brain. 2008;131(pt 4):1035.
White OB, Saint-Cyr JA, Tomlinson RD, Sharpe JA. Ocular motor deficits in Parkinson’s disease. II. Control of the saccadic and smooth pursuit systems. Brain. 1983;106:925.
Barker RA, Michell AW. “The eyes have it”. Saccadometry and Parkinson’s disease. Exp Neurol. 2009;219(2):382.
Sharpe JA, Lo AW, Rabinovitch HE. Control of the saccadic and smooth pursuit systems after cerebral hemidecortication. Brain. 1979;102:387.
Deng S-Y, Goldberg ME, Segraves MA, et al. The effect of unilateral ablation of the frontal eye fields of saccadic performance in the monkey. In: Keller EL, Zee DS, eds. Adaptive Processes in Visual and Oculomotor Systems. Oxford, England: Peramon Press; 1986.
Guitton O, Buchtel HA, Douglas RM. Frontal lobe lesions in man cause difficulties in suppressing reflex- ive glances and in generating goal-directed saccades. Exp Brain Res. 1985;58:455.
Lasker AG, Zee DS. Saccades in Huntington’s dis- ease: initiation defects and distractibility. Neurology. 1987;37:364.
Zee DS, Yee RD, Singer HS. Congenital ocular motor apraxia. Brain. 1977;100:581.
Baloh RW, Yee RD, Boder E. Ataxia-telangiectasia: quantitative analysis of eye movements in six cases. Neurology. 1978;28:1099.
Baloh RW, Langhofer L, Honrubia V, Yee RD. On-line analysis of eye movements using a digital computer. Aviat Space Environ Med. 1980;51:563.
Zackon DH, Sharpe JA. Smooth pursuit in senes- cence: effects of target velocity and acceleration. Acta Otolaryngol. 1987;104:290.
Kaufman SR, Abel LA. The effects of distraction on smooth pursuit in normal subjects. Acta Otolaryngol (Stockh). 1986;102:57.
Baloh RW, Honrubia V, Sills A. Eye-tracking and optokinetic nystagmus. Results of quantitative testing in patients with well-defined nervous system lesions. Ann Otol Rhinol Laryngol. 1977;86:108.
Holzman PS, Levy DL, Uhlenhuth EH, Proctor LR, Freedman DX. Smooth-pursuit eye move- ments, and diazepam, CPZ, and secobarbital. Psychopharmacologia. 1975;44:111.
Fletcher WA, Sharpe JA. Smooth pursuit dysfunction in Alzheimer’s disease. Neurology. 1988;38:272.
Dejong JD, Melvill Jones G. Akinesia, hypokinesia and bradykinesia in the oculomotor system of patients with Parkinson’s disease. Exp Neurol. 1971;32:58.
Baloh RW, Yee RD, Honrubia V. Optokinetic nys- tagmus and parietal lobe lesions. Ann Neurol. 1980;7:269.
Leigh RJ, Fusa EW. Disturbance of smooth pursuit caused by infarction of parieto-occipital cortex. Ann Neurol. 1985;17:185.
Uemura T, Suzuki J, Hozawa J, Highstein S. Neuro- otological Examination. Baltimore: University Park Press; 1977.
Yee RD, Baloh RW, Honrubia V, et al. Slow buildup of optokinetic nystagmus associated with downbeat nys- tagmus. Invest Ophthalmol Vis Sci. 1979;18:622.
Lafortune S, Ireland DJ, Jell RM, Duval L. Human optokinetic after nystagmus. Acta Otolaryngol (Stockh). 1986;101:183.
Baloh RW, Sills AW, Honrubia V. Impulsive and sinu- soidal rotatory testing. A comparison with results of caloric testing. Laryngoscope. 1973;89:646.
Sills AW, Honrubia V, Baloh RW. Is the adaptation model a valid description of the vestibulo-ocular reflex? Biol Cybern. 1978;30:209.
Baloh RW, Honrubia V, Yee RD, Hess K. Changes in the human vestibulo-ocular reflex after loss of periph- eral sensitivity. Ann Neurol. 1984;16:222.
Aw ST, Haslwanter T, Halmagyi GM, et al. Three- dimensional vector analysis of the normal human vestibuloocular reflex in response to high-accelera- tion head rotations. 1. Responses in normal subjects. J Neurophysiol. 1996;76:4009.
Crane BT, Demer JL. Human horizontal vestibulo-ocular reflex initiation: effects of acceleration, target distance, and unilateral deafferentation. J Neurophysiol. 1998;80:1151.
Aw ST, Halmagyi GM, Black RA, et al. Head impulses reveal loss of individual semicircular canal function. J Vestib Res. 1999;9:173.
Collewijn H, Van Der Mark F, Jansen TC. Precise recording of human eye movements. Visi Res. 1974;15:447.
Weber KP, MacDougall HG, Halmagyi GM, Curthoys IS. Impulsive testing of semicircular-canal func- tion using video-oculography. Ann NY Acad Sci. 2009;1164:486.
Tomlinson RD, Saunders GE, Schwarz DWF. Analysis of human vestibulo-ocular reflex during active head movements. Acta Otolaryngol (Stockh). 1980;90:184.
O’Leary DP, Davis LL. High-frequency autorota- tional testing of the vestibulo-ocular reflex. Neurol Clin. 1990;8:297.
Blatt PJ, Schubert MC, Roach KE, Tusa RJ. The reliability of the Vestibular Autorotation Test (VAT) in patients with dizziness. J Neurol Phys Ther. 2008;32(2):70.
Furman JM, Durrant JD. Somatosensory cueing of head-only rotational testing. J Vestib Res. 1999;9:189.
Grossman GE, Leigh RJ, Abel LA, Lanska DJ, Thurston SE. Frequency and velocity of rotational head perturbations during locomotion. Exp Brain Res. 1988;70:470.
Tirelli G, Bigarini S, Russolo M, Giacomarra V, Sasso
F. Test-retest reliability of the VOR as measured via Vorteq in healthy subjects. Acta Otorhinolaryngol Ital. 2004;24(2):58.
Demer JL, Oas JG, Baloh RW. visual–vestibular interaction in humans during active and passive, ver- tical head movement. J Vestib Res. 1993;3:101.
Della Santina CC, Cremer PD, Carey JP, Minor LB. Comparison of head thrust test with head autoro- tation test reveals that the vestibulo-ocular reflex is enhanced during voluntary head movements. Arch Otolaryngol Head Neck Surg. 2002;128(9): 1044.
Guyot J-P, Psillas G. Test–retest reliability of ves- tibular autorotation testing in healthy subjects. Otolaryngol Head Neck Surg. 1997;117:704.
Baloh RW, Honrubia V, Konrad HR. Ewald’s sec- ond law reevaluated. Acta Otolaryngol (Stockh). 1977;83:475.
Jenkins HR, Honrubia V, Baloh RW. Evaluation of multiple frequency rotatory testing in patients with peripheral labyrinthine weakness. Am J Otolaryngol. 1982;3:182.
Aw ST, Halmagyi GM, Haslwanter T, et al. Three- dimensional vector analysis of the normal human ves- tibuloocular reflex in response to high-acceleration head rotations. 2. Responses in subjects with unilat- eral vestibular loss and selective semicircular canal occlusion. J Neurophysiol. 1996;76:4021.
Foster GA, Demer JL, Morrow MJ, Baloh RW. Deficits of gaze stability in multiple axes follow- ing unilateral vestibular lesions. Exp Brain Res. 1997;116:501.
Baloh RW, Jacobson KM, Beykirch K, Honrubia
V. Horizontal vestibulo-ocular reflex after acute peripheral lesions. Acta Otolaryngol Suppl (Stockh). 1989;468:323.
Honrubia V, Marco J, Andrews J, et al. Vestibulo- ocular reflexes in peripheral labyrinthine lesions. III. Bilateral dysfunction. Am J Otolaryngol. 1985;6:342.
Hyden D, Larsby B, Schwarz DW, Odkvist LM. Quantification of slow compensatory eye movements in patients with bilateral vestibular loss. A study with a broad frequency-band rotatory test. Acta Otolaryngol (Stockh). 1983;96:199.
Baloh RW, Jacobson K, Honrubia V. Idiopathic bilat- eral vestibulopathy. Neurology. 1989;39:272.
Wiest G, Tian J, Baloh RW, et al. Vestibular function in severe bilateral vestibulopathy. J Neurol Neurosurg Psychiatry. 2001;71:53.
Thursion SF, Leigh RR, Abel LA, Dell’Osso LF. Hyperactive vestibulo-ocular reflex in cerebellar degeneration. Neurology. 1987;37:53.
Baloh RW, Yee RD, Honrubia V. Internuclear oph- thalmoplegia. I. Saccades and dissociated nystagmus. Arch Neurol. 1978;35:484.
Baloh RW, Yee RD, Honrubia V. Internuclear ophthalmoplegia. II. Pursuit’ optokinetic nystag- mus and the vestibulo-ocular reflex. Arch Neurol. 1978;35:490.
Baloh RW, Sakala SM, Yee RD, et al. Quantitative vestibular testing. Otolaryngol Head Neck Surg. 1984;92:145.
Hyden D, Istl YE, Schwarz DWF. Human visuoves- tibular interaction as a basis for quantitative clinical diagnosis. Acta Otolaryngol (Stokch). 1982;94:53.
Baloh RW, Yee RD, Honrubia V. Eye movements in patients with Wallenberg’s syndrome. Ann NY Acad Sci. 1981;374:600.
Baloh RW, Yee RD, Kimm J, Honrubia V. The ves- tibulo-ocular reflex in patients with lesions of the ves- tibulocerebellum. Exp Neurol. 1981;72:141.
Diamond SG, Markham CH, Simpson NE, Curthoys IS. Binocular counterrolling in humans dur- ing dynamic rotation. Acta Otolaryngol (Stockh). 1979;87:490.
Kirienko NM, Money KE, Landolt JR, et al. Clinical testing of the otoliths: a critical assessment of ocular counterrolling. J Otolaryngol. 1984;13:281.
Nelson JR, House WF. Ocular countertorsion as an indicator of otolith function: effects of unilat- eral vestibular lesions. Trans Am Acad Ophthalmol Otolaryngol. 1971;75:1313.
Diamond SG, Markham CH, Furuya N. Binocular counterrolling during sustained body tilt in normal humans and in a patient with unilateral vestibular nerve section. Ann Otol. 1982;91:225.
Diamond SG, Markham CH, Baloh RW. Ocular counterrolling abnormalities in spasmodic torticollis. Arch Neurol. 1988;45:164.
Markham CH, Diamond SG. Distinctive counter- rolling disruption caused by brainstem compres- sion. In: Kunze K, Zangemeister WH, Arlt A, eds. Clinical Problems of Brainstem Disorders. Stuttgart, Germany: Georg Thieme Verlag; 1986.
Gresty MA, Bronstein AM. Otolith stimulation evokes compensatory reflex eye movements of high velocity when linear motion of the head is combined with con- current angular motion. Neurosci Lett. 1986;65:149.
Crane BT, Viirre ES, Demer JL. T he human horizon- tal vestibulo-ocular reflex during combined linear and angular acceleration. Exp Brain Res. 1997;114:304.
Gresty MA, Bronstein AM, Barratt H. Eye movement responses to combined linear and angular head move- ment. Exp Brain Res. 1987;65:377.
Dai MJ, Curthoys IS, Halmagyi GM. Linear acceler- ation—perception in the roll plane before and after unilateral vestibular neurectomy. Exp Brain Res. 1989;77:315.
Böhmer A, Mast F. Chronic unilateral loss of otolith function revealed by the subjective visual vertical during off center yaw rotation. J Vestib Res. 1999;9:413.
Böhmer A, Mast F. The subjective visual vertical dur- ing off-center angular rotation: a parameter to reveal chronic unilateral loss of vestibular function? J Vestib Res. 1996;6:20.
Benson AJ. Modification of the response to angular accelerations by linear accelerations. In: Kornhuber HH, ed. Handbook of Sensory Physiology: Vestibular System. Vol 6. Pt 2. Berlin, Germany: Springer- Verlag; 1974.
Stockwell CW, Turnipseed GT, Guedry FE. Nystagmus responses during rotation about a tilted axis. Pensacola, FL: Naval Aerospace Medical Research Lab; 1971.
Wall CD, Furman JM. Visual–vestibular interaction in humans during earth-horizontal axis rotation. Acta Otolaryngol (Stockh). 1990;109:337.
Furman, JM, Schor RH, Schumann TL. Off-vertical axis rotation: a test of the otolith–ocular reflex. Ann Otol Rhinol Laryngol. 1992;101:643.
Furman JMR, Wall C III, Kamerer DB. Earth hori- zontal axis rotational responses in patients with uni- lateral peripheral vestibular deficits. Ann Otol Rhinol Laryngol. 1989;98:551.
Niven JI, Hixon WC, Correia MJ. Elicitation of hori- zontal nystagmus by periodic linear acceleration. Acta Otolaryngol (Stockh). 1966;62:429.
Buizza A, Schmid R, Droulez J. Influence of linear acceleration on oculomotor control. In: Fuchs AF, Becker V, eds. Progress in Oculomotor Research. New York: Elsevier/North Holland; 1981.
Baloh RW, Beykirch K, Honrubia V, Yee RD. Eye movements induced by linear acceleration on a paral- lel swing. J Neurophysiol. 1988;60:2000.
Bronstein AM, Gresty MA. Short latency compensa- tory eye movement responses to transient linear head acceleration: a specific function of the otolith–ocular reflex. Exp Brain Res. 1988;71:406.
Bronstein A, Gresty MA, Brookes GB. Compensatory otolithic slow phase eye movement responses to abrupt linear head motion in the lateral direction. Acta Otolaryngol Suppl (Stockh). 1991;418:42.
Lempert T, Gianna C, Brookes G, Bronstein A, Gresty M. Horizontal otolith–oculalr responses in humans after unilateral vestibular deafferentation. Exp Brain Res. 1998;118:533.
Baloh RW, Fife TD, Zwerling L, et al. Comparison of static and dynamic posturography in young and older normal people. J Am Geriatr Soc. 1994;42:405.
Baloh RW, Corona S, Jacobson KM, Enrietto JA, Bell
T. A prospective study of posturography in normal older people. J Am Geriatr Soc. 1998;43:438.
Baloh RW, Jacobson KM, Beykirch K, Honrubia
V. Static and dynamic posturography in patients with vestibular and cerebellar lesions. Arch Neurol. 1998;55:649.
Nashner LM. A model describing vestibular detec- tion of body sway motion. Acta Otolaryngol (Stockh). 1971;72:429.
Nashner LM, Black FO, Wall C III. Adaptation to altered support and visual conditions during stance: patients with vestibular deficits. J Neurosci. 1982;2:536.
Allum JHJ, Pfaltz CR. Visual and vestibular contribu- tions to pitch sway stabilization in the ankle muscles of normals and patients with bilateral peripheral ves- tibular deficits. Exp Brain Res. 1985;58:82.
Black FO, Wall C III, Nashner LM. Effect of visual and support surface references upon postural con- trol in vestibular deficit subjects. Acta Otolaryngol (Stockh). 1983;95:199.
Baloh RW, Jacobson KM, Enrietto JA, Corona S, Honrubia V. Balance disorders in older people: quan- tification with posturography. Otolaryngol Head Neck Surg. 1998;119:89.
Furman JM, Baloh RW, Kamran B, et al. Assessment: posturography. Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 1996;46:1763.
Piirtola M, Era P. Force platform measurements as predictors of falls among older people: a review. Gerontology. 2006;52:1.
Gianoli G, McWilliams S, Soileau J, Belafsky P. Posturographic performance in patients with the potential for secondary gain. Otolaryngol Head Neck Surg. 2000;122:11.
Elidan J, Sohmer H. Vestibular tests in evolution. III. Vestibular evoked potentials. In: Baloh RW, Halmagyi GM, eds. Disorders of the Vestibular System. New York: Oxford University Press; 1996: 274.
Hofferberth B. Evoked potentials to rotatory stimula- tion. Acta Otolaryngol Suppl (Stockh). 1984;406:134.
Pirodda E, Ghedini S, Zanetti MA. Investigations into vestibular-evoked responses. Acta Otolaryngol (Stockh). 1987;104:77.
Bickford RG, Jacobson JL, Cody DT. Nature of aver- age evoked potentials to sound and other stimuli in man. Ann NY Acad Sci. 1964;112:204.
Colebatch JG, Halmagyi GM. Vestibular evoked potentials in human neck muscles before and after unilateral vestibular deafferentation. Neurology. 1992;42:1635.
Ferber-Viart C, Dubreuil C, Duclaux R. Vestibular evoked myogenic potentials in humans: a review. Acta Otolaryngol (Stockh). 1999;119:6.
Curthoys IS, Kim J, McPhedran SK, Camp AJ. Bone conducted vibration selectively activates irregular pri- mary otolithic vestibular neurons in the guinea pig. Exp Brain Res. 2006;175(2):256.
Colebatch JG, Halmagyi GM, Skuse NF. Myogenic potentials generated by a click-evoked vestibu- locollic reflex. J Neurol Neurosurg Psychiatry. 1994;57(2):190.
Uchino Y, Sato H, Sasaki M. Sacculocollic reflex arcs in cats. J Neurophysiol. 1997;77(6):3003.
Colebatch JG, Rothwell JC. Motor unit excitabil- ity changes mediating vestibulocollic reflexes in the sternocleidomastoid muscle. Clin Neurophysiol. 2004;115(11):2567.
Todd NP, Rosengren SM, Aw ST, Colebatch JG. Ocular vestibular evoked myogenic poten- tials (OVEMPs) produced by air- and bone-cond ucted sound. Clin Neurophysiol. 2007;118(2): 381.
Todd NP, Rosengren SM, Colebatch JG. A utricu- lar origin of frequency tuning to low-frequency
vibration in the human vestibular system? Neurosci Lett. 2009;451(3):175.
Iwasaki S, Chihara Y, Smulders YE. The role of the superior vestibular nerve in generating ocular vestib- ular-evoked myogenic potentials to bone conducted vibration at Fz. Clin Neurophysiol. 2009;120(3): 588.
Rosengren SM, Aw ST, Halmagyi GM, Todd NP, Colebatch JG. Ocular vestibular evoked myogenic potentials in superior canal dehiscence. J Neurol Neurosurg Psychiatry. 2008;79(5):559.
Brantberg K. Vestibular evoked myogenic potentials (VEMPs): usefulness in clinical neurotology. Semin Neurol. 2009;29(5):541.
de Waele C, Huy PT, Diard JP, Freyss G, Vidal PP. Saccular dysfunction in Meniere’s disease. Am J Otol. 1999;20(2):223.
Murofushi T, Shimizu K, Takegoshi H, Cheng PW. Diagnostic value of prolonged latencies in the ves- tibular evoked myogenic potential. Arch Otolaryngol Head Neck Surg. 2001;127(9):1069.
Helling K, Schönfeld U, Clarke AH. Treatment of Ménière’s disease by low-dosage intratympanic gentamicin application: effect on otolith function. Laryngoscope. 2007;117(12):2244.
Patko T, Vidal PP, Vibert N, Tran Ba Huy P, de Waele C. Vestibular evoked myogenic potentials in patients suffering from an unilateral acoustic neu- roma: a study of 170 patients. Clin Neurophysiol. 2003;114(7):1344.
Suzuki M, Yamada C, Inoue R, Kashio A, Saito Y, Nakanishi W. Analysis of vestibular testing in patients with vestibular schwannoma based on the nerve of origin, the localization, and the size of the tumor. Otol Neurotol. 2008;29(7):1029.
Ushio M, Iwasaki S, Chihara Y. Is the nerve origin of the vestibular schwannoma correlated with vestibular evoked myogenic potential, caloric test, and auditory brainstem response? Acta Otolaryngol. ePub ahead of print, November 26, 2008.
Murofushi T, Halmagyi GM, Yavor RA, Colebatch JG. Absent vestibular evoked myogenic potentials in vestibular neurolabyrinthitis. An indicator of infe- rior vestibular nerve involvement? Arch Otolaryngol Head Neck Surg. 1996;122(8):845.
Kim HA, Hong JH, Lee H. Otolith dysfunction in vestibular neuritis: recovery pattern and a predictor of symptom recovery. Neurology. 2008;70(6):449.
Brantberg K, Bergenius J, Tribukait A. Vestibular- evoked myogenic potentials in patients with dehis- cence of the superior semicircular canal. Acta Otolaryngol. 1999;119(6):633.
Watson SRD, Halmagyi GM, Colebatch JG. Vestibular hypersensitivity to sound (Tullio phe- nomenon): structural and functional assessment. Neurology. 2000;54(3):722.
Brantberg K, Verrecchia L. Testing vestibular-evoked myogenic potentials with 90-dB clicks is effective in the diagnosis of superior canal dehiscence syndrome. Audiol Neurootol. 2009;14(1):54.
This page intentionally left blank
![]()
TYPES OF HEARING DISORDERS
Conductive Sensorineural
Central Hearing Disorders BEDSIDE TESTS OF HEARING BEHAVIORAL AUDIOMETRY
The Audiogram
Speech Recognition Tests Stenger Test
IMPEDANCE AUDIOMETRY
TYMPANOMETRY
The Acoustic Reflex
AUDITORY-EVOKED RESPONSES
Electrocochleography
Brainstem Auditory-Evoked Response
GENERATING POTENTIALS TEST METHODOLOGY RESULTS IN PATIENTS
CENTRAL AUDITORY SPEECH TESTS SUMMARY OF AUDITORY TEST RESULTS
Hearing disorders can be classified as conduc- tive, sensorineural, and central, based on the anatomic site of lesion. A battery of audiologi- cal tests can help localize the lesion within the anatomical pathways.1,2
Conductive
Conductive hearing loss results from lesions involving the external or middle ear. The tympanic membrane and ossicles act as a trans- former amplifying airborne sound and effi- ciently transferring it to the inner ear fluid (see “Middle Ear” in Chapter 2). If this normal pathway is obstructed, transmission may occur across the skin and through the bones of the skull (bone conduction), but at the cost of con- siderable energy loss.
The most common cause of conductive hear- ing loss is impacted cerumen in the external auditory canal. This benign condition is often first noticed after bathing or swimming, when water closes the remaining tiny passageway.
The most common serious cause of conductive hearing loss is inflammation of the middle ear, or otitis media. Either infected (suppurative oti- tis) or noninfected (serous otitis) fluid accumu- lates in the middle ear, impairing the conduc- tion of airborne sound. Otosclerosis produces progressive conductive hearing loss by immobi- lizing the stapes with new bone growth in front of and below the oval window. Other causes of conductive hearing loss include large tympanic membrane perforations, trauma, congenital malformations of the external and middle ears, and tumors of the temporal bone.
Sensorineural
Sensorineural hearing loss results from lesions of the cochlea and/or the auditory division of the eighth cranial nerve.3 The spiral cochlea mechanically analyzes the frequency content of sound. For high-frequency tones, only sensory cells in the basal turn are activated, whereas for low-frequency tone maximum stimulation occurs at the apex, even though all or nearly all sensory cells are activated with loud sounds.
219
Therefore, with lesions of the cochlea and its afferent nerve, hearing levels for different fre- quencies are often unequal, and the timing (phase) relationship between different fre- quencies may be altered. Patients with sen- sorineural hearing loss often have difficulty hearing speech that is mixed with background noise and may be annoyed by loud speech.
Distortion of sound is common with sen- sorineural hearing loss. A pure tone may be heard as noisy, rough, or buzzing, or it may be distorted so that it sounds like a complex mixture of tones. Binaural diplacusis occurs when the two ears are affected unequally so that the same frequency has a different pitch in each ear; that is, the patient hears different sounds in each ear. Monaural dipla- cusis occurs when two tones or a tone and noise are heard simultaneously in one ear. With recruitment there is an abnormally rapid growth in the sensation of loudness as the intensity of a sound is increased so that faint or moderate sounds cannot be heard, whereas there is little or no change in the loudness of loud sounds.
Sudden unilateral deafness is a common syn- drome with ill-defined pathophysiology.4 Probably most cases are due to viral infections involving either the cochlea or auditory nerve.5 Viral infections can be part of a systemic viral illness such as measles, mumps, and infectious mononucleosis or an isolated infection of the labyrinth or eighth nerve without systemic symptoms. Mumps is a particularly common cause of unilateral hearing loss in school-aged children. Other common causes of acute uni- lateral hearing loss are head trauma and vascu- lar occlusive disease.
Relapsing unilateral sensorineural hearing loss associated with tinnitus, ear fullness, and vertigo is typical of Meniere’s syndrome. Ototoxic drugs produce a bilateral subacute hearing loss, and acoustic neuromas (vestibular schwannomas) characteristically produce a slowly progressive unilateral sensorineural hearing loss. The chronic progressive bilateral hearing loss associated with advancing age is called presbycusis. It may include conductive and central dysfunction, but the most consis- tent effect of aging is on the sensory cells and neurons of the cochlea.5 Genetic disorders account for the majority of cases of sensorineu- ral hearing loss in children (see “Hereditary Disorders” in Chapter 18).
Central Hearing Disorders
Central hearing disorders result from lesions of the central auditory pathways: the cochlear and dorsal olivary nuclear complexes, inferior col- liculi, medial geniculate bodies, and auditory cortex in the temporal lobes and their intercon- necting afferent and efferent fiber tracts. As a rule, patients with central lesions do not have impaired hearing levels for pure tones, and they understand speech if it is clearly spoken in a quiet environment. If the listener’s task is made more difficult with the introduction of background or competing messages, perfor- mance deteriorates in patients with central lesions more than in normal subjects. Lesions involving the nerve root entry zone or cochlear nucleus can result in unilateral hearing loss for pure tones (e.g., demyelination or infarction of the lateral pontomedullary region). Because at least 50% of afferent nerve fibers cross central to the cochlear nucleus, this is the most central structure in which a lesion can result in a uni- lateral hearing loss.
A quick test for hearing loss in the speech range is to observe the response to spoken commands at different intensities (whisper, conversation, shouting).6 The examiner stands behind the patient to prevent lip reading and occludes and masks the non-test ear by moving a finger back and forth in the patient’s external ear canal. Finger rubs at different intensities and dis- tances from the ear have been shown in a rigor- ous study to be a rapid, reliable, and valid screening test for hearing loss in the frequency range of speech.7 If a patient can hear a faint finger rub stimulus at a distance of 70 cm (approximately one arm’s length) from one ear, then a hearing loss on that side—as defined by a gold standard audiogram threshold of >25 dB at 1000, 2000, and 4000 Hz—is highly unlikely. On the other hand, if a patient cannot hear a strong finger rub stimulus at 70 cm, then a hear- ing loss on that side is highly likely.7 Tuning fork tests permit a rough assessment of the hearing level for pure tones of known frequency. Clinicians can use their own hearing level as a reference standard. The Rinne test compares the patient’s hearing by air conduction with that
by bone conduction. The fork (256 or 512 Hz) is first held against the mastoid process until the sound fades. It is then placed 1 inch from the ear. Normal subjects can hear the fork longer by air than by bone conduction. If bone con- duction is greater than air conduction, a con- ductive hearing loss is suggested. The Weber test compares the patient’s hearing by bone conduction in the two ears. The fork is placed at the center of the forehead or on a central inci- sor and the patient is asked where he or she hears the tone. Normal subjects hear it in the center of the head, patients with unilateral con- ductive loss hear it on the affected side, and patients with unilateral sensorineural loss hear it on the side opposite the loss. Though these tuning fork tests can be useful in some clinical settings, they are not considered to be reliable or valid screening tests.6
Audiometry typically consists of a battery of tests, the differential results of which provide site-of-lesion information.1 It typically begins with pure-tone threshold testing to compare a subject’s hearing sensitivity to norms at selected frequencies (the audiogram).
The Audiogram
Pure tones are defined by their frequency and their intensity. To quantify the magnitude of hearing loss, normal hearing levels have been established. These levels approximate the inten- sity of the faintest sounds that can be heard by normal ears. A patient’s hearing level (HL) is the difference in decibel (dB) between the faintest pure tone that the patient can hear and the normal reference level given by the stan- dard, where 0 dB HL is the sound pressure level (SPL) at which listeners with normal hearing are able to perceive the signal 50% of the time. Brief-duration tones at selected frequencies are presented by earphones (air conduction) and a vibrator pressed against the mastoid portion of the temporal bone (bone conduction). The results of air and bone conduction testing are plotted on a graph from which the magnitude of the sensitivity loss (in dB) as a function of fre- quency is determined (Fig. 8–1). A simple way to summarize the severity of hearing loss is to calculate the pure tone average (PTA) = the average hearing level at 500, 1000, and 2000 Hz. For example, if the hearing level is 40 dB, 50 dB, and 60 dB at 500, 1000, and 2000 Hz, respectively, then the PTA is 50 dB, considered a moderate hearing loss (Table 8–1).
Frequency in Hz
125 250 500 1000 2000 4000 8000
Hearing level in dB re ANSI, 1969
Hearing level in dB re ANSI, 1969
0
10
20
30
40
50
60
70
80
90
100
110
125 250 500 1000 2000 4000

0
10
20
30
40
50
60
70
80
90
100
110
8000
Air | Bone | |
Right ear | ||
Left ear |
Air | Bone | |
Right ear | ||
Left ear |
(with masking)
![]()
![]()
![]()
![]()
Figure 8–1. Pure-tone audiogram: left ear, normal; right ear, conductive hearing loss due to otosclerosis.
Table 8–1 Severity of hearing loss based on the pure tone average (PTA)
![]()
Normal 0-25 dB
Mild Hearing Loss 26-40 dB
Moderate Hearing Loss 41-55 dB Moderate Severe Hearing Loss 56-70 dB Severe Hearing Loss 71-90 dB
Profound Hearing Loss >90 dB
![]()
PTA = Average of hearing levels at 500, 1000 and 2000 Hz.
With a conductive hearing loss, air conduction is impaired while bone conduction remains nor- mal (i.e., an air-bone gap on the audiogram, Fig. 8–1, right ear). Measurement of bone conduc- tion requires careful masking of the nontest ear. Masking involves introducing airborne noise into the nontest ear to eliminate cross-hearing via
bone conduction. There is a <5 dB attenuation between the two ears for a bone conduction receiver placed on any part of the skull. Therefore, a nonhearing ear may appear to have nearly nor- mal hearing via bone conduction if the normal ear is not properly masked. The best masking sound for pure tones is a narrow band of white noise centered about the pure tone being tested. Lesions producing sensorineural hearing loss impair both air and bone conduction, often with changing pure tone levels at different fre- quencies. Although the audiogram does not provide specific diagnostic information to iden- tify the site of lesion, certain patterns suggest specific diagnoses. Typical audiograms seen in patients with four common causes of sen- sorineural hearing loss are shown in Figure 8–2. None of these patterns is pathognomonic of a given disorder, but they occur often enough to
be of diagnostic value.
Frequency in Hz
A 125
0
10
20
30
40
50
60
Hearing level in dB re ANSI, 1969
Hearing level in dB re ANSI, 1969
70
80
90
100
110
250 500 1000 2000 4000 8000

B 125 250 500 1000 2000 4000 8000
0

10
20
30
40
50
60
70
80
90
100
110
C 125 250 500 1000 2000 4000 8000
0

10
20
30
40
50
60
70
80
90
100
110
D 125 250 500 1000 2000 4000 8000
0

10
20
30
40
50
60
70
80
90
100
110
Figure 8–2. Audiograms illustrating four common patterns of sensorineural hearing loss. (A) Notched pattern of noise- induced hearing loss. (B) Downward sloping pattern of presbycusis. (C) Low-frequency trough of Meniere’s syndrome. (D) V-pattern of congenital hearing loss.
Two categories of tests are used to determine the patient’s ability to hear and understand speech: (1) the speech reception threshold and
speech discrimination.
The speech reception threshold (SRT) is the intensity at which the patient can correctly repeat 50% of highly familiar two-syllable words (e.g., airplane, cowboy, sidewalk). The SRT is an estimate of the minimum level of conversation that a person can hear. It provides a check on the validity of the pure- tone audiogram, as it should agree (±5 dB) with an average of the two best pure-tone thresholds in the speech range (500 to 2000 Hz). It is not a test of discrimination, but it does provide information about a patient’s abil- ity to recognize and respond to speech. Occasionally, a patient who is profoundly deaf or aphasic cannot repeat words. In such cases a speech detection threshold (SDT) is obtained by having the patient respond to a speech signal.
The speech discrimination test is a measure of the patient’s ability to understand speech when it is presented at a level that is easily heard. For this test, the patient is usually pre- sented with 50 phonetically balanced monosyl- labic words at a comfortable listening level (typically 35 to 40 dB above the SRT). Each word is presented with a carrier phrase, such as “you will say ” or “say the word ” The test is scored as a percentage of the correct responses (e.g., 49 out of 50 correct = 98% speech discrimination). In patients with eighth nerve lesions, speech discrimination can be severely reduced even when pure-tone levels are normal or near normal, whereas in patients with cochlear lesions, discrimination tends to be proportional to the magnitude of hearing loss.8
Stenger Test
The Stenger test is helpful for determining whether a patient is deliberately exaggerating or feigning a hearing loss.9 It is based on the psychophysical observation that a tone pre- sented to both ears in the same phase at differ- ent intensities above threshold is perceived only in the ear with better hearing. Tones are presented simultaneously to both good and
suspect ears. The signals are turned on together and the patient is asked to respond as long as the tone is on. As long as the tone is below the actual threshold in the suspect ear, the patient will hear the sound in the other ear and respond. If the tone in the suspect ear is then increased above its true threshold, the patient will either fail to respond or show confusion, being unaware of the sound in the good ear and not wanting the examiner to know that he hears the sound in the suspect ear. A patient with true unilateral loss will of course only hear the sound presented in the good ear and quickly respond. Although screening tests are usually done at one intensity, a more complete test can be done at multiple intensities or using the same procedure with spondee words. Of course, the Stenger test works only with a unilateral loss, as a minimum 20 dB threshold difference is required at the test frequency.10
Impedance may be defined as the resistance of a given system to the flow of energy. Acoustic impedance refers to the resistance of the mid- dle ear system to the passage of sound. Its reciprocal, acoustic compliance, refers to the ease of sound transmission through the middle ear system. In simplified terms, acoustic imped- ance is related to the elasticity and stiffness of the middle ear conduction system. Acoustic impedance measurements are based on the principle that energy that is not absorbed by the ear is reflected.11 By measuring the differ- ence between the intensity of a sound going into the external auditory canal and that reflected from the tympanic membrane, one can estimate the impedance (or compliance) of the middle ear system.
Acoustic impedance measurements are made by a probe tip hermetically inserted into the ear canal. The probe tip contains three openings: (1) one for air pressure generation and measurement, (2) one for probe tone gen- eration, and (3) one for pickup of sound waves reflected off the tympanic membrane. A sche- matic drawing of the impedance measurement system is shown in Figure 8–3. With this sys- tem the difference between generated and reflected sound is systematically measured at different external ear pressure levels.
Loudspeaker
Sound generator 220 Hz
Air pressure generation and measurement
Air pump
Intensity knob
Manometer
Rectifier
Rectifier
AMP
AMP
Compliance change meter
Probe
DC source (18 V)
DC source (18 V)
SPL & compliance sensing & measurement
Microphone (sound reflecting off
tympanic membrane to microphone pickup)
Figure 8–3. Schematic drawing of acoustic impedance measuring system (see text for details). SPL, sound pressure level. (From Goodhill V. Ear Diseases, Deafness and Dizziness. Harper & Row, Hagerstown, 1979, with permission.)
Tympanometry is a method for evaluating changes in acoustic impedance by producing systematic changes in air pressure in the exter- nal ear canal. A plot of compliance change ver- sus air pressure (the tympanogram) is made by first introducing a positive pressure into the external canal (usually equivalent to +200 mm of water) and then decreasing the pressure to approximately −200 mm of water (although the negative range can be extended to −600 mm of water). As the pressure is changed, compliance will change if the conduction system is normal. The shape of the normal tympanogram resem- bles a teepee (Fig. 8–4). The peak of the teepee represents the point of maximum com- pliance at which the air pressure in the middle ear equals the air pressure in the external audi- tory canal. Tympanometry can provide useful information about (1) mobility of the tympanic membrane, (2) perforations of the tympanic membrane, (3) pressure within the middle ear, and (4) patency and dynamic function of the eustachian tubes. Artifactual responses can result from inappropriate placement of the probe (against the canal wall or into impacted cerumen) or from an inadequate seal.
Four characteristic abnormal tympanographic patterns should be recognized (see Fig. 8–4).12
A restricted tympanogram implies normal mid- dle ear pressure and limited compliance relative to normal mobility. It is typically seen in advanced cases of otosclerosis, lateral fixation of the ossicular chain, tympanic membrane fibro- sis, and middle ear tympanosclerosis. A hyper- mobile tympanogram indicates a flaccid tym- panic membrane. It occurs with ossicular chain discontinuity and with partial atrophy of the tympanic membrane. A flat tympanogram means that there is little or no change in middle ear compliance when ear pressure is varied in the external ear canal. This pattern is more commonly seen with secratory otitis media but can also be seen with congenital malforma- tions of the middle ear and occlusion of the external ear canal by cerumen, epithelium, and foreign bodies. Finally, with a retracted tym- panogram, the maximum compliance occurs at negative pressures greater than −100 mm of water. It implies a negative middle ear pressure with a retracted tympanic membrane. This pat- tern is most commonly seen with poor Eustachian tube function.
The Acoustic Reflex
The acoustic reflex refers to contraction of the stapedius muscle in response to a loud sound.

5 A B
Compliance change
Compliance change
0
5

5 C D
0
–300
–100 0 +100 +300 –300 –100 0 +100 +300
Air pressure, mm H2O
Figure 8–4. Four characteristic abnormal tympanograms. (A) Restricted. (B) Hypermobile. (C) Flat. (D) Retracted. Blue area, normal range.
It is measured by monitoring the change in acoustic impedance in response to a loud sound introduced into either ear. The stapedius mus- cle contracts bilaterally, regardless of which ear is stimulated. Contraction of the stapedius muscle produces stiffening of the tympanic membrane and thus an increase in acoustic impedance. This results in an attenuation of sound transmitted to the cochlea by about 10 dB. In a normal subject the acoustic reflex will be observed when a pure-tone signal is pre- sented between 70 and 90 dB above hearing level (median value 82 dB), and when a white noise stimulus is presented at 65 dB above hearing level.
Patients with conductive hearing loss often have an absent acoustic reflex because the lesion prevents a change in compliance with stapedius muscle contraction. An air bone gap as small as 5 dB may obscure the acoustic reflex. Acoustic reflex testing is particularly useful for identify- ing a false or so-called intralabyrinthine conduc- tive hearing loss (see Semicircular Canal Dehiscence Syndrome, Chapter 16). A normal acoustic reflex in a patient with an apparent con- ductive hearing loss suggests the likelihood of superior semicircular dehiscence. The acoustic reflex can also be useful for identifying the site of lesion for different types of sensorineural hearing loss. With cochlear lesions the acoustic reflex often can be demonstrated at a sensation
level <60 dB above the auditory pure-tone threshold. This is another form of abnormal loudness growth or recruitment. A cochlear hearing loss must be severe before the acoustic reflex is lost. An absent reflex is rarely associated with a cochlear hearing loss <50 dB, and only when the hearing loss exceeds 85 dB is the reflex absent in 50% of patients. By contrast, patients with eighth nerve lesions often have either nor- mal hearing or only mildly impaired hearing (<20 dB), yet they have an abnormal acoustic reflex. The reflex may be absent, exhibit an ele- vated threshold, or exhibit abnormal decay. Reflex decay is present if the amplitude decreases to one-half of its original size within 10 sec of tonal stimulation. However, the test has not been proven to be a highly accurate test for discriminating between these causes, or even identifying an eighth nerve lesion.32
The advent of averaging by computer made it possible to collect and analyze a variety of evoked electrical potentials from the auditory system.14,15 Repetitive sounds are delivered to the external ear and an “averaged” series of specific brain wave potentials are recorded with disk electrodes for up to 500 msec after
signal onset. The latencies (relative to signal onset) of each of the potentials are used as the most reliable means by which generator sources for the potentials are identified. Electrical events in the cochlea (so-called electrococh- leography) can best be monitored by an elec- trode inside the external canal or from a tran- stympanic electrode on the cochlear promontory.16,17 A series of waves in the first 5 msec after the stimulus reflects the cochlear microphonics and the compound cochlear action potential (Fig. 8–5).18 Other evoked potentials from the auditory nerve and central nervous system (CNS) can be recorded with differential scalp electrodes.19 One electrode is placed at the vertex (the positive electrode) and another on the earlobe or mastoid (the negative electrode) ipsilateral to the acoustic stimulation. A third electrode (contralateral mastoid or earlobe) serves as the ground. Acoustic signals are presented via earphones, or bone vibrator and responses are amplified, filtered, and averaged. The early (0 to 10 msec) evoked responses reflect the far-field represen- tation of electrical events generated at points from the periphery (eighth nerve action poten- tial) to the level of the brain stem. The five to seven waves in the first 10 msec are referred to as the brainstem–evoked response. The mid- dle latency responses (12 to 50 msec) have received less systematic study but probably reflect electrical activity in the upper brain stem and in the primary and nearby secondary auditory projection areas. The late evoked responses (50 to 300 msec) reflect cortical electrical activity.
Electrocochleography
Up to the present, this technique has been most useful for identifying cochlear responses in infants or in very young children who are unable to cooperate with behavioral testing.20 A rough estimate of the cochlear response at multiple frequencies and intensities can be obtained. Of course, the presence of these responses does not indicate that the subject actually “hears”; rather, it documents that electrical activity is being generated within the cochlea. The presence of cochlear microphonics and summating potentials (see Fig. 8–5) in the absence of a compound action potential suggests a denervated cochlea. Electrocochleography is useful in conjunction with brainstem auditory-evoked responses (BAER) in adults when one is attempting to identify whether a hearing loss is cochlear or retrocochlear. The presence of a normal elec- trocochleogram in a patient with an absent BAER suggests an auditory neuropathy.21 Absence of both can occur with either cochlear or retrocochlear lesions. Electrocochleography is also used to assess the likelihood of Meniere’s syndrome, though it is not a very accurate test (see Chapter 11).
Brainstem Auditory-Evoked Response
As noted above, the BAER reflects the far-field representation of electrical events occurring in the eighth nerve and brain stem.15 It is highly reproducible in a given subject, and the latency of the various waves shows little variation
1
1
N
Stimulus
N
N
CM
2
among normal subjects.22 With rare exceptions, the BAER is not affected by inattention to the stimulus, alterations in the level of conscious- ness, or drugs. For this reason it can be used to test the integrity of the peripheral and brain- stem auditory pathways in patients who cannot cooperate with subjective auditory testing (e.g., infants, comatose patients).23
0 1 2 3 4 5
msec
Figure 8–5. Electrical response of the cochlea to a tone burst. An electrode near the round window records the cochlear microphonic potential (CM) and a compound action potential (N1 and N2). (Adapted from Sohmer H, Feinmesser M. Electrocochleography in clinical audiologi- cal diagnosis. Arch Otorhinolaryngol (Berl). 1974;206:91.)
A schematic drawing of a BAER and the neural centers that are thought to generate each component of the response is shown
0 1 2 3 4 5 6 7 8 9 10
![]()
60 dBHL 1.9 3.0 4.1 5.2
+
+
Cz
5.9 7.6 9.2
5.8
IV
V
0.1 µV units
0.1 µV units
II III
I
VII
VI
Lateral lemniscus
z
z
C – Cochlea, acoustic n. terminals
Cochlear nucleus, Superior olivary complex
Inferior colliculus
Medial geniculate
Auditory radiations
Figure 8–6. Normal brain stem auditory evoked response evoked by clicks of 60 dB HL (60 dB above normal hearing threshold) at a rate of 10/sec. Normal mean latencies for waves I through VII are shown on the time scale (the intermedi- ate latency, 5.8 msec, between waves IV and V, is the mean peak latency of a fused wave IV/V when present). The neural centers thought to be responsible for generating each wave are shown at the bottom. (Adapted from Stockard JJ, Stockard JE, Sharbrough FW. Detection and localization of occult lesions with brain stem auditory responses. Mayo Clin Proc. 1977;52:761.)
in Figure 8–6.31 Wave I (average latency 1.9 msec) results from activation of the eighth nerve terminals within the cochlea, whereas wave II (average latency 3.0 msec) results from activation of the central portions of the eighth nerve. The remainder of the waves are gener- ated by the brainstem auditory nuclei and path- ways.24,25 Although a useful working tool, this schematic electroanatomic correlation is an oversimplification. Clearly, each vertex-positive and vertex-negative potential after wave I reflects simultaneous activity in multiple brain- stem loci. The more caudal generators, such as the eighth nerve, cochlear nuclei, and superior olivary complex, contribute to the response beyond waves II and III. By the time waves VI and VII appear, the summation of potentials from the different neural centers is so complex that the concept of single principle contribu- tors to individual waves no longer applies.
The standard stimulus for eliciting a BAER is a click caused by a very short (<1 msec) pulse.
This signal produces a spectrally diffused acoustic stimulus, with most of the energy con- centrated in the high frequencies (around 1–4K Hz with most of the commercially available speakers). The absolute latency of the BAER waves is dependent on the intensity of the click stimulus. The BAERs to unfiltered clicks pre- sented at various intensity levels from a normal subject are shown in Figure 8–7. Wave V is most robust, often being identifiable at only 10–20 dB above hearing level. At 60 dB above hearing level, all waves are identifiable. Such latency intensity functions provide a basis for estimating the degree of hearing loss in patients who cannot cooperate with standard pure-tone behavioral testing.26 Because of this latency- intensity relationship, however, BAERs must be interpreted with caution in patients with severe conductive or cochlear hearing loss (particularly if it involves high frequencies).
In practice, only waves I, III, and V are used to define response abnormalities. Waves II, IV,
IV
200 nV ![]() I II III
I II III
VI
dB SL 70
50
Right ear
II III
I
IV - V
![]()
30 0 2
Left ear I
10
4 6 8 10
IV - V
III
0 5 10 15
msec
Figure 8–7. Brain stem auditory evoked responses in a
normal subject that are induced by clicks at varying inten-
sity levels (10 to 70 dB sensation level [SL]).
VI, and VII are sufficiently variable in the nor- mal population to preclude their routine use in defining response abnormalities on the basis of a single recording. Often waves IV and V fuse into a single complex, which is designated as wave IV/V. Because wave I disappears before wave IV/V with decreasing stimulus intensity (e.g., see Fig. 8–7), the absence of wave IV/V in the presence of wave I suggests a retrocochlear lesion. The absence of all waves, on the other hand, often reflects a peripheral lesion (con- ductive or cochlear) or a technical problem.27 If peak I occurs at normal latency, then prolonga- tion of the I–III or I–IV/V interval suggests a lesion of the eighth nerve or brain stem.
In clinical neurotology BAERs have been used mostly in (1) evaluating the cause and reversibility of coma, (2) assessing the likeli- hood of multiple sclerosis (MS), and (3) assessing the likelihood of a structural lesion involving the eighth nerve (e.g., vestibular schwannomas) (Fig. 8–8).25 Because BAERs are relatively resistant to metabolic insults, they can help differentiate between metabolic and structural causes of brainstem dysfunction. The presence of an intact BAER in a patient with global brainstem dysfunction on neurologic examination suggests the possibility of a meta- bolic, reversible cause of coma. The absence of waves III and/or V in the presence of wave I, however, suggests widespread structural dam- age of the brainstem and implies a poor prog- nosis.28,29 The complete absence of a BAER may have a similar poor prognosis but requires
0 2 4 6 8 10
Time in msec
Figure 8–8. Brain stem auditory evoked responses in a patient with a left acoustic neuroma. Dashed lines indicate repeat test. Wave I occurs at normal latency on both sides, but the I–III and I–V intervals are prolonged on the left side.
more cautious interpretation, as there may be other explanations for the lack of response (technical or otologic problems).
In patients suspected of having MS who present with lesions outside of the brain stem (e.g., optic nerve or spinal cord), an abnormal BAER supports the likely diagnosis (the impor- tant second lesion).27,30 Obviously, if the initial lesion involves the brain stem, the finding of an abnormal BAER provides little additional information. In the past brainstem auditory- evoked responses were used to screen for ves- tibular schwannomas but with the improved resolution and reduction in costs of MR imag- ing the BAER can no longer be recommended for primary screening for these tumors (see Chapter 15).32
Patients with lesions of the central auditory pathways usually have normal pure-tone hearing levels. Routine speech tests are also usually normal because speech contains a great deal of redundancy. Within the central auditory
pathways, redundancy is enhanced by the mul- tiple crossings and interactions. One of the few diagnostically useful central audiologic findings is the reduced ability of patients with temporal lobe lesions to discriminate speech in the ear contralateral to the lesion when the task is complicated by distorting the speech.33 Apparently, by making the speech less redun- dant, heavier demands are placed on the inte- grating, synthesizing function of the auditory cortex.
There are several varieties of central audi- tory speech tests currently in use, each involv- ing methods of presenting distorted speech. Portions of the frequency spectrum of speech can be filtered, the speech can be time com- pressed, it can be presented at very low intensi- ties, and the speech can be interrupted at irregular intervals. Dichotic stimulation involves presenting two different messages to each ear.34 Both monosyllabic and spondee words can be used. As with distorted speech tests, when a temporal lobe lesion exists, the ear contralateral to the lesion performs poorer than the ear ipsilateral to the lesion.
SUMMARY OF AUDITORY TEST RESULTS
Hearing loss for pure tones is typically divided into conductive and sensorineural;
central lesions rarely produce such hearing loss (Table 8–2). Conductive and sensorineu- ral loss can usually be differentiated with an audiogram based on the finding of an air- bone gap with the former and a characteris- tic frequency intensity profile with the latter. The causes of a conductive hearing loss can be differentiated with tympanometry. There are several test results that help distinguish between a cochlear and retrocochlear sen- sorineural hearing loss. Speech discrimina- tion is relatively preserved with a cochlear loss, whereas it is impaired early and dispro- portionately to any pure tone loss with a sensorineural loss. Loudness recruitment is commonly seen with a cochlear loss, whereas tone decay is a common feature of a sen- sorineural loss. Abnormalities of the stapedius reflex and BAER provide an objective differentiation between cochlear and retrocochlear lesions. With cochlear hearing loss these measurements remain normal unless the loss is profound. On the other hand, the stapedius reflex and BAER are usually abnormal with only mild to moderate hearing loss due to retrocochlear damage. Central hearing disorders may show abnormalities of the late waves of the BAER or of cortical-evoked responses or in discriminating speech when the task is com- plicated by distorting the content of the speech.
Table 8–2 Summary of Auditory Test Results with Lesions at Different Locations
![]()
SENSORINEURAL
![]()
![]()
Test Conductive Cochlea Nerve Central
Speech
discrimination
Normal Relatively preserved
Abnormal early Normal
Tympanometry Abn: restricted,
hypermobile, flat
Normal Normal Normal
Stapedius reflex Absent Normal Absent or decay Normal
BAER Normal Normal Absent, delayed I–III, I–V
Delayed III–V
Special tests — Loudness recruitment
![]()
Abn, abnormal.
Tone decay Dichotic listening, alter- nating speech, filtered speech
REFERENCES
Katz J, Medwetsky L, Burkard RF, et al. eds. Handbook of Clinical Audiology. 6th ed. Baltimore: Lippincott Williams & Wilkins; 2009.
Kileny PR, Zwolan TA. Diagnostic and rehabilitative audiology. In: Cummings CW, Flint PW, Haughey BH, et al. eds. Otolaryngology Head and Neck Surgery. 4th ed. St Louis, MO: CV Mosby; 2005.
Arts HA. Sensorineural hearing loss: Evaluation and management in adults. In: Cummings CW, Flint PW, Haughey BH, et al. eds. Otolaryngology Head and Neck Surgery. 4th ed. St Louis, MO: CV Mosby; 2005.
Rauch SD. Idiopathic sudden sensorineural hearing loss. N Engl J Med. 2008;359:833.
Schuknecht HL. Pathology of the Ear. 2nd ed. Philadelphia: Lea & Febiger; 1993.
Bagai A, Thavendiranathan P, Detsky AS. Does this patient have hearing impairment? JAMA. 2006;295:416.
Torres-Russotto D, Landau WM, Harding GW, Bohne BA, Sun K, Sinatra PM. Calibrated finger rub auditory screening test (CALFRAST). Neurology. 2009;72(18):1595.
Jerger J, Jerger S. Diagnostic significance of PB word functions. Arch Otolaryngol. 1971;93:573.
Durmaz A, Karahatay S, Satar B, Birkent H, Hidir
Y. Efficiency of Stenger test in confirming pro- found, unilateral pseudohypacusis. J Laryngol Otol. 2009;123(8):840.
Kintsler DP, Phelan JG, Lavender RB. Efficiency of the Stenger and speech Stenger tests in functional hearing loss. Audiology. 1972;11:187.
Jerger J, Jerger S, Maulden L. Studies in impedance audiometry. I. Normal and S-N ears. Arch Otolaryngol. 1972;96:513.
Liden G, PedersonJL, Bjorkman G. Tympanometry.
Arch Otolaryngol. 1970;92:248.
Jerger J, Anthon L, Jerqer S, et al. Studies in imped- ance audiometry. III. Middle ear disorders. Arch Otolaryngol. 1974;99:165.
Kileny PR, Young KE, Niparko JK. Acoustic and elec- trical assessment of the auditory pathway. In: Jackler RK, Brackmann DE, eds. Neurotology. St. Louis, MO: CV Mosby; 1993.
Stone JL, Calderon-Arnulphi M, Watson KS, et al. Brainstem auditory evoked potentials–a review and modified studies in healthy subjects. J Clin Neurophysiol. 2009;26(3):167.
Sohmer H, Feinmesser M. Cochlear action poten- tials recorded from the external ear in man. Ann Otol Rhinol Laryngol. 1967;76:427.
Margolis RH, Rieks D, Fournier EM, Levine SE. Tympanic cochleography for diagnosis of Meniere’s disease. Arch Otolaryngol Head Neck Surg. 1995; 121:44.
Sohmer H, Feinmesser M. Electrocochleography in clinical audiological diagnosis. Arch Otorhino-laryngol (Berl). 1974;206:91.
Møller AR, Janetta PJ. Neural generators of the audi- tory brainstem response. In: Jacobson JT, ed. The Auditory Brainstem Response. Boston: College-Hill Press; 1985.
Northern JL, Downs MP. Hearing in Children. Baltimore: Williams & Wilkins; 1978.
Vlastarakos PV, Nikolopoulos TP, Tavoulari E, Papacharalambous G, Korres S. Auditory neuropathy: endocochlear lesion or temporal processing impair- ment? Implications for diagnosis and management. Int J Pediatr Otorhinolaryngol. 2008;72(8):1135.
Musiek FE, Kibbe-Michal K, Ceurkink NA, et al. ABR results inpatients with posterior fossa tumors and nor- mal pure tone hearing. Otolaryngol Head Neck Surg. 1986;94:568.
Wilkinson AR, Jiang ZD. Brainstem auditory evoked response in neonatal neurology. Semin Fetal Neonatal Med. 2006;11(6):444.
Møller AR, Janetta PJ, Møller MB. Neural generators of brain stem evoked potentials. Results from human intracranial recordings. Ann Otol Rhinol Laryngol. 1981;90:591.
Walsh P, Kane N, Butler S. The clinical role of evoked potentials. J Neurol Neurosurg Psychiatry. 2005;76(suppl 2):ii,16.
Don M, Eggermont JJ, Brachmann DE. Reconstruction of the audiogram using brain stem responses and high-pass noise masking. Ann Otol Rhinol Laryngol. 1979;88(suppl 57):1.
Coats AC. Human auditory nerve action potentials and brain stem evoked responses. Arch Otolaryngol. 1978;104:799.
Coldie WD, Chiappa KH, Young RR, Brooks EB. Brain stem auditory and short somatosensory evoked responses in brain death. Neurology. 1981;31:248.
Hall JW, Mackey-Hargadine JR, Kim EE. Auditory brain stem response in determination of brain death. Arch Otolaryngol. 1985;111:613.
Paludetti G, Ottaviani F, Gallai V, et al. Auditory brain stem responses (ABR) in multiple sclerosis. Scand Audiol. 1985;14:27.
Stockard JJ, Stockard JE, Sharbrough FW. Brain stem auditory evoked potentials in neurology: methodology, interpretation, and clinical application. In: Aminoff MJ, ed. Electrodiagnosis in Clinical Neurology. New York: Churchill Livingstone; 1986.
Fortnum H, O’Neill C, Taylor R, et al. The role of magnetic resonance imaging in the identification of suspected acoustic neuroma: a systematic review of clinical and cost effectiveness and natural history. Health Technol Assess. 2009;13(18):iii, ix, 1.
Berlin C, Lowe-Bell S, Janetta P, Kline D. Central auditory deficits after temporal lobectomy. Arch Otolaryngol. 1972;96:4.
Denes H, Cariezel F. Dichotic listening in crossed aphasia. Arch Neurol. 1981;38:182.
![]()
Diagnosis and Management of Common Neurotologic
![]()
This page intentionally left blank
![]()
ACUTE OTITIS MEDIA AND OTOMASTOIDITIS
CHRONIC MASTOIDITIS AND CHOLESTEATOMA
Diagnosis Management
BACTERIAL LABYRINTHITIS
Diagnosis Management PETROSITIS
Diagnosis Management
INTRACRANIAL EXTENSION OF EAR INFECTIONS
Routes of Spread Meningitis Epidural Abscess
Lateral Sinus Thrombophlebitis Brain Abscess
Otitic Hydrocephalus Diagnosis Management
MALIGNANT EXTERNAL OTITIS
Diagnosis Management
VIRAL INFECTIONS OF THE INNER EAR
Clinical Syndromes Diagnosis
Viral versus Other Causes of Peripheral Cochleovestibular Loss
Management
SYPHILITIC INFECTIONS OF THE EAR
Diagnosis Management
TUBERCULOSIS AND MYCOTIC INFECTIONS OF THE INNER EAR
Tuberculous Mastoiditis Mycotic Mastoiditis Basilar Meningitis
Bacterial infections involving the audio-vestibular structures result from the spread of an infec- tion from another region of the body, typically the middle ear or the meninges. As with most bacterial infectious disorders, the advent of antibiotics substantially changed the preva- lence and scope of complications from infec- tious diseases. Prior to antibiotics, it was not uncommon for the common variety of middle ear infection, otitis media, to evolve into a chronic infection that eventually eroded into the inner ear. However, with antibiotics it is now quite rare for this to occur. Thus, at this point viral infections—or at least presumed viral infections—are the most common type
of infectious process involving the audio- vestibular system.
In this chapter we discuss infectious diseases that can involve the audio-vestibular system.
ACUTE OTITIS MEDIA AND OTOMASTOIDITIS
Infection of the middle ear (otitis media) is the most common disease treated with antibiotics in the United States. Because the air cavity of the middle ear is in direct communication with the mastoid air cells, infection can spread
233
Upper respiratory tract infection
Acute tubotympanitis
Secretory otitis media
Secretory otitis media
Acute otitis media
Acute otitis media
Acute otomastoiditis
Polypoid granuloma
Polypoid granuloma
Chronic otomastoiditis
Perilymph fistula
Labyrinthitis
Perilymph fistula
Labyrinthitis
Cholesteatoma
Cholesteatoma
Figure 9–1. Patterns of progression of middle ear infections.
throughout the pneumatized parts of the tem- poral bone (see Fig. 2–2). Typical patterns of progression of middle ear infections are sum- marized in Figure 9–1. The most common complication of otitis media is mastoiditis.1
Eighty percent of children between the ages of 1 and 6 years have at least one bout of acute otitis media.2 The symptoms typically include unilateral otalgia, fever, and hearing loss. Vertigo is not a common symptom since the infection in uncomplicated cases is restricted to the middle ear. The peak incidence occurs during the first year of life, reaching an inci- dence rate of almost 50% and then gradually decreasing to an incidence rate of <10% beyond age 6.3 With acute otitis media, there is a middle-ear effusion that can be determined by the appearance of the tympanic membrane (i.e., fullness or bulging of the membrane along with cloudiness or redness), pneumatic otos- copy, or tympanometry. Spontaneous perfora- tion can occur, allowing the purulent effusion to drain into the external ear (i.e., otorrhea). Following resolution of the acute infection, a serous or mucinous effusion can persist for months (i.e., chronic otitis media with effusion). Persistent pain and swelling behind the ear may indicate mastioditis.4 However, the incidence of mastoiditis is very low (< 1%) even among
patients with acute otitis media managed initially with observation rather than antibiot- ics.5 The majority of patients who develop mas- toiditis are receiving antibiotic therapy for acute otitis media, so antibiotics are not a safe- guard against developing acute mastoiditis.
Diagnosis and Management
The diagnosis of acute otitis media rests on find- ing the characteristic changes in the tympanic membrane in a patient complaining of acute otalgia and hearing loss. The most common organisms associated with acute otitis media worldwide are Streptococcus pneumoniae, Hemophilus influenza, Moraxella catarrhalis, and group A Streptococcus.6 Once the clinical diagnosis is made, the patient can either be managed with observation and conservative management when the case is uncomplicated (mild otalgia and fever <39°C) or with antibi- otic therapy, typically amoxicillin.5 An observa- tion period is reasonable because in some cases the cause will be viral rather than bacterial and because randomized placebo-controlled trials have shown that most patients do well even without antibiotics.5,7 If there is clinical wors- ening or no improvement in 48 to 72 hours,
then antibiotics should be initiated. If the infection fails to resolve within a few days of antibiotic therapy, then the antibiotic is typi- cally changed to amoxicillin-clavulante. If acute otitis media persists, then a myringotomy can be performed to obtain a sample of the fluid for culture and also to treat otalgia. The antibiotics are then adjusted on the basis of the results of culture. Signs of acute mastoiditis include pos- tauricular swelling, erythema, tenderness, and protrusion of the auricle. The causative organ- isms in acute mastoiditis are often different from those causing acute otitis media. Since potential drug-resistant staphylococcus and pneumococcus infections are common with mastoiditis, obtaining a specimen for culture is important for directing antibiotic treatment.8
CHRONIC MASTOIDITIS AND CHOLESTEATOMA
Chronic mastoiditis results from an untreated or nonresponsive acute mastoiditis or otitis media.2,3 Pathology is characterized by thick- ened edematous mucosa with obliteration of the mastoid air cell lumen, perivascular fibro- sis, and osteitis. Chronic obstruction of the mastoid antrum leads to irreversible changes in the mucosa and bone of the mastoid. Polypoid granulomas composed of hyperplastic mucosa may fill the mastoid antrum, extend into the middle ear, and extrude through a tym- panic membrane perforation into the external auditory canal.
Another consequence of chronic otitis media is a cholesteatoma, which is a collection of ker- atinized squamous epithelium that can invade the middle ear and other pneumatized areas of the temporal bone through the tympanic mem- brane.9 The term cholesteatoma is a misnomer because it does not contain cholesterol and is not a neoplasm. Cholesteatomas usually develop in the epitympanic space of the middle ear following a perforation in the pars flaccida region of the tympanic membrane (see Fig. 6–1B). From here they extend posteriorly into the antrum, into the central mastoid tract, or inferiorly into the middle ear to erode the ossi- cles and bony labyrinth, producing a mixed conductive sensorineural hearing loss and ver- tigo. The mechanism by which a cholesteatoma erodes bone is not entirely clear, but it is not a
simple pressure necrosis. Ultra-structural stud- ies in humans and experimental studies in ani- mals suggest that the bone resorption is caused primarily by activation of multinucleated osteo- clasts in the bone.10 Ultimately, a cholestea- toma may erode through the temporal bone into the intracranial cavity, producing central nervous system (CNS) symptoms and signs.
Cholesteatomas themselves are prone to recurrent infection, because they contain kera- tin debris enclosed in a tissue space. The bacte- ria seen with chronic cholesteatomas are dif- ferent from those seen with acute otitic infections, with the most common aerobe being Pseudomonas aeruginosa, followed by Staphylococcus aureus.11 The most common anaerobes are Proteus and bacteroides. When acutely infected, cholesteatomas can rapidly cause bone destruction. With chronic mastoidi- tis and cholesteatoma formation, a fistula may develop in the bony labyrinth, producing an artificial communication between the peri- lymph and the middle ear.12 The fistula can be caused by either progressive rarefying osteitis or erosion by the cholesteatoma. Patients with a perilymph fistula experience severe episodes of vertigo when they sneeze or cough because the sudden change of pressure in the middle ear is transmitted directly to the inner ear.
Diagnosis
Patients with chronic mastoiditis typically pres- ent with painless purulent otorrhea. Otoscopic examination of the tympanic membrane may reveal evidence of a perforation, particularly in the pars flaccida region, and a cholesteatoma or granuloma may be visible in the epitympanic region of the middle ear. The otoscopic appear- ance of a cholesteatoma can be quite variable. The typical attic retraction cholesteatoma appears as a “pearly tumor” in the posterior superior portion of the tympanic membrane. In other cases, a cholesteatoma develops at the margin of a perforation and migrates into the middle ear. Cholesteatomas sometimes appear behind or within an intact tympanic membrane (so-called primary cholesteatomas). Rarely, a cholesteatoma is not seen otoscopically but is discovered at the time of mastoid surgery. A perilymph fistula can be identified on examina- tion by transiently changing the pressure in the external canal using a pneumatic bulb attached
Figure 9–2. Computed tomography scan of the temporal bone in a patient with a cholesteatoma eroding the wall of the horizontal semicircular canal (A) and the facial canal (B). Arrows point toward area of bony erosion. FC, facial canal; HC, horizontal semicircular canal; IAC, internal auditory canal; V, vestibule.
to the otoscope (see Chapter 6). With a positive fistula test the patient develops vertigo and nystagmus lasting 10 to 20 sec. The nystagmus can be in either direction but usually is in the same direction with both positive and negative pressure. Computed tomography (CT) scan- ning of the temporal bone may reveal a non- pneumatized or poorly pneumatized mastoid, haziness of air spaces, or bony erosion from a cholesteatoma or osteitis (Fig. 9–2). One can also identify erosion of the bony wall of the horizontal semicircular canal or the facial canal.
Management
Initial management is directed at medical treat- ment of the chronic infection.3,13 Chronic oto- mastoiditis that is unresponsive to medical management requires surgical eradication of all diseased tissue. A complete transcortical mastoidectomy is performed with attention to establishing good communication from the mastoid into the middle ear space. A bony fis- tula requires microsurgical removal of the lesion and closure with either perichondrium or fascia. Damage to the ossicular chain can usually be repaired.
Labyrinthitis refers to an inflammatory process of the labyrinth. Bacterial infections initially affect the otic capsule from which they extend into the perilymphatic space and ultimately involve the membranous labyrinth. There are two types of labyrinthitis associated with acute and chronic bacterial infections of the tempo- ral bone: (1) serous or toxic labyrinthitis, in which bacterial toxins or chemical products invade the inner ear; and (2) suppurative laby- rinthitis, in which bacteria invade the inner ear. The former often leads to only subtle symptoms such as an insidious high-frequency sensorineu- ral hearing loss, whereas the latter typically leads to a profound combined auditory and vestibular loss with little or no recovery.
Serous labyrinthitis is a common complica- tion of acute or chronic middle ear infections. With acute otitis media, small molecules such as bacterial toxins or inflammatory mediators such as cytokines and nitric oxide rapidly dif- fuse through the round window into the scala tympani.14 Acute and chronic inflammatory cells also infiltrate the round window, and a fine serofibrinous precipitate forms just medial to the round window membrane. The toxins and inflammatory mediators may penetrate the
basilar membrane and invade the endolymph at the basal turn of the cochlea. Such changes could explain the high incidence of high- frequency sensorineural hearing loss in patients with chronic otitis media.15 A more rapid onset of serous labyrinthitis results in a more com- plete sensorineural hearing loss along with ves- tibular symptoms, including episodic vertigo and unsteadiness.16
The most common port of entry of bacteria into the inner ear (i.e. suppurative labyrinthi- tis) is from the spinal fluid in patients with meningitis (which may or may not be a compli- cation of middle ear infection).17 Children with bacterial meningitis are particularly susceptible to developing suppurative labyrinthitis with residual severe hearing loss occurring in about 11%.18 The prevalence of residual vestibulopa- thy has not been well defined. Patients with bacterial meningitis develop labyrinthitis when bacteria enter the perilymphatic space by way of the cochlear aqueduct or internal auditory canal. Meningogenic bacterial labyrinthitis is usually bilateral, whereas direct invasion from a chronic otitic infection is almost always uni- lateral. The most common route for a direct bacterial invasion of the labyrinth is via a horizontal semicircular canal fistula from a cholesteatoma. Endolymphatic hydrops can be a sequela of both serous and suppurative labyrinthitis.19
Diagnosis
Acute suppurative labyrinthitis has become relatively rare since the introduction of antibi- otics because patients at high risk of develop- ing it (e.g., those with meningitis or severe middle ear infections) are typically started on treatment early in the course of the infection before extension to the inner ear occurs. Symptoms to indicate involvement of the inner ear include the sudden onset of severe vertigo, nausea, vomiting, and unilateral or bilateral hearing loss. The diagnosis of bacterial labyrin- thitis in association with acute or chronic ear infections is based on finding the characteristic symptoms and signs of inner ear dysfunction. As noted earlier, serous labyrinthitis most com- monly produces a slowly progressive, insidious high-frequency sensorineural hearing loss, which may be discovered only with audiomet- ric testing. Suppurative labyrinthitis produces
a much more fulminant course, with both audi- tory and vestibular loss. Intermediate clinical pictures are not uncommon, however, and it may be impossible to differentiate between a toxic and suppurative labyrinthitis on the basis of the symptoms alone. Bacterial labyrinthitis should be considered in any patient with acute or chronic ear infection or meningitis who develops a sudden or progressive sensorineural hearing loss and vestibular symptoms. A posi- tive fistula test suggests that the chronic inner ear infection has eroded into the horizontal semicircular canal.
Management
Management of labyrinthitis is directed at the associated infection of the middle ear, mastoid, and, if present, meninges. Any patient with acute or chronic bacterial ear disease associ- ated with sudden or rapidly progressive inner ear symptoms should be hospitalized and the primary source (typically otitis media or men- ingitis) should be identified. The appropriate course of antibiotics should then be started.13 If mastoiditis occurs, then surgical intervention to eradicate the middle ear and mastoid infection is often required after a few days of antibiotic treatment. If bacterial meningitis is diagnosed, then corticosteroids as adjuvant therapy can reduce the incidence of long-term hearing loss.18,20 In a rat model of meningitis-associated hearing loss, antioxidant therapy plus antibiot- ics was more effective in preventing hearing loss than antibiotics alone.21 If the labyrinthitis and meningitis are secondary to chronic ear infection, a labyrinthectomy might be indicated if the meningitis fails to respond to adequate medical treatment. A resistant or recurrent meningitis may result from unrecognized posterior fossa epidural abscesses with dural perforation or from congenital direct commu- nications with the cerebrospinal fluid (CSF).
Infections involving the perilabyrinthine bone may extend into the apical regions of the petrous bone, producing petrositis. Although only about 30% of petrous bones are pneuma- tized into the petrous apex, when infection
does spread into this region, management can be difficult because drainage is more restricted and the proximity of the apical air cells to dip- loic spaces predisposes to osteomyelitis.13,22 Because of these problems, petrositis is com- monly associated with both bacterial labyrin- thitis and with intracranial extension of the infection.
Diagnosis
The main symptom of an indolent infection in the petrous bone is a deep, boring pain. With infection in the perilabyrinthine region, pain is often referred to the occipital, parietal, or temporal regions. With infection confined to the petrous apex, pain is referred to the deep retro-orbital area. In 1904, Gradenigo described a classic triad associated with lesions of the petrous apex: (1) deep retro- orbital pain, (2) paralysis of the ipsilateral lat- eral rectus muscle from involvement of the abducens nerve as it crosses the petrous bone, and (3) otitic infection with purulent discharge from the ear.23 The syndrome may be associ- ated with vertigo and hearing loss, either from a concomitant bacterial labyrinthitis or from involvement of the eighth nerve in its bony canal. Radiologic evidence for infection of the petrous apex can be difficult to establish even with high-resolution CT and magnetic reso- nance imaging (MRI). The difficultly with radiologic diagnosis is that abnormalities in this region can be missed, and fluid accumula- tion in the petrous apex can be a benign finding even earning the label “leave alone lesions.”24–26
Management
Management is usually a combined medical– surgical approach. After appropriate antibiotic therapy, the mucosal and bone infection must be removed. Surgically, one usually begins with a radical mastoidectomy and dissection along the cell tracts to the petrous bone. Osteomyelitis of the petrous apex and cranial base can be life threatening. The surgical goal is to remove the maximum amount of infected temporal bone while preserving the seventh and eighth cranial nerves and the labyrinth.27
INTRACRANIAL EXTENSION OF EAR INFECTIONS
Extension of infection from the temporal bone into the cranial cavity demands rapid diagnosis and effective therapy to prevent permanent neurologic sequelae or death. Complications can result directly from either acute otitis media and mastoiditis or with chronic otomas- toiditis and bone destruction. In a patient with an otitic infection who is febrile and continues to complain of severe ear and mastoid pain or headache despite appropriate antibiotic ther- apy, intracranial extension of the infection should be considered. Localized neurologic signs frequently do not develop until late in the disease process, and the diagnosis should be considered before focal signs develop.
Although the incidence of morbidity and mortality with CNS complications of ear infec- tions has markedly decreased since the antibi- otic era, these disorders have not disappeared and still remain a major problem in the twenty- first century. In a recent review of 3364 patients with bacterial otitis media admitted to a ter- tiary referral university hospital, 422 (12.5%) presented with complications (extracranial/ intracranial ratio 1:1).1 Mastoiditis was the most common extracranial complication and lateral sinus thrombophlebitis the most common intracranial complication. In those with com- plications the mortality rate was 1.4% and the morbidity rate 3.8%.
Routes of Spread
Infection within the temporal bone can reach the intracranial space via three routes: (1) direct extension, (2) hematogenous spread, and
thrombophlebitis. Extracranial subpe- riosteal abscesses, intracranial extradural abscesses, and sigmoid sinus thrombophlebitis almost always result from a direct extension of the temporal bone infection. Subdural and brain abscesses may also result from direct extension along the soft tissue planes through the petromastoid canal to the poste- rior fossa or along the petrosquamous suture line to the middle fossa. There is a rich network of veins in and around the temporal bone, and these veins directly communicate with extra- cranial, intracranial, and cranial diploic veins.
Meningitis
Meningitis secondary to ear disease is primarily a disease of infants with acute otitis media.28 As a general rule, the bacteria causing the menin- gitis are similar to those causing the acute ear infection. With chronic ear infections and cholesteatoma, however, multiple microorgan- isms may be involved in the meningitis. Recurrent meningitis associated with middle ear infections suggests a CSF fistula, with
pneumoniae being the most common organ- ism responsible.29
Epidural Abscess
A potential intracranial complication of chronic otitic infections is extradural abscess, a collection of purulent fluid between the dura mater and bone of the middle or posterior fossa.19,29 The dura mater is usually an effective barrier, and the infection remains localized outside the nervous system. Extradural abscesses are frequently asymptomatic and are discovered incidentally on CT or during mas- toidectomies for acute or chronic disease.30 Extradural abscesses in the middle fossa may become large and compress the temporal lobe, whereas abscesses in the posterior fossa remain small because of the tight attachments of the dura. The initial symptoms of fever, severe headache, and vomiting without focal neuro- logic signs can create a diagnostic and thera- peutic dilemma.
Lateral Sinus Thrombophlebitis
Of the three dural sinuses intimately connected with the temporal bone, the lateral sinus is most commonly affected by acute or chronic temporal bone infection.31 Inflammation in the extradural space adjacent to the lateral sinus causes a local phlebitis and formation of a mural thrombus. The thrombus enlarges within the lumen of the vessel and may occlude it or become infected. A bland or infected throm- bus may propagate in either direction and become organized. When infected, septic emboli are released into the bloodstream, caus- ing septicemia and its systemic manifestations (i.e., fever, chills). When the cerebral veins are involved, the patient may develop focal or gen- eralized seizures.
Brain Abscess
Although the overall incidence of otitic brain abscesses has decreased due to early treatment with antibiotics, atypical brain abscesses in immunocompromised patients are becoming more common.32 As noted earlier, brain abscesses associated with ear infections pre- dominantly originate from venous throm- bophlebitis rather than direct extension through the dura mater.33 The temporal lobe is most commonly involved, followed by the cerebel- lum. Both aerobic and anaerobic organisms are found in pure or mixed cultures within brain abscesses. Multiple organisms are found in more than half of the cases.34 Surprisingly, although H. influenza and Pseudomonas spe- cies are common organisms with ear infections, they are rare with brain abscesses.
Neurologic signs associated with temporal lobe abscess are often subtle, particularly in an immunocompromised patient or if the patient has received inadequate antibiotic therapy.35 An upper-quadrant hemianopia can result from involvement of the optic radiations on either side, and when the abscess is in the dominant hemisphere, speech may be abnormal. Usually some weakness of the contralateral face and arm occurs, but gross paralysis is rare. The signs of a cerebellar abscess are usually more prominent. The patient complains of severe neck stiffness and holds the head rigid in a tilted position. Neurologic examination reveals
Otitic Hydrocephalus
In 1931, Symonds36 coined the term otitic hydrocephalus to describe the syndrome of increased intracranial pressure without evi- dence of meningitis or brain abscess in patients with chronic ear infections (i.e., pseudotumor cerebri). Symonds later hypothesized that the
brain edema resulted from thrombosis of the superior sagittal sinus, leading to impaired CSF resorption. More recent MRI studies show that thrombosis of the lateral venous sinus alone can impede venous draining and produce a rise in CSF pressure.37 Complications include chronic headaches and visual loss due to optic nerve compression.38
Diagnosis
An intracranial complication of an otitic infec- tion should be considered in any patient with a known ear infection who continues to com- plain of severe pain and headache despite appropriate antibiotic therapy (Fig. 9–3). The presence of fever, neck rigidity, and a positive
History
Persistent pain and drainage despite appropriate antibiotics
Examination
Persistent fever and neck stiffness Meningismus
Focal neurologic signs
CT scan with contrast
Negative
MRI MRV
Negative
LP
Positive
Positive
Positive
Mastoiditis Cholesteatoma Petrositis Abscess
Venous thrombosis
Small epidural or subdural effusion Cerebritis
Venous thrombosis
Meningitis Epidural effusion Cerebritis
Meningitis Epidural effusion Cerebritis
Negative
Continued observation
Continued observation
Figure 9–3. Algorithm for the diagnosis of intracranial complications of ear infections. CT, computerized tomography; LP, lumbar puncture; MRI, magnetic resonance imaging; MRV, magnetic resonance venography.
Kernig’s or Brudzinski’s sign supports the ini- tial impression. As noted earlier, focal neuro- logic signs develop late in the course, even with localized brain abscesses, so one must have a high degree of suspicion based on the clinical presentation. After a detailed history and a careful physical examination, a CT scan with contrast is the initial diagnostic test. The CT scan will show bone erosion, collections of pus within the intracranial cavity, and can identify thrombosis of the venous sinuses.13 It can miss small collections of extradural or subdural pus and early stages of brain abscess formation, however, which might be identified with MRI. Venous thrombosis is best identified by MRI with venography (MRV).31 If a mass lesion has been ruled out, a lumbar puncture is performed for analysis of the CSF. The characteristic profile of bacterial meningitis (pleocytosis, decreased glucose, and increased protein) is readily identified. More subtle changes (mono- cytic pleocytosis and mildly increased protein) can be seen with collections of pus in the epi- dural spaces or with brain abscess. The finding of increased intracranial pressure without a structural lesion suggests the possibility of a sinus thrombosis.
Management
Treatment of the intracranial complications secondary to ear infections is directed along two lines: (1) eradication of the infection with appropriate antibiotics and (2) establishing adequate drainage and excision of infected tis- sue when necessary.13 Complications due to acute otitis media are usually effectively con- trolled by a myringotomy and adequate paren- teral antibiotics. Complications associated with bone destruction from chronic otitis media and mastoiditis usually require some type of mastoidectomy along with parenteral antibiot- ics. As a general rule, when there is a collection of pus within the intracranial cavity, treatment is directed first to reduce intracranial pressure—usually by evacuating the empyema. Appropriate management of meningitis con- sists of first identifying the offending organism, beginning appropriate intravenous antibiotics, and periodic monitoring of the progress by lumbar puncture.39 Otitic hydrocephales typi- cally requires long-term CSF drainage with a ventriculoperitoneal shunt.38
Otitis externa, usually a benign disorder, can lead to a debilitating disease called malignant external otitis in elderly diabetic patients.40,41 It typically begins with a nonspecific infection of the external canal, resulting in complaints of pain, drainage, and fullness of the ear. The pain becomes severe and continuous as the infec- tion spreads to contiguous soft tissues and adja- cent bony structures. The invading organism is almost always P. aeruginosa. The organism invades the junction of the cartilaginous and osseous portions of the external auditory canal and spreads to the temporo-occipital bones. The most common neurologic sequela is involvement of the facial nerve in the fallopian canal or at the stylomastoid foramen.42 Occasionally, multiple cranial nerves are com- pressed extradurally, and in rare cases, the infection spreads across the dura to produce a purulent meningitis.
Diagnosis
Diagnosis rests on finding the characteristic granulation tissue in the external canal along with a positive culture for P. aeruginosa (Fig. 9–4). A CT scan of the temporal bone may reveal (1) a soft tissue mass in the external canal, (2) clouding of the mastoid air cells, (3) sequestra of the bony canal, (4) erosion of bony structures at the base of the skull, and (5) soft tissue masses within the parapharynx and nasopharynx.43 Initial findings on CT can often predict the likely clinical course.44 An MRI is useful on follow-up to identify soft tissue evo- lution and meningeal enhancement.45
Management
Initial local treatment consists of removal of all accumulated debris and granulation tissue from the external canal and instillation of a gentamicin wick (gentamicin sulfate drops on 1/2-inch gauze packing) in the external canal. This, along with strict control of the underlying diabetes, can be sufficient treatment in some patients with early stages of disease. In patients with more advanced disease, including bony erosion, or in those who fail to respond promptly to this initial therapy, a bone scan is indicated
External otitis with extuberant tissue in a diabetic patient
Biopsy and culture
Malignant external otitis
Computed tomography (CT)
Initial therapy: | |
Persistence | |
Debridement
Gentamicin wick
Strict control of diabetes
Initial therapy: | |
Persistence | |
Debridement
Gentamicin wick
Strict control of diabetes
Bone scan
Osteitis, osteomyelitis
Initial therapy plus:
Parenteral antibiotics
Surgery (sequestrectomy, drain abscesses)
Continue until local disease clears
Bone erosion, abscesses
combination of auditory and vestibular symp- toms. They may be part of a systemic viral illness such as measles, mumps, and infectious mono- nucleosis, or more commonly it is an isolated infection of the labyrinth and/or eighth nerve without systemic involvement. In the latter case, the infecting agent is rarely identified, but an iso- lated viral infection of the inner ear is presumed based on the typical self-limited clinical course and prior pathologic evidence to support the link between the sudden onset of hearing loss and/or vertigo and a viral infection.46 Similar assump- tions about a viral etiology are made with idio- pathic facial nerve palsy (i.e., Bell’s palsy), which is also a typically self-limited cranial neuritis.
Of the thousands of infants born deaf every year, about 20% of cases are thought to be the result of congenital viral infections.47 CMV infections are most common.48 In adults, two relatively common presumed viral disorders are vestibular neuritis and sudden sensorineu- ral hearing loss (SSNHL) or “sudden deaf- ness.”49,50 Despite this strong suspicion of a viral origin for these common neurotologic syndromes, proof of a viral pathophysiology in individual cases is rare. Epidemiologic evi- dence supports a viral cause in most patients with either sudden deafness or acute prolonged vertigo. A large percentage of such patients report an upper respiratory tract illness within 1 to 2 weeks prior to the onset of symptoms. Both syndromes occur in epidemics, may affect several members of the same family, and erupt more commonly in the spring and early summer.51–53A list of the viruses that have been clinically associated with cases of deafness and/
Figure 9–4. Algorithm for the management of malignant external otitis. (Adapted from Smith PG, Lucente FE. External ear: Infections. In: Cummings CW, Fredrickson JM, Harker LA, Krause CJ and Schuller DE (eds). Otolaryngology—Head and Neck Surgery, CV Mosby, St. Louis, 1986, with permission.)
to assess the possibility of osteitis or osteomy- elitis. If positive, the patient will require long- term parenteral antibiotic therapy dependent on the microbial findings and local cleansing and debridement, as required (Fig. 9–4).
VIRAL INFECTIONS OF THE INNER EAR
Viral infections of the inner ear can present with sudden deafness, acute vertigo, or with some
or vertigo is given in Table 9–1.54 In most cases, however, proof that these viruses caused the symptoms is circumstantial.
Table 9–1 Viruses Clinically Associated with Hearing Loss and/or Vertigo
![]()
Cytomegalovirus Hepatitis
Rubella Adenovirus
Mumps Influenza
Rubeola Parainfluenza
Varicella-zoster Poliomyelitis
Herpes simplex Coxsackie
Epstein-Barr Lymphocytic choriomeningitis
Variola Yellow fever
![]()
Adapted from Davis and Johnson.54

Figure 9–5. Pathologie findings in a patient with vestibular neuritis. Sections through comparable areas of Scarpa’s ganglia on the normal side (A) and the side with absent caloric response (B) stained with toluidine blue. Only a few small neurons remain in B (bar = 100 µm). (From Baloh RW et al. Vestibular neuritis: clinical pathological correlation. Otolaryngol Head Neck Surg. 1996;114:586–592, with permission.)
Possibly, the most convincing evidence for a viral cause of these auditory and vestibular syndromes comes from the temporal bone studies of Schuknecht and colleagues.46,52,55 These and other investigators have reported pathologic evidence for isolated viral involvement of the cochlea and auditory nerve in patients with sudden deafness and of the vestibular end organs and vestibular nerve in patients with isolated sudden vertigo (Fig. 9–5).56,57 The atrophy of the nerves and end organs is identical to that associated with well-documented viral disorders (such as mumps or measles).58 In all of these cases the vasculature was intact; there was no evidence of a vascular cause for the sudden deafness or vertigo.
Clinical Syndromes
HERPES ZOSTER OTICUS
A clear example of a viral syndrome involving the eighth nerve is herpes zoster oticus (also known as the Ramsay Hunt syndrome).59 Presumably, the zoster virus remains dormant in the ganglia associated with the seventh and eighth nerves and is reactivated during a period of lowered immunity. The patient initially develops a deep burning pain in the ear followed a few days later by vesicular eruption in the external auditory canal and concha. At some time after the onset of pain, either before or after the vesicular eruption, the patient may develop hearing loss, vertigo, and
facial weakness. About half have cochleoves- tibular symptoms and facial paralysis and half have just facial paralysis.60 The pathologic find- ings in patients with herpes zoster oticus con- sist of perivascular, perineural, and intraneural round cell infiltration in the seventh nerve and in both divisions of the eighth nerve.61
VESTIBULAR NEURITIS
Vestibular neuritis is typically manifested by the gradual onset of vertigo, nausea, and vomit- ing over several hours.62 Occasionally patients have prodromal dizzy spells for a day or so before onset.63 The symptoms usually reach a peak within 24 hr and then gradually resolve over several weeks. During the first day there is severe truncal unsteadiness and imbalance to the point that falling toward the affected side when trying to walk is common.64 Patients also often report difficulty focusing because of the spontaneous nystagmus. In most patients the course is self-limited with complete func- tional recovery within 3 months.65 There are important exceptions to this rule, however. Occasionally patients—particularly the elderly—will have intractable dizziness that persists for years.66 Recurrences are relatively rare.67 A small percentage of these patients will have multiple recurrent episodes of vertigo leading to a profound bilateral vestibulopathy (so-called bilateral sequential vestibular neuri- tis).68 The episodic vertigo is eventually replaced by persistent disequilibrium and oscillopsia.
Experimental studies in animals have shown that several viruses will selectively infect the vestibularnerveandlabyrinth.54,69 Furthermore, differential susceptibility to viral infection and antigen expression has been observed in differ- ent cell types within the inner ear.70 Herpes simplex virus type 1 (HSV-1) can produce selective involvement of the vestibular nerve and labyrinth when inoculated into the ear of a guinea pig.69 The HSV is a DNA-containing virus that establishes a latent infection in neu- rons by incorporating part of its DNA into the nucleus of the neuron. Although the molecular mechanisms involved in the establishment and maintenance of latency and reactivation from the latent state are only partially understood, part of the HSV genome, called LAT for latency-associated transcript, can be identified in latently infected cells using molecular
hybridization methods. Presumably, LAT codes for a protein that blocks the replication cycle of the latent virus at some early point so that the infected cells are not damaged and are able to maintain the latent state. Since reactiva- tion of a latent HSV-1 infection in the genicu- late ganglion is thought to be a common cause of Bell’s palsy,71 reactivation of a latent HSV-1 infection in Scarpa’s ganglia might be the cause of vestibular neuritis. HSV-1 has been identi- fied in Scarpa’s ganglia72,73and in the vestibular labyrinth74 of autopsied adults without a prior history of vertigo, but there is still no conclu- sive proof that reactivation of latent HSV-1 virus is the cause of vestibular neuritis.
A unique feature with most cases of vestibu- lar neuritis is selective damage to the superior part of the vestibular labyrinth with sparing of the inferior part. Since the posterior semicircu- lar canal remains functional, patients often develop benign paroxysmal positional vertigo as a sequela even though they have no remain- ing horizontal or anterior semicircular canal function. Rotational studies with measurement of three-dimensional eye movements in patients with vestibular neuritis indicates that the posterior semicircular canal is typically spared even though anterior and horizontal semicircular canal function is absent.75,76 How the presumed viral etiology can lead to selec- tive involvement of the superior vestibular labyrinth is unknown. There is an anastamosis between the facial nerve and the superior ves- tibular nerve, but HSV-1 is found equally in the superior and inferior vestibular ganglia.77 A few cases have been reported in which selec- tive involvement of the inferior division of the vestibular nerve appeared to be the cause.73,78,79 In these cases, the caloric responses were normal (indicating normal horizontal canal function, and thus intact superior vestibular nerve) but the VEMP response—a method to measure the function of the saccule and thus inferior vestibular nerve—was abnormal, or an impulse test in the plane of the posterior canal was abnormal.76,78,79 The clinical presentation of inferior vestibular neuritis remains ill- defined, however. Though one would expect a spontaneous downbeating and torsional nys- tagmus in the acute phase, none of the reports claiming a diagnosis of inferior vestibular neuritis describe this pattern of nystagmus.76,78,79 In fact, most reported cases of inferior vestibular
neuritis report no nystagmus at all, even in the acute phase and even after provocative tests.76,78,79 In addition, some of the cases reported are very nonspecific presentations rather than the typical sudden and constant acute vestibular syndrome presentation.78
SUDDEN SENSORINEURAL HEARING LOSS (SUDDEN DEAFNESS)
Although the term sudden deafness is com- monly used, the hearing loss due to viral infec- tion usually comes on over several hours and may even extend over several days.46,80 The loss is usually unilateral, often profound, and may be permanent, although it reverses at least par- tially in most cases. It returns to normal in more than 50% of patients (with or without treatment).81 Prognosis is best when the hear- ing loss is mild and begins to recover within 2 weeks. Severe hearing loss (>90 dB), advanced age, and presence of vertigo are poor prognos- tic factors. Tinnitus and fullness in the involved ear are common.82
As with vestibular neuritis a viral cause is likely with most cases of sudden deafness, although it is rare that a viral cause can be proven in an individual case.83 Several viruses will selectively infect the cochlea and spiral ganglia in animal models.84,85 Latent HSV1 has been identified in the spiral ganglia of autop- sied adults without a history of hearing loss85 so like vestibular neuritis reactivation of a latent herpes infection may be a cause of sudden deafness.
Diagnosis
The diagnosis of Herpes zoster oticus rests on finding the characteristic cutaneous eruptions about the auricle and external ear canal. A spe- cific diagnosis can be made using either direct fluorescent antigen assay (DFA) or polymerase chain reaction (PCR). For DFA testing, a vesi- cle should be unroofed and then the base of the lesion rubbed with a cotton-tipped swab. The swab is then rubbed on a glass slide and sent for DFA testing.86 For PCR testing, either vesicular fluid can be collected or material from the swab can be used.87,88 PCR can also be performed using saliva, CSF, or crusts or
skin scrapings from maculopapular lesions. Although the herpetic external otitis is self- limiting, the seventh and eighth nerve damage is often profound and nonreversible. An MRI with contrast will often show diffuse enhance- ment of the seventh and eighth cranial nerves.89,90
The diagnosis of vestibular neuritis or sud- den deafness rests on finding the characteristic clinical presentation and audio-vestibular sys- tem examination findings in the absence of neurologic symptoms and signs (See Video 6–2, Video 6–3, Video 6–4, Video 6–5, and Video 6–6). Routine otologic examination is usually unremarkable, and CT scanning of the tempo- ral bone reveals only a normal-appearing bony labyrinth. Occasionally, the eighth nerve or membranous labyrinth will be enhanced after contrast on MRI, but this only indicates inflam- mation of unknown cause. Serologic studies can demonstrate that a virus infected the patient, but they do not prove that the infec- tious agent caused the inner ear damage. Furthermore, isolation of an infectious agent from the nasopharynx or other tissue other than the membranous labyrinth does not prove a causal relationship between the virus and the inner ear disease. Although a few viruses have been cultured directly from perilymph samples in infected ears, this is not a practical method for routine diagnosis of viral infections of the inner ear.54
Vestibular neuritis presents with signs of both canal and otolith dysfunction.91,92 Nearly all have a unilateral caloric paresis, a positive head-thrust sign, and tilt of the subjective visual vertical. Between 30% to 50% have a decreased vestibular evoked myogenic potential (VEMP) on presentation. Otolith-related test abnormal- ities improve more rapidly than canal-related abnormalities, and patients with a persistently positive head-thrust sign at follow-up are more likely to continue to complain of dizziness than patients in whom the head-thrust sign returns to normal.92 Patients with vestibular neuritis may have hearing loss in the ultrahigh- frequency range, which suggests that the audi- tory end organ may be involved to a minor degree even though it is clinically silent.93,94 With sudden deafness the hearing loss tends to be most prominent in the high frequencies, a finding consistent with neuropathologic studies demonstrating the greatest degree of damage
in the basilar turn of the cochlea.80 Brainstem auditory-evoked (BAER) studies are usually normal, consistent with a cochlear site of pathology. Vestibular abnormalities have been identified on electronystagmography (ENG) in patients with sudden deafness but without associated vestibular symptoms.95
Viral versus Other Causes of Peripheral Cochleovestibular Loss
Viral infections of the inner ear must be dif- ferentiated from other forms of labyrinthitis (bacterial and syphilitic) and from acute laby- rinthine ischemia and from perilymph fistula (Table 9–2). As noted earlier, bacterial labyrin- thitis is typically associated with acute and chronic otomastoiditis, which should be easily identified on examination of the ear and with CT of the temporal bone, or meningitis. Suppurative labyrinthitis invariably results in a fulminate profound loss of both auditory and vestibular function, usually with only
minimal recovery. Serous labyrinthitis may produce only a high-frequency sensorineural hearing loss if the toxic products remain con- fined to the basilar region of the cochlea. Syphilitic labyrinthitis might initially be con- fused with viral neurolabyrinthitis, but the for- mer leads to recurrent episodes of vertigo and hearing loss, usually progressing to severe bilat- eral dysfunction over a period of months. Unlike the gradual onset of symptoms over hours with viral syndromes, infarction of the labyrinth results in a sudden profound loss of auditory and vestibular function—often in a setting of prior episodes of transient ischemia within the vertebrobasilar system (see Chapter 14). Infarction of the labyrinth, lateral brain stem, or cerebellum should be considered a potential cause in patients who present with acute onset vertigo and imbalance. The proba- bility of having a vascular etiology drops sub- stantially when the symptoms are isolated audio-vestibular symptoms,96 and it becomes remarkably low when the key examination fea- tures also suggest a peripheral etiology.64
Table 9–2 Differential Diagnosis of Acute Peripheral Vestibulopathy
![]()
![]()
History Examination Laboratory
Viral
neurolaby- rinthitis
Bacterial labyrinthitis
Syphilitic labyrinthitis
Labyrinthine ischemia
Perilymph fistula
Developing over hours, resolving over days, prior flu-like illness
Abrupt onset, associated hearing loss, prior ear infections
Recurrent episodes, associated tinnitus and hearing loss, prior congenital or acquired syphilis
Abrupt onset, usually associated neurologic symptoms, prior vascular disease
Abrupt onset associated with head trauma, barotrauma, or sudden strain during heavy lifting, coughing, or sneezing; chronic otomastoiditis with cholesteatoma
Normal except for signs of acute unilateral vestibular loss
Signs of otitis media or meningitis
May be stigmata of congenital syphilis, rarely associated signs of neurosyphilis
May be signs of brain stem or cerebellar infarction
Positive fistula test, may be chronic otitis with tympanic membrane perforation
ENG: caloric hypoexcitability Audio: may show ultrahigh-
frequency loss
ENG: absent caloric response Audio: profound sensorineural
hearing loss CSF: pleocytosis
ENG: caloric hypoexcitability Audio: low-frequency loss Serology: positive FTA-ABS CSF: usually normal
ENG: absent caloric response Audio: profound sensorineural
hearing loss
Neuroimaging: may show brain infarction
ENG: caloric hypoexcitability Audio: usually sensorineural loss CT of temporal bone may show
erosion from cholesteatoma
![]()
CSF, cerebrospinal fluid; CT, computed tomography; ENG, electronystagmography; FTA-ABS, fluorescent treponemal antibody absorption (test).
Like viral infections of the inner ear, peri- lymph fistulae can present with hearing loss, vertigo, or a combination of auditory and ves- tibular symptoms. With the latter, however, the onset is usually abrupt, and there is nearly always a precipitating event, such as head trauma, barotrauma, or a sudden strain during heavy lifting, coughing, or sneezing. Perilymph fistulae are particularly common in patients who have undergone stapedectomy for otoscle- rosis. Most perilymph fistulae are self-limited, so it is presumed that the fistulae heal. Indications for exploratory surgery include ongoing symptoms suspicious for a perilymph fistulae (episodes induced by coughing or sneezing) and/or a positive fistula test (see Chapter 16).
Management
During the acute stages of herpes zoster oticus, warm, moist compresses applied locally can provide symptomatic relief, although often sys- temic analgesics are required. Based on treat- ment of zoster infections in other parts of the body a combination of high-dose corticoster- oids (e.g., prednisione 60 mg/d with taper over 10–14 days) and antiviral drugs (e.g., valacycol- vir 1 g three times a day for 7–10 days) are typically used for treatment.97 However, there remains uncertainty because of a lack of ade- quate trials98 in Ramsay Hunt syndrome and also because the beneficial effects of corticos- teroids and antivirals in the most common her- pes zoster disorder (i.e., shingles) is the time to pain recovery and time to skin lesion healing, without evidence of a beneficial long-term out- come.97 In Ramsay Hunt syndrome the most important outcomes are facial nerve recovery and recovery of audio-vestibular function.
The management of patients who present with an isolated episode of auditory and vestibu- lar loss is controversial because the pathophysi- ology is often uncertain. As suggested earlier, unless there is convincing evidence to suspect a
vascular or nonviral infectious cause, the patient’s symptoms should be managed as a pre- sumed viral neurolabyrinthitis. Symptomatic treatment with antivertiginous and antiemetic medications are useful to treat acute vertigo with nausea and vomiting (see Chapter 19). Vestibular physical therapy is appropriate for patients with acute vestibular neuritis, though most of the randomized trials of vestibular phys- ical therapy were conducted in patients with surgical lesions or groups of mixed or vaguely stated etiologies99 (see Chapter 20). Steroids have been recommended for their anti- inflammatory effect based on a few controlled studies.100–104 One large randomized placebo- controlled trial of oral corticosteroids and valcy- clovir (2 by 2 factorial design) found that on average patients treated with corticosteroids within 3 days of onset had a superior improve- ment in vestibular recovery as measured by the surrogate outcome of caloric response at 12 months compared to placebo.102 Valcyclovir did not demonstrate an average beneficial effect. However, it is not known if corticosteroids improve functional outcome since functional outcome was not assessed in this trial and many patients with a chronic caloric asymmetry are asymptomatic. High doses of prednisone or methylprednisone are given for 3 days to a week and then rapidly tapered. A course of corticoste- riods is also generally recommended for patients with sudden deafness though adequate trials are lacking.105,106 Interest has increased in the use of intratympanic (IT) corticosteroids injection in sudden sensorineural hearing loss, particularly when used as a “salvage” therapy following failed oral treatment,107,108 but large and rigorous trials are still necessary to establish the effect. Numerous so-called vasodilating regimens have been proposed, but they would have little effect on the presumed viral pathophysiology. Although many patients with vestibular neuritis are left with a permanent loss of vestibular function (as documented by serial caloric examinations), the CNS is able to adapt to the vestibular loss, and residual symptoms are usually minimal once the compensation has occurred (see Chapter 20).
Although antiviral agents such as cytosine arabinase and acyclovir have been used for treating systemic viral illnesses in children, it is unclear whether the hearing loss that is often associated with disorders such as cytomegalovi- rus and rubella infections is altered by such treatment.55
SYPHILITIC INFECTIONS OF THE EAR
Involvement of the eighth nerve and/or laby- rinth can be an early or late manifestation of both congenital and acquired syphilis. About one in three patients with congenital syphilis develops otologic manifestations. Although the number of new cases of congenital syphilis pro- gressively declined from 1930 to 1968, the inci- dence of new cases appears to have stabilized since 1968. From 1981 through 1989, the over- all incidence of syphilis in the United States increased by 34%.109 More recently the inci- dence of syphilis is again on the rise due to widespread human immunodeficiency virus (HIV) infections.110 Syphilitic infections pro- duce auditory and vestibular symptoms by two different pathophysiologic mechanisms: (1) meningitis with involvement of the eighth nerve and (2) osteitis of the temporal bone with associated labyrinthitis.111,112 The former typi- cally occurs as an early manifestation of acquired syphilis, whereas the latter occurs as a late manifestation of both congenital and acquired syphilis. With early congenital syphi- lis there may be a lymphocytic infiltration of both the membranous labyrinth and eighth nerve, leading to profound bilateral deafness. Spirochetes have been demonstrated in tem- poral bones obtained at autopsy in such patients. With early acquired syphilis, the pre- dominant pathologic finding is basilar meningi- tis affecting the eighth nerve, particularly the auditory branch. The hearing loss typically occurs with the rash and lymphadenopathy of secondary syphilis. It is usually abrupt in onset, tends to be bilateral, and is rapidly progressive. Vestibular symptoms are often absent. Patients demonstrate symptoms and signs of meningi- tis, including headache, stiff neck, cranial nerve palsies, and optic neuritis. With late syphilis most patients have a combination of hearing loss, tinnitus, and vertigo.113
Both congenital and acquired syphilitic infec- tions produce temporal bone osteitis and laby- rinthitis as a late manifestation. The congenital variety is approximately three times as common as the acquired variety.114 The time of onset of congenital syphilitic labyrinthitis is anywhere from the first to seventh decades, with the peak incidence in the fourth and filth decades, whereas acquired syphilitic labyrinthitis rarely occurs before the fourth decade and has a peak
incidence in the fifth and sixth decades. The congenital variety is often associated with other stigmata of congenital syphilis, such as intersti- tial keratitis, Hutchinson’s teeth, saddle nose, frontal bossing, and rhagades. Of these associ- ated signs, interstitial keratitis is by far the most common, occurring in approximately 90% of patients.114 Pathologic changes in the labyrinth are similar in the congenital and acquired vari- ety, consisting of inflammatory infiltration of the membranous labyrinth and osteitis of all three layers of the otic capsule.19 A combination of hydrops of the membranous labyrinth and atrophy of the cochlear and vestibular end organs resembles the pathologic findings in idiopathic Meniere’s syndrome.
The natural history of syphilitic labyrinthitis is a slow, relentless progression to profound or total bilateral loss of vestibular and auditory function.115,116 This progression is marked by episodes of sudden deafness and vertigo and fluctuation in the magnitude of hearing loss and tinnitus.
Diagnosis
Infants with congenital syphilis exhibit a pro- found bilateral sensorineural hearing loss along with extensive damage to multiple organs (Table 9–3). The hearing loss associated with early acquired syphilis may be the only mani- festation of a basilar meningitis, or it may be associated with headaches, stiff neck, and mul- tiple neurologic findings. The rash of second- ary syphilis may precede or accompany the onset of hearing loss. A CSF examination is invariably abnormal with a pleocytosis and ele- vated protein.112 The CSF venereal disease research laboratory (VDRL) test may or may not be positive.
The diagnosis of syphilitic labyrinthitis as a late manifestation of either congenital or acquired syphilis is based on the finding of a positive serum fluorescent treponemal anti- body absorption (FTA-ABS) test in a patient with the typical clinical history of fluctuating hearing loss and vertigo (see Table 9–3).117 The serum VDRL is positive in only about 75% of cases, making it an unreliable test for syphilitic labyrinthitis.114 As noted earlier, patients with congenital syphilitic labyrinthitis often have associated stigmata of congenital syphilis, whereas patients with the acquired variety may
Table 9–3 Differential Features of Different Otologic Manifestations of Syphilis
![]()
EARLY LATE
![]()
![]()
Congenital Acquired Congenital Acquired
Pathophysiology Inflammation of
membranous labyrinth and eighth nerve
Meningitis with involvement of eighth nerve
Temporal bone osteitis and petrositis with secondary degeneration of membranous labyrinth leading to endolymphatic hydrops
Hearing loss Congenital deafness Abrupt onset,
bilateral, progressive
Begins unilateral, fluctuating, progresses to bilateral over months; associated tinnitus and ear pressure
Vertigo No Infrequent Episodic (hours) with fluctuating hearing loss
Age of peak incidence
At birth 20–30 years 30–50 years 40–60 years
Infection involving multiple organs
Rash of
secondary syphilis
Interstitial keratitis, other stigmata of congenital syphilis
Other
features of tertiary syphilis
Cerebrospinal fluid
Usually abnormal (pleocytosis, elevated protein, VDRL ±)
Usually normal Usually normal
Treatment IV aqueous penicillin, 20 million U/day ×
2 weeks
IM benzathine penicillin, 2.4 million U, weekly × 3 months; prednisone 30–60 mg qod, 3–6 months, slow tapering
![]()
IM, intramuscular; IV, intravenous; VDRL, venereal disease research laboratory (test).
have other clinical symptoms and signs of ter- tiary syphilis. The CSF examination is usually normal in both the congenital and acquired varieties of syphilitic labyrinthitis.113
Management
Penicillin is still the treatment of choice for the otologic manifestations of syphilis, although the optimal regimen for each variety remains uncertain.116 Because CSF infection accompa- nies the early manifestations of both congenital and acquired syphilis, high-dose intravenous penicillin seems appropriate. Prognosis follow- ing treatment is poor in the early congenital form, but it is excellent in the early acquired variety. Complete recovery of both hearing and vestibular function usually occurs with the latter.112
For the late manifestations of both congeni- tal and acquired syphilitic labyrinthitis, the combination of steroids and penicillin appears to be superior to penicillin alone.115,116 Numerous penicillin regimens have been used, with the most popular being benzathine penicillin
(2.4 million units) given weekly for 6 weeks to 3 months. Along with the penicillin, predni- sone, beginning at a dose of 60 mg/day on an alternate day regimen, is given for 3 months, followed by a slow tapering. If symptoms recur during the tapering, a more long-term mainte- nance dosage of prednisone may be required. Most patients can be expected to stabilize or improve on this therapeutic regimen.115
TUBERCULOSIS AND MYCOTIC INFECTIONS OF THE INNER EAR
Tuberculous Mastoiditis
The tuberculosis pandemic along with the development of resistant strains has become a global public health problem.118 Tuberculous mastoiditis accompanies both pulmonary and nonpulmonary infections, presumably because of hematogenous spread. In contrast to bacte- rial infections, tuberculosis of the temporal bone runs an indolent course and usually pro- duces little pain.119 The classical presentation is
multiple perforations of the tympanic mem- brane, ear discharge, and progressive hearing loss. The foul-smelling otorrhea, when cul- tured, typically shows nonspecific mixed organ- isms. It may mimic chronic otomastoiditis with cholesteatoma formation because there is often tympanic membrane perforation and promi- nent granulomatous tissue. A CT scan of the temporal bone shows an irregular, punched- out lesion resembling a cholesteatoma. Tuberculosis mastoiditis can usually be differ- entiated from bacterial mastoiditis by the pres- ence of soft tissue in the entire middle ear cavity, preservation of mastoid air cells without sclerosis, and soft tissue extension or mucosal thickening in the external ear canal.120 Diagnosis rests on culturing tuberculosis from the otorrhea or by histologic examination of the mastoid granulomatous tissue. Management consists of antituberculous drugs and surgical eradication of the lesion.
Mycotic Mastoiditis
characteristic CSF profile of lymphocytic pleocytosis, elevated protein, and decreased glucose. Often, there are associated systemic symptoms and signs, including multiple pulmonary lesions. Mycobacterium tuberculosis may be seen on acid-fast smears and cultured from the CSF. The diagnosis of fungal menin- gitis relies on identifying the appropriate anti- gen in the CSF.
REFERENCES
Mostafa BE, El Fiky LM, El Sharnouby MM. Complications of suppurative otitis media: still a prob- lem in the 21st century. ORL J Otorhinolaryngol Relat Spec. 2009;71(2):87.
Chole RA, Choo M-J. Chronic otitis media, mastoidi- tis and petrositis. In: Cummings CW, Fredrickson JM, Harker LA, Krause CJ, Schuller DE, eds. Otolaryngology – Head and Neck Surgery. 3rd ed. St. Louis, MO: CV Mosby; 1998.
Tarlow M. Otitis media: pathogenesis and medical sequelae. Ear Nose Throat J. 1998;77(suppl 6):3.
van den Aardweg MT, Rovers MM, de Ru JA, Albers FW, Schilder AG. A systematic review of diagnostic criteria for acute mastoiditis in children. Otol Neurotol.
Primary mycotic infections such as actinomy-
2008;29(6):751.
Subcommitte on Management of Acute Otitis Media.
cosis and coccidioidomycosis can occur in the temporal bone. Their manifestations are com- parable to those of tuberculosis. Although the mucorales group of fungi are usually of low virulence, mucormycosis of the mastoid bone
Diagnosis and management of acute otitis media.
Pediatrics. 2004;113:1451.
Vergison A. Microbiology of otitis media: a moving tar- get. Vaccine. 2008;26 Suppl 7:G5.
Powers JH. Diagnosis and treatment of acute otitis
can lead to a life-threatening illness in patients who are chronically ill (particularly with diabe- tes or malignancy) or are receiving chemother- apy or broad-spectrum antibiotic therapy.121,122
media: evaluating the evidence. Infect Dis Clin North
Am. 2007;21(2):409, vi.
Luntz M, Brodsky A, Nusem S, et al. Acute mas- toiditis—the antibiotic era: a multicenter study. Int J Pediatr Otorhinolaryngol. 2001;57(1):1.
Albino AP, Kimmelman CP, Parisier SC.
The organism enters the sinuses from the nose and penetrates the muscular wall of arteries, inciting thrombosis and infarction of tissue. The infection may then spread to the petrous apices, the middle and inner ears, and into the intracranial cavity. Thrombosis of the major cerebral arteries often develops despite ther- apy with amphotericin.
Basilar Meningitis
Tuberculosis, cryptococcosis, and coccidioido- mycosis produce basilar meningitis with involvement of multiple cranial nerves, includ- ing the eighth nerve.123 The clinical picture is that of an insidious febrile illness associated with progressive bilateral sensorineural hear- ing loss. The diagnosis rests on finding the
Cholesteatoma: a molecular and cellular puzzle. Am J Otol. 1998;19:7.
Chole RA. Cellular and subcellular events of bone resorption in human and experimental cholesteatoma: the role of osteoclasts. Laryngoscope. 1984;95:76.
Erkan M, Asian T, Sevuk E, Guney E. Bacteriology of chronic suppurative otitis media. Ann Otol Rhinol Laryngol. 1994;103:771.
Busaba NY. Clinical presentation and management of labyrinthine fistula caused by chronic otitis media. Ann Otol Rhinol Laryngol. 1999;108:435.
Dew LA, Shelton C. Complications of temporal bone infections. In: Cummings CW, Fredrickson JM, Harker LA, Krause CJ, Schuller DE, eds. Otolaryngology Head and Neck Surgery. 3rd ed. St. Louis, MO: CV Mosby; 1998.
Cureoglu S, Schachern PA, Rinaldo A, Tsuprun V, Ferlito A, Paparella MM. Round window membrane and labyrinthine pathological changes: an overview. Acta Otolaryngol. 2005;125(1):9.
Mac Andie C, O’Reilly BF. Sensorineural hear- ing loss in chronic otitis media. Clin Otolaryngol. 1999;24:220.
Hydén D, Akerlind B, Peebo M. Inner ear and facial nerve complications of acute otitis media with focus on bacteriology and virology. Acta Otolaryngol. 2006;126(5):460.
Merchant SN, Gopen Q. A human temporal bone study of acute bacterial meningogenic labyrinthitis. Am J Otol. 1996;17:375.
Van de Beek D, de Gans J, McIntyre P, Prasad
K. Corticosteriods for acute bacterial meningitis.
Cochrane Database Sys Rev. 2007;1:CD004405.
Schuknecht HE. Pathology of the Ear. Philadelphia: Lea & Febiger; 1993.
Ruben R. Bacterial meningitic deafness: historical development of epidemiology and cellular pathology. Acta Otolaryngol. 2008;128(4):388.
Klein M, Koedel U, Pfister HW, Kastenbauer S. Meningitis-associated hearing loss: protection by adjunctive antioxidant therapy. Ann Neurol. 2003;54(4):451.
Goldstein NA, Casselbrant ML, Bluestone CD, Kurs- Lasky M. Intratemporal complications of acute otitis media in infants and children. Otolaryngol Head Neck Surg. 1998;119:444.
Gradenigo G. Sulla leptomeningite circoscritta e sulfa paralisi dell’ abducente di origine otitica. G Accad Med Torino. 1904;10:59.
Murakami T, Tsubaki J, Tahara Y, Nagashima T. Gradenigo’s syndrome: CT and MRI findings. Pediatr Radiol. 1996;26:684.
Connor SE, Leung R, Natas S. Imaging of the petrous apex: a pictorial review. Br J Radiol. 2008;81(965): 427.
Arriga MA. Petrous apex effusion:a clinical disorder.
Laryngoscope. 2006;116:1349.
Visosky AM, Isaacson B, Oghalai JS. Circumferential petrosectomy for petrous apicitis and cranial base osteomyelitis. Otol Neurotol. 2006;27(7):1003.
Gower D, McGuirt WF. Intracranial complications of acute and chronic infectious ear disease: a problem still with us. Laryngoscope. 1983;93:1028.
Barry B, Delattre J, Vie F, Bedos JP, Gehanno
P. Otogenic intracranial infections in adults.
Laryngoscope. 1999;109:483.
Bizakis JG, Velegrakis GA, Papadakis CE, Karampekios SK, Helidonis ES. The silent epidural abscess as a complication of acute otitis media in children. Int J Pediatr Otorhinolaryngol. 1998;45:163.
Christensen N, Wayman J, Spencer J. Lateral sinus thrombosis: a review of seven cases and proposal of a management algorithm. Int J Pediatr Otorhinolaryngol. 2009;73(4):581.
Frazier JL, Ahn ES, Jallo GI. Management of brain abscesses in children. Neurosurg Focus. 2008;24(6):E8.
Hirsch JF, Roux FX, Saint-Rose C, et al. Brain abscess in childhood. Childs Brain. 1983;10:251.
Harrison MJG. The clinical presentation of intracra- nial abscesses. Q J Med. 1982;204:461.
Staecker H, Nadol JB, Ojeman R, McKenna MJ. Delayed intracranial abscess after acoustic neuroma surgery: a report of 2 cases. Am J Otol. 1999;20:369.
Symonds CP. Otitic hydrocephalus. Brain. 1931;54:55.
Tomkinson A, Mills RG, Cantrell PJ. The pathophysi- ology of otitic hydrocephalus. J Laryngol Otol. 1997;111:757.
Durairaj VD, Andrews B, Rao RR, Chan KH. Morbid complications of otitic hydrocephalus. Orbit. 2008;27(1):51.
Hasbun R, Aronin SI, Quagliarello VJ. Treatment of bacterial meningitis. Compr Ther. 1999;25:73.
Morosa L, Modugno GC, Pirodda A. Malignant external otitis: review and personal experience. Acta Otolaryngol Suppl (Stockh). 1996;521:3.
Balkany TJ, Ress BD. Infections of the external ear. In: Cummings CW, Fredrickson JM, Harker LA, Krause CJ, Richardson MA, Schuller DE, eds. Otolaryngology Head and Neck Surgery. 3rd ed. St. Louis, MO: CV Mosby; 1998.
Mani N, Sudhoff H, Rajagopal S, Moffat D, Axon PR. Cranial nerve involvement in malignant external oti- tis: implications for clinical outcome. Laryngoscope. 2007;117(5):907.
Gold S, Som PM, Lucente FE, et al. Radiographic findings in progressive necrotizing “malignant” exter- nal otitis. Laryngoscope. 1984;94:363.
Peleg U, Perez R, Raveh D, Berelowitz D, Cohen D. Stratification for malignant external otitis. Otolaryngol Head Neck Surg. 2007;137(2):301.
Grandis JR, Curtin HD, Yu VL. Necrotizing (malig- nant) external otitis: prospective comparison of CT and MR imaging in diagnosis and follow-up. Radiology. 1995;196:499.
Schuknecht HE. Neurolabyrinthitis. Viral infections of the peripheral auditory an vestibular systems. In: Nomura Y, ed. Hearing Loss and Dizziness. Tokyo, Japan: Igaku-Shoin; 1985:1.
Pappas DG. Hearing impairments and vestibu- lar abnormalities among children with subclinical cytomegalovirus. Ann Otol Rhinol Laryngol. 1983; 92: 552.
Samileh N, Ahmad S, Mohammad F, Framarz M, Azardokht T, Jomeht E. Role of cytomegalovirus in sensorineural hearing loss of children: a case-control study Tehran, Iran. Int J Pediatr Otorhinolaryngol. 2008;72(2):203.
Wilson WR, Veltri RW, Laird N, Sprinkle PM. Viral and epidemiologic studies of idiopathic sudden hearing loss. Otolaryngol Head Neck Surg. 1983;91:653.
Schuknecht HF, Kitamura K. Vestibular neuritis. Ann Otol Rhinol Laryngol. 1981;90 (suppl):1.
Dishoeck H, Van Bierman T. Sudden perceptive deaf- ness and viral infection (report of first one hundred patients). Ann Otol Rhinol Laryngol. 1957;66:963.
Hart C. Vestibular paralysis of sudden onset and probable viral etiology. Ann Otol Rhinol Laryngol. 1965;74:33.
Merilield D. Self-limited idiopathic vertigo (epidemic vertigo). Arch Otolaryngol. 1965;81:355.
Davis LE, Johnsson LG. Viral infections of the inner ear: clinical, virologic and pathologic studies in humans and animals. Am J Otolaryngol. 1983;4:347.
Schuknecht HF, Kimura RR, Nanfal PM. The pathol- ogy of idiopathic sensorineural hearing loss. Arch Otorhinolaryngol. 1986;243:1.
Baloh RW, Lopez I, lshiyama A, Wackym PA, Honrubia V Vestibular neuritis: clinical–pathological correlation. Otolaryngol Head Neck Surg. 1996;114:586.
Rauch SD. Vestibular histopathology of the human temporal bone. What can we learn? Ann NY Acad Sci. 2001;942:25.
Khetarpal U, Nadol JB, Jr., Glynn RJ. Idiopathic sud- den sensorineural hearing loss and postnatal viral labyrinthitis: a statistical comparison of temporal bone findings. Ann Otol Rhinol Laryngol. 1990;99:969.
Robillard RB, Hilsinger RL, Adour KK. Ramsay Hunt facial paralysis: clinical analysis of 185 patients. Otolaryngol Head Neck Surg. 1986;95:292.
Ohtani F, Furuta Y, Aizawa H, Fukuda S. Varicella- zoster virus load and cochleovestibular symptoms in Ramsay Hunt syndrome. Ann Otol Rhinol Laryngol. 2006;115(3):233.
Zajtchuk J, Matz C, Lindsay J. Temporal bone pathol- ogy in herpes oticus. Ann Otol Rhinol Laryngol. 1972;81:331.
Baloh RW. Vestibular neuritis. N Engl J Med. 2003;348:1027.
Lee H, Kim BK, Park HJ, Koo JW, Kim JS. Prodromal dizziness in vestibular neuritis: frequency and clini- cal implication. J Neurol Neurosurg Psychiatry. 2009;80(3):355.
Newman-Toker DE, Kattach JC, Alvernia JE, Wang DZ. Normal head impulse test differentiates acute cerebellar strokes from vestibular neuritis. Neurology. 2008;70:2378.
Kammerlind AC, Ledin T, Skargren E, Odkvist LM. Long-term follow-up after acute unilateral vestibular loss and comparison between subjects with and with- out remaining symptoms. Acta Oto-Laryngologica. 2005;125:946.
Kammerlind AC, Ledin T, Odkvist LM, Skargren E. Influence of asymmetry of vestibular caloric response and age on balance and perceived symptoms after acute unilateral vestibular loss. Clin Rehabil. 2006;20:142.
Huppert D, Strupp M, Theil D, Glaser M, Brandt T. Low recurrence rate of vestibular neuritis: a long-term follow-up. Neurology. 2006;67(10):1870.
Schuknecht HF, Witt RL. Acute bilateral sequential vestibular neuritis. Am J Otolaryngol. 1985;6:255.
Stokroos RJ, Albers FW, Schirm J. The etiology of idiopathic sudden sensorineural hearing loss. Experimental herpes simplex virus infection of the inner ear. Am J Otol. 1998;19:447.
Keithley EM, Sharp R Woolf NK, Harris JP. Temporal sequence of viral antigen expression in the cochlea induced by cytomegalovirus. Acta Otolaryngol (Stockh). 1988;106:46.
Murakami S, Mizobuchi M, Nakashiro Y, Doi T, Hato N, Yanagihara N. Bell palsy and herpes simplex virus: identification of viral DNA in endoneural fluid and muscle. Ann Intern Med. 1996;124:27.
Furuta Y, Takasu T, Fukuda S, Inuyama Y, Sato S, Nagashima K. Latent herpes simplex virus type 1 in human vestibular ganglia. Acta Otolaryngol Suppl (Stockh). 1993;503:85.
Theil D, Arbusow V, Derfuss T, Strupp M, Pfeiffer M, Mascolo A, Brandt T. Prevalence of HSV-1 LAT in human trigeminal, geniculate, and vestibular ganglia and its implication for cranial nerve syndromes. Brain Pathol. 2001;11(4):408.
Arbusow V, Theil D, Strupp M, Mascolo A, Brandt
T. HSV-1 not only in human vestibular ganglia but also in the vestibular labyrinth. Audiol Neurootol. 2001;6(5):259.
Fetter M, Dichgans J. Vestibular neuritis spares the inferior division of the vestibular nerve. Brain. 1996;119:755.
Aw ST, Fetter M, Cremer PD, Karlberg M, Halmagyi GM. Individual semicircular canal function in supe- rior and inferior vestibular neuritis. Neurology. 2001;57:768.
Arbusow V, Schulz P, Strupp M, et al. Distribution of herpes simplex virus type 1 in human geniculate and vestibular ganglia: implications for vestibular neuritis. Ann Neurol. 1999;46:416.
Halmagyi GM, Aw ST, Karlberg M, Curthoys IS, Todd MJ. Inferior vestibular neuritis. Ann NY Acad Sci. 2002;956:306.
Monstad P, Økstad S, Mygland A. Inferior vestibular neuritis: 3 cases with clinical features of acute vestibu- lar neuritis, normal calorics but indications of saccular failure. BMC Neurol. 2006;6:45.
Stokroos RJ, Albers FWJ. The etiology of idiopathic sudden sensorineural hearing loss, a review of the lit- erature. Acta Otolaryngol Belg. 1996;50:69.
Laird N, Wilson WR. Predicting recovery from idiopathic sudden hearing loss. Am J Otolaryngol. 1983;4:161.
Tamhankar M, Solomon D. Acute hearing loss. Curr Treat Options Neurol. 2004;6(1):55.
Schattner A, Halperin D, Wolf D, Zimhony
O. Enteroviruses and sudden deafness. CMAJ. 2003;168(11):1421.
Nomura Y, Hara M, Kurata T Experimental herpes simplex virus and cytomegalovirus labyrinthitis. Acta Otolaryngol Suppl (Stockh). 1989;457:57.
Fukuda S, Furuta Y, Takasu T, et al. The significance of herpes viral latency in the spiral ganglia. Acta Otolaryngol Suppl (Stockh). 1994;514:108.
Dahl H, Marcoccia J, Linde A. Antigen detection: the method of choice in comparison with virus isolation and serology for laboratory diangnosis of herpes zoster in human immunodeficiency virus-infected patients. J Clin Microbiol. 1997; 35:347.
Vazquez M, LaRussa PS, Gershon AA, et al. The effec- tiveness of the varicella vaccine in clinical practice. N Engl J Med. 2001;344:955.
Harbecke R, Oxman NM, Arnold BA, et al. A real time PCR assay to identify and discriminate among wild type and vaccine strains of varicella-zoster virus and herpes simplex virus in clinical specimen, and comparison with the clinical diagnosis. J Med Virol. 2009;81:1310.
Kuo MJ, Drago PC, Proops DW, Chavda SV. Early diagnosis and treatment of Ramsay Hunt syndrome: the role of magnetic resonance imaging. J Laryngol Otol. 1995;109:777.
Berretini S, Bianchi MC, Segnini G, et al. Herpes zoster oticus: correlations between clinical and MRI findings. Eur Neurol. 1998;39:26.
Faralli M, Molini E, Ricci G, et al. Study of vestibu- lar evoked myogenic potentials in unilateral vestibu- lopathy: Otolithic versus canal function testing. Otol Neurotol. 2006;27(8):1115.
Kim HA, Hong JH, Lee H, et al. Otolith dysfunction in vestibular neuritis: recovery pattern and a predictor of symptom recovery. Neurology. 2008;70(6):449.
Bergenius J, Borg E, Audio-vestibular findings in patients with vestibular neuronitis. Acta Otolaryngol (Stockh). 1983;906:389.
Rahko T, Karma P. New clinical finding in vestibular neuritis: high frequency audiometry hearing loss in the affected ear. Laryngoscope. 1986;96:198.
Wilson WR, Laird N, Kavesh DA. Electronystagmographic findings in idiopathic sud- den hearing loss. Am J Otolaryngol. 1982;3:279.
Kerber KA, Brown DL, Lisabeth LD, Smith MA, Morgenstern LB. Stroke among patients with diz- ziness, vertigo, and imbalance in the emergency department: a population-based study. Stroke. 2006;37:2484.
Whitley RJ. A 70 year old woman with shingles: review of herpes zoster. JAMA. 2009;309:73.
Uscategui T. Doree C, Chamberlain IJ, Burton MJ. Antiviral therapy for Ramsay Hunt syndrome in adults. Cochrane Database Sys Rev. 2008:CD006851.
Strupp M, Arbusow V, Maag KP, Gall C, Brandt T. Vestibular exercises improve central vestibulospinal compensation after vestibular neuritis. Neurology. 1998;51:838.
Wilson W, Byl FM, Laird N. The efficacy of steroids in the treatment of idiopathic sudden hearing loss: a double-blind clinical study. Arch Otolaryngol. 1980;106:772.
Ariyasu L, Byl FM, Sprague MS, Adour KK. The beneficial effect of methylprednisone in acute ves- tibular vertigo. Arch Otolaryngol Head Neck Surg. 1990;116:700.
Strupp M, Zingler VC, Arbusow V, et al. Methylprednisolone, valacyclovir, or the com- bination for vestibular neuritis. N Engl J Med. 2004;351(4):354.
Shupak A, Issa A, Golz A, Margalit Kaminer, Braverman I. Prednisone treatment for vestibular neuritis. Otol Neurotol. 2008;29(3):368.
Walker MF. Treatment of vestibular neuritis. Curr Treat Options Neurol. 2009;11(1):41.
Rauch SD. Clinical practice. Idiopathic sudden sen- sorineural hearing loss. N Engl J Med. 2008;359: 833.
Wei BP, Mubiru S, O’Leary S. Steroids for idio- pathic sudden sensorineural hearing loss. Cochrane Database Sys Rev. 2006:CD003998.
Plontke SK, Lowenheim H, Mertens J, et al. Randomized, double blind, placebo controlled trial on the safety and efficacy of continuous intratym- panic dexamethasone delivered via a round window catheter for severe to profound sudden idiopathic sensorineural hearing loss after failure of systemic therapy. Laryngoscope. 2009;119(2):359.
Ho HG, Lin HC, Shu MT, Yang CC, Tsai HT. Effectiveness of intratympanic dexamethasone injection
in sudden-deafness patients as salvage treatment.
Laryngoscope. 2004;114:1184.
Rolfs RT, Nakashima AK. Epidemiology of primary and secondary syphilis in the United States, 1981 though 1989. JAMA. 1990;264:1432.
Mishra S, Walmsley SL, Loutfy MR, Kaul R, Logue KJ, Gold WL. Otosyphilis in HIV-coinfected indi- viduals: a case series from Toronto, Canada. AIDS Patient Care STDS. 2008;22(3):213.
Karmody C, Schuknecht H. Deafness in congenital syphilis. Arch Otolaryngol. 1966;83:18.
Saltiel P, Melmed CA, Portnoy D. Sensorineural deafness in early acquired syphilis. Can J Neurol Sci. 1983;10:114.
Yimtae K, Srirompotong S, Lertsukprasert K. Otosyphilis: a review of 85 cases. Otolaryngol Head Neck Surg. 2007;136(1):67.
Morrison AW. Management of Sensorineural Deafness. Boston: Butterworh; 1975.
Steckelberg JM, McDonald TJ. Otologic involvement in late syphilis. Laryngoscope. 1984;94:753.
Amenta CA, III, Dayal VS, Flaherty J, Weil RJ. Luetic endolymphatic hydrops: diagnosis and treatment. Am J Otol. 1992;13:516.
Hughes GB, Rutherford I. Predictive value of sero- logic tests for syphilis in otology. Ann Otol Rhinol Laryngol. 1986;95:250.
Nicolau Y, Northrop C, Eavey R. Tuberculous otitis in infants: temporal bone histopathology and clinical extrapolation. Otol Neurotol. 2006;27(5):667.
Jeanes A, Friedmann I. Tuberculosis of the middle ear. Tubercle, J Br Tuber Assoc. 1960;41:109.
Rho MH, Kim DW, Kim SS, Sung YS, Kwon JS, Lee SW. Tuberculous otomastoiditis on high-resolution temporal bone CT: comparison with nontuberculous otomastoiditis with and without cholesteatoma. AJNR Am J Neuroradiol. 2007;28(3):493.
Olalla I, Ortin M, Hermida G, et al. Autologous peripheral blood stem cell transplantation in a patient with previous invasive middle ear mucomycosis. Bone Marrow Transpl. 1996;18:1183.
Slack CL, Watson DW, Abzug MJ, Shaw C, Chan KH. Fungal mastoiditis in immunocompromised children. Arch Otolaryngol Head Neck Surg. 1999;125(1): 73.
Yaramis A, Girkan F, Elevli M, et al. Central nervous system tuberculosis in children: a review of 214 cases. Pediatrics. 1998;102:E49.
This page intentionally left blank
![]()
HISTORICAL BACKGROUND CAUSES OF BENIGN POSITIONAL
POSTERIOR CANAL VARIANT OF BENIGN POSITIONAL VERTIGO
Clinical Features Pathophysiology
Diagnosis Management
OTHER VARIANTS OF BENIGN POSITIONAL VERTIGO
Horizontal Canal Benign Positional Vertigo Anterior Canal Benign Positional Vertigo Mimics of Benign Positional Vertigo
Benign positional vertigo (BPV), also called benign positioning vertigo and benign paroxys- mal positional vertigo, is a common inner ear disorder resulting from abnormal stimulation of the semicircular canals (usually the poste- rior). The direction of the provocative move- ment and the appearance of the induced eye movements (nystagmus) identify the involved canal(s). The abnormal stimulation is due to the presence of detached otoconia (canaliths) moving in the canal endolymph under the influence of gravity. In order for positional ver- tigo to occur, two events must happen: (1) oto- conia must be dislodged from the utricular macule, and (2) the head must be held in a critical position that allows the otoconia to enter a semicircular canal. BPV is not a disease; rather, it is a syndrome that can have multiple causes of the detached otoconia.
The basic features of BPV and the associated paroxysmal positional nystagmus were first described in a single patient by Bárány in 1921,1 but it was not until 1952 that Dix and Hallpike described the provocative positioning
maneuver and clearly defined the clinical syndrome.2 Bárány speculated that the parox- ysmal positional nystagmus was caused by a lesion of the otolith organs, as it was induced by a change in head position relative to gravity. Dix and Hallpike came to a similar conclusion after reviewing the clinical features of 100 patients with BPV and identifying unilateral degeneration of the utricular macule at necropsy in a typical case.
In 1961, Schuknecht reviewed the temporal bone specimens from three patients who had been reported to have BPV and was struck by the remarkable similarity of the pathologic changes.3 Each had a selective degeneration of the superior part of the labyrinth, including the superior branch of the vestibular nerve, the utricle, and the crista of the horizontal and anterior semicircular canals. He concluded that in each case the damage to the labyrinth resulted from occlusion of the anterior vestibu- lar artery, the branch of the internal auditory artery that supplies these organs. Schuknecht felt that the paroxysmal positional nystagmus that occurred in these cases must have origi- nated from the posterior semicircular canal since it was the only peripheral sensory organ capable of generating nystagmus that was still functioning. He did not feel that the intact
255
saccular macule was capable of generating nystagmus.
With this hypothesis in mind, Schuknecht attempted to produce paroxysmal positional nystagmus in four cats by cutting off the blood supply in the left anterior vestibular artery.3 Each animal developed the expected acute vestibular syndrome with horizontal nystagmus and imbalance in the immediate postoperative period. These symptoms gradually subsided over several days. One of the animals, however, developed typical benign paroxysmal positional nystagmus 3 months after the operation, which persisted until termination of the experiments at 7 months. Schuknecht theorized that loose otoconia from the degenerating utricular mac- ule came in contact with the cupula of the pos- terior semicircular canal, causing it to move in the plane of the canal after position changes.
In 1969, Schuknecht found basophilic depos- its on the cupulae of the posterior semicircular canals in two patients who manifested isolated BPV without any other ear symptoms prior to death from unrelated disease (Fig. 10–1).4 The deposits were present on the side that was undermost when the paroxysmal positional nystagmus was induced. These findings sup- ported his earlier hypothesis and, even though he observed particles in the canal in addition to those attached to the cupula, he coined the term cupulolithiasis.
The cupulolithiasis theory of Schuknecht gained general acceptance, but there were fea- tures of the characteristic paroxysmal positional nystagmusthatwerenotadequatelyexplained— the transient duration and the fatigability with repeated positioning. If the particles are adherent to the cupula, transient duration and
A Deposit
on cupula
Normal cupula
Left
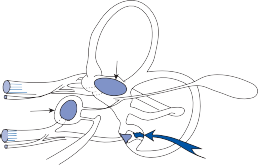
B
Superior division
Saccule
Inferior division
Macule of the utricle
Right
Deposit on the cupula of the posterior semicircular canal
Figure 10–1. Basophilic deposits on the cupula in benign positional vertigo. A Histopathological section through the crista of the posterior semicircular canals of a patient who exhibited typical benign paroxysmal positional nystagmus in the head- hanging left position prior to death from unrelated causes. Attached to the cupula is a granular, basophilic staining deposit. B Drawing illustrating relationship between the macule of the utricle and the ampulla of the posterior semicircular canal when the head is erect. (From Schuknecht HF. Pathology of the Ear. Harvard University Press, Cambridge, 1993, with permission.)
fatigability would not be expected. In his initial report,3 Schuknecht suggested that the otoco- nial debris might dislodge from the cupula and float away, allowing the cupula to return to its normal position, and that the particles might become dispersed within the endolymph with repeated positionings. However, when his sub- sequent postmortem studies showed that the otolithic debris was firmly attached to the cup- ula of the posterior semicircular canal, he felt that these possibilities were less likely.4
In the late 1970s, McClure began to experi- ment with mechanical models of the inner ear to see if these problems could be resolved. His simple model consisted of a water bottle repre- senting the utricular chamber and two attached rounded tubes representing the posterior and anterior semicircular canals. He placed mer- cury in the canals to represent the otoconial debris and noted that repeated positional changes from sitting to head-hanging position caused the mercury to enter the utricle. He recognized that freely floating debris in the posterior semicircular canal could readily explain both the paroxysmal nature of the nys- tagmus and the fatigability. In 1979, along with Hall and Ruby5, McClure published his model and suggested that benign paroxysmal posi- tional nystagmus could be divided into two types: type A, a nonfatigable form caused by debris adherent to the cupula (i.e., cupulo- lithiasis), and type B, a more common fatigable form caused by free-floating particles in the endolymph, canalithiasis. This work served as the theoretical basis for the subsequent use of positional exercises and positional maneuvers to treat BPV.
Epley did not distinguish between cupulo- lithiasis and canalithiasis but rather suggested that debris was both attached to the cupula and free floating in the canal in most cases of BPV.6 He recognized that debris moving within the narrow semicircular canal would be much more effective in deviating the cupula than an equal amount of debris floating within the ampulla next to the cupula. He saw the analogy of the bolus of particles acting as a piston mov- ing within the narrow confines of the semicir- cular canal, which, according to Pascal’s formula, would lead to a magnified force acting on the cupula. Epley argued that if this bolus of debris could move so easily with position change, it should be possible to move it around and out of the posterior semicircular canal and
into the utricle with appropriate positional changes. In a case series report in 1992, he reported that a simple particle repositioning maneuver cured most patients with BPV.7 Later, randomized placebo-controlled (sham positional maneuvers) trials confirmed the treatment effect of what has become known as the Epley maneuver.8
At about the same time of Epley’s original reports, Parnes and McClure described a sur- gical procedure for blocking the posterior semicircular canal with a bony plug that was highly effective in curing intractable cases of BPV.9 During the process of exposing the membranous labyrinth of the posterior semicircular canal for the plugging operation, Parnes and McClure observed a chalky white substance within the endolymph of the posterior semicircular canal. Their obser- vation supported the canalithiasis model pro- posed by Hall et al.5 and was consistent with the success of Epley’s particle repositioning maneuver.
CAUSES OF BENIGN POSITIONAL VERTIGO
The first event that must happen for BPV to occur is that otoconia must be free floating in the utricule. However, in most cases, there is no easily explainable reason for why the otoco- nia are released. The attacks of positionally triggered vertigo are not typically preceded by a specific event. However, the prevalence of the disorder dramatically increases with increasing age so that release of the otoconia could be an effect of the aging process. Osteopenia and osteoporosis are also more common in patients with idiopathic BPV than in controls, suggesting that abnormal calcium metabolism is an important etiological factor.10–12 Both osteopenia and BPV are more common in women and older people. In osteopenic rats, otoconia have decreased density and increased size compared to con- trols so the otoconia may be more easily dislodged.13 Furthermore, degeneration of otoconia is seen in older rats regardless of osteopenia.14
Another important association with BPV is migraine. Migraine is more common in patients with idiopathic BPV than in controls
particularly in patients under the age of 50.15,16 The reason for this association is not clear, but it is possible that migranous vasospasm could lead to dislodged otoconia.
Head trauma is the most common identifi- able cause for dislodging otoconia. The most common cause of head trauma is motor vehicle accidents followed by common falls.17 BPV after head trauma tends to be more severe and more protracted than BPV from other causes. Infection and ischemia of the inner ear are the next most common causes for free-floating oto- conia. In animal models, brief periods of isch- emia result in prominent release of otoconia from the utricular macule (see Anatomy of Otolith Organs, Chapter 2). Nearly half of patients with vestibular neuritis have BPV as a sequela (see Chapter 9). Vigorous exercise such as aerobics, mountain biking, and swim- ming are associated with recurrent BPV.18–20 Presumably repeated abrupt accelerations can dislodge otoconia. Surgeries involving drilling of the temporal bone such as with cochlear implants can dislodge otoconia and predespose to BPV.21
The second event that must happen for BPV to occur is the otoconial debris must enter a semicircular canal. Free-floating otoconia in the utricle are not believed to cause symptoms. It is only when the otoconia enter a semicircu- lar canal that symptoms occur. Because of its position in the head the posterior semicircular canal is most vulnerable. Loose otoconia in the utricle will fall into the posterior semicircular canal and become trapped if the head is held backward below the horizontal plane for a few minutes. This explains why BPV often first occurs after situations such as going to a hair- dresser, a dentist, or fixing a leaky kitchen sink. People with certain occupations, such as auto mechanics and plumbers, are particularly prone to developing recurrent BPV. It is not too farfetched to assume that otoconial debris is floating freely within the endolymph in nearly everyone, particularly older people.22 It is just a matter of whether the head is held in the criti- cal position long enough for the particles to drop into the posterior semicircular canal. Benign positional vertigo commonly occurs after surgical procedures regardless of the nature or location of the surgery. Initially this was thought to result from some metabolic changes associated with the trauma of surgery or from the anesthesia, but more likely the BPV
is secondary to prolonged positioning with the head back in people who already have free- floating debris in the vestibule. Positioning for dental and radiological procedures is another common precipitating factor for BPV.23,24 Interestingly BPV occurs more commonly on the right side compared to the left, possibly because people tend to sleep more on the right side than the left.25,26
POSTERIOR CANAL VARIANT OF BENIGN POSITIONAL VERTIGO
Clinical Features
By far the most common variety of BPV is that associated with debris in the posterior semicir- cular canal (PC-BPV).27 Patients develop brief episodes of vertigo and nystagmus (usually last- ing <30 sec) with position change: typically when turning over in bed, getting in and out of bed, bending over and straightening up, or extending the neck to look up.28 (See Video 6–12 and Video 6–13) Importantly, the vertigo of BPV is triggered by certain position changes. Any patient with vertigo will report worsening of the vertigo in certain positions, but patients with BPV will report that the vertigo is triggered by the position change. “Top shelf vertigo,” in which a patient experiences an episode of vertigo while reaching for something on a high shelf, is nearly always due to PC- BPV. Since the most common provocative movements for inducing PC-BPV occur in bed or when getting in and out of bed, patients report that the vertigo is severe in the morning and tends to disappear once they are up. The common report of having attacks mostly in the morning may also be because the particles have had a chance to accumulate in one part of the canal while the patient is lying still sleeping. Turning the head from side to side while erect does not induce nystagmus because this movement does not stimulate the posterior semicircular canal. Often after a flurry of episodes, patients com- plain of more prolonged nonspecific dizziness (a motion-sick sensation) that may last hours or even persist throughout the day. Vertigo can awaken the patient; presumably, positional ver- tigo occurs when they turn over while sleeping, and some patients experience severe nausea
and vomiting, which can be even more bother- some than the vertigo.
As the name implies, BPV is a benign disor- der meaning that it is not progressive and in most cases will eventually remit spontane- ously.29 However, spontaneous remission can take months or longer, and there are some patients who report having symptoms for many years and are only cured after a canalith repositioning maneuver. In a longitudinal cohort study of patients who presented to an Otolaryngology clinic, the mean time to symp- tom resolution was 39 days with a wide standard deviation of 47 days.29 In a population- based telephone survey study, patients with BPV reported a mean symptom duration of 2 weeks (range 0.5 days to 104 weeks), with about one-third reporting symptoms more than 1 month.30,31 The duration of symptoms in con- trol groups of randomized trials was generally more than 1 month for about 80% of the patients.32 The “benign” label also cannot be mistaken to mean that the symptoms are trivial. Patients with BPV have an increased incidence of falls, depression, and impairment of daily activities.33–35 During periods of symptomatic BPV, 24% of patients reported giving up driv- ing a car and 18% avoided leaving their home.31 Also reflecting the burden of BPV on patients, in the population-based study nearly 80% of BPV patients reported a visit to a medical doc- tor.31 More than half of patients with PC-BPV have at least one recurrence after a remis- sion.15,27,31 In some patients, bouts of PC-BPV are intermixed with variable periods of remis- sion over many years.
In their initial description of the syndrome in 1952, Dix and Hallpike found evidence of ear disease in approximately two-thirds of their 100 patients with BPV.2 When the ear disease was unilateral, the nystagmus was usually induced when the abnormal ear was under- most. Subsequent reports confirmed the benign nature of the disorder but suggested that most cases were unassociated with identifiable lesions.28,36,37 Despite its common occurrence, there have been relatively few reports of large series of patients with BPV.
We reviewed the clinical features of 240 cases of BPV, each with a typical clinical his- tory and the stereotyped paroxysmal positional nystagmus.28 The mean age of onset was 54 years (range 11 to 84 years). The two largest diagnostic categories were posttraumatic and
Table 10–1 Diagnoses in 240 Patients with Benign Paroxysmal Positional Vertigo28
![]()
Idiopathic 118
Post-traumatic 43
Viral neurolabyrinthitis 37
Basilar vertebral insufficiency 11
Meniere’s disease 5
Postsurgery (general) 5
Postsurgery (ear) 5
Ototoxicity 4
Luetic labyrinthitis 2
Chronic otomastoiditis 2
![]()
Other 8
postviral (Table 10–1). Patients with the for- mer had the onset of BPV within 3 days of well- documented head trauma. Patients in the viral group reported a prior episode of acute vertigo gradually resolving over 1 to 2 weeks. In 25 of 37 postviral patients, there was an associated sudden hearing loss that improved as vertigo subsided, although most patients were left with a residual unilateral sensorineural hearing loss. The associated hearing loss suggests that the viral process targeted the labyrinth (i.e., “laby- rinthitis”) rather than the more common tar- get, the vestibular nerve (i.e., “vestibular neuri- tis”). Episodes of BPV can begin as soon as 1 week and long as 8 years after the acute viral syndrome. Most patients reported a cold or flu- like illness within 2 weeks of the acute vertigi- nous episode. Eleven patients reported typical symptoms of vertebrobasilar insufficiency (in addition to vertigo) prior to the onset of BPV. In these cases, the BPV could have resulted from ischemic damage to the labyrinth. It is important to recognize that these episodes of positional vertigo in such patients do not indi- cate recurrent vascular ischemia.
Females outnumbered males by a ratio of 1.6:1, combining all diagnostic categories. This ratio was approximately 2:1 if only the idio- pathic and miscellaneous groups were consid- ered. Other investigators have reported an even higher female-to-male preponderance with idiopathic BPV.37 The age of onset peaked in the sixth decade in the idiopathic group, in the fourth and fifth decades in the postviral group, and was evenly distributed over the sec- ond to sixth decades in the posttraumatic group.
As noted earlier, the most likely explanation for PC-BPV is canalithiasis involving the posterior semicircular canal.7,38,39 With the patient sitting upright, a clot of calcium carbonate crystals forms at the most dependent portion of the posterior canal (Fig. 10–2A). Movement of the head back and to the side in the plane of that posterior canal (such as with the standard positioning test) causes the clot to move in an ampullofugal direction, producing ampullofu- gal displacement of the cupula due to the “plunger” effect of the clot moving within the narrow canal (Fig. 10–2B). Fatigability with repeated positioning is explained by dispersion of particles from the clot, making the plunger less effective. Reactivation of the positional vertigo after prolonged bed rest is also explained as the particles reform into a clot. The induced
vertigo and nystagmus are brief in duration because once the clot reaches its lowest posi- tion in the canal with respect to the earth’s sur- face, the cupula returns to its primary position with its usual time constant determined pri- marily by cupular elasticity. The typical latency of about 5 seconds before onset of nystagmus is explained by the delay in setting the clot into motion. It also explains why slow positioning does not induce vertigo or nystagmus, as the clot would move slowly along the undermost wall of the canal without producing a plunger effect.
Probably the most convincing argument for the canalithiasis theory is the dramatic response of PC-BPV to positional maneuvers designed to move the clot around the posterior semicircular canal into the utricle.7,27 By rotating the subject about the plane of the posterior canal, the clot moves from a position next to the cupula, as
![]()
![]()
A B C

Anterior
Horizontal
Utricle Anterior
Posterior
Horizontal
Posterior
Clot
Horizontal
Posterior
Anterior
![]()
Posterior
D E
Anterior Horizontal
Horizontal
Anterior
Posterior
Endolymphatic duct
Figure 10–2. Pathophysiology of the posterior canal variant of benign positional vertigo. In the sitting position (A) the clot of calcium carbonate crystals lies at the bottom-most position within the posterior canal. Movement to the head-hanging position (B) causes the clot to move away from the cupula, producing an excitatory burst of activity in the ampullary nerve from the posterior canal (ampullofugal displacement of the cupula). Rolling across to the opposite side in the plane of the posterior canal (C and D) causes the clot to enter the common crus of the posterior and anterior semicircular canals. Finally, when the patient sits up (E) and the clot falls into the utricle. (Adapted from Epley JM. The canalith reposition- ing procedure: For treatment of benign paroxysmal positional vertigo. Otolaryngol Head Neck Surg. 1992;107:199, with permission).
Diagnosis
The diagnosis of PC-BPV rests on finding the characteristic paroxysmal positional nystagmus in a patient with a typical history of positional vertigo. The nystagmus is induced by rapidly moving the patient from the sitting to head- hanging position as originally described by Dix and Hallpike (see Fig. 6–6 in Chapter 6). It is important to prepare the patient in advance by explaining that the goal is to induce positional vertigo and that the patient must cooperate by keeping the eyes open and avoid blinking as much as possible. The typical nystagmus has tor- sional and vertical components. The eyes beat upward (toward the forehead) with the upper poles beating toward the ground (thus an upbeat- torsional nystagmus is seen).40,41 The vertical component is larger in the contralateral eye and the torsional component is larger in the ipsilat- eral eye, consistent with the known excitatory connections from the posterior semicircular canal to the eye muscles (see Fig. 3–8c in Chapter 3). A reverse nystagmus (downbeat and torsional) of lesser magnitude usually occurs if the patient is brought directly back up from the head-hanging position to the sitting position, a finding which is also highly supportive of free-floating particles in the canal because sit- ting up reverses the movement of the particles in the canal, thus triggering nystagmus in the reverse direction. The nystagmus triggered in the head-hanging position fatigues (decreases with repeated positioning) in more than 90% of patients, but there are occasional patients with
otherwise typical BPV and nystagmus who do not show fatigue with repeated positioning. There is usually a latency from the time the head-hanging position is achieved to the onset of positioning nystagmus but, as with fatigability, it is not an absolute feature. The presence of posi- tioning nystagmus correlates with the clinical symptoms. Unless the patient is tested during a period when he or she is having acute episodes of vertigo, the positioning nystagmus may not be observed. Patients with BPV may have negative positional testing if the particles are dispersed within the canal rather than coalescent as a clot. With the development of particle reposition- ing maneuvers designed to remove debris from the posterior semicircular canal, treating the patients with these maneuvers also serves to confirm the diagnosis.27 Once the typical posi- tioning nystagmus is induced, we proceed with a positional maneuver to cure the condition (see later discussion). If the positioning nystag- mus and symptoms are gone after completing
the maneuver, the diagnosis was correct.
Since BPV can generally be diagnosed and cured at the time of the office visit, ancillary testing is rarely indicated. Laboratory tests are generally not helpful for the diagnosis of BPV. Patients with recurrent BPV might be screened for osteopenia, although it is unproven whether correcting osteopenia decreases the likelihood of recurrent BPV. ENG and VNG are not help- ful since there is usually a minimal horizontal component to record and the prominent tor- sional component cannot easily be recorded. VNG allows one to actually view the nystagmus so that the torsional component can be appre- ciated, but the pattern of nystagmus is readily identifiable at the bedside without the need for VNG. The characteristic nystagmus is easily observed visually even if the patient attempts to fixate (i.e., there is no need for Frenzel lenses or infrared video recordings). MRI of the brain is not indicated, but high resolution MRI of the inner ear might be informative for visualizing the otoconia debris and canal structure in very rare atypical or refractory cases.42,43
Management
Once BPV is diagnosed, a simple explanation of the nature of the disorder and its favorable prognosis can help relieve the patient’s anxiety. Because of the dramatic nature of the episodes

1
sion.31 The likelihood of a recurrence should be explained to patients so they are not unduly frightened if it occurs.
We typically perform a particle repositioning maneuver designed to liberate the clot from the posterior semicircular canal immediately after the diagnosis is confirmed with the Dix-Hallpike positioning test. We use a modified Epley maneuver, shown in Fig. 10–3.

2
![]()
3
5
4
Figure 10–3. Modified Eply maneuver for treating the posterior canal variant of benign positional vertigo. After turning the head toward the affected side (right side in this case) the patient moves from a sitting to head hanging position (The Dix-Hallpike test, 1–2); once the nystagmus and vertigo have subsided the patient rolls across to the opposite side, nose facing the ground, all in one motion (2–4); finally after about 30 seconds the patient returns to the sitting position (4–5). (From Fife TD et al. Quality Standards Subcommittee, American Academy of Neurology. Practice parameter: therapies for benign paroxysmal positional vertigo (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2008;70:2067 with permission).
2 3
Figure 10–4. Modified Seymont maneuver for treating the posterior canal variant of benign positional vertigo. After turn- ing the head toward the unaffected side (left side in this case) the patient moves from a sitting to right lateral position (1–2); while maintaining the head in the same position the patient moves across to the opposite lateral position, in the plane of the right posterior canal (2–3); after vertigo and nystagmus subsides the patient returns to the sitting position. (From Fife TD et al. Quality Standards Subcommittee, American Academy of Neurology. Practice parameter: therapies for benign paroxysmal positional vertigo (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2008;70:2067 with permission).
(See Video 10–1) The Semont maneuver (Fig. 10–4) may be equally effective, although there have been fewer controlled studies using this maneuver.32 The key feature of the particle repositioning maneuvers is to move the patient in the plane of the posterior semicircular canal to allow the clot of debris to rotate around the canal and enter the utricle.
The Epley maneuver is among the most efficacious interventions in all of clinical medicine. The maneuver has been tested in numerous randomized placebo (i.e., sham pro- cedures) controlled trials. Trial quality has been rigorously scrutinized independently by the Cochrane Collaboration,8 the American Academy of Neurology Quality Standards Subcommittee,32 a multidisciplinary guideline development panel chosen by the American Academy of Otolaryngology–Head and Neck Surgery Foundation,44 and other independent groups.45,46 The summary results of all the valid randomized controlled trials indicate that the Epley maneuver has a large effect size in treating patients with BPV. In these studies, 61%–80% of patients treated with the maneu- ver had resolution of BPV compared with only
10%–20% of patients in the control groups with outcomes measured from 1 day to 1 month. These effect sizes translate into a “number needed to treat” (NNT) of 1.43 to 2.44. The NNT is a statistical measure that indicates the number of patients that had to have treatment to achieve the beneficial outcome in one patient. Thus, approximately two patients with BPV require treatment with the Epley maneu- ver to eliminate the symptoms in one patient; this is among the largest effect sizes achievable in clinical medicine, and it is particularly impressive considering that the outcome used was elimination of symptoms (i.e., cure of symptoms) as opposed to only an improvement in symptoms.
Despite the large effect size of the Epley maneuver for BPV, the maneuver appears to be substantially underutilized in routine prac- tice. Of patients with BPV who presented to a physician, only 27% reported undergoing diag- nostic positional testing (i.e., the Dix-Hallpike test) and only 10% underwent a particle repo- sitioning maneuver of any type.31
It is doubtful that physicians are just instruct- ing patients to avoid provocative positions and await spontaneous resolution because about
75% of patients with BPV who present to a physician end up having at least one diagnostic test ordered, including imaging studies, ves- tibular tests, an audiogram, and blood tests.31,47 Although most patients are cured after a single particle repositioning maneuver, the cure rate is improved by repeating the proce- dure until no vertigo or nystagmus occurs in any position. Occasionally a patient will develop severe nausea and have to be rescheduled and premedicated with a vestibular suppressant drug. Although not routinely recommended, occasionally, vibration applied to the mastoid region is useful if, rather than a burst of nystag- mus with position change, the patient develops a slow, persistent nystagmus, suggesting that the otolithic debris is stuck to the wall of the semicircular canal or to the cupula and not freely moving. A sign that the modified Epley maneuver is going to be successful is the pro- duction of a second identical burst of positional nystagmus when the patient is moved from the initial head-hanging position across to the opposite head-hanging position.48 This indi- cates that the particles are moving along in the canal in the correct direction toward the utri- cle. If, on the other hand, a burst of nystagmus in the reverse direction occurs when moving from one head-hanging position to the other, the particles are most likely moving in the wrong direction back toward the cupula, a sign that the particle repositioning maneuver will be unsuccessful. When this occurs, the patient most likely elevated the head while rolling over from the head-hanging position to the other side because this can cause the particles to move back in the opposite direction because of gravity. It is critical that the head stay down during this phase of the positioning maneuver. When returning to the sitting position at the end of the particle repositioning maneuver, patients may have a brief but severe burst of vertigo (falling sensation) as late as 30 min after assuming the position.49 This delayed vertigo in the sitting position presumably occurs as the bolus of otolithic debris drops out of the common crus of the posterior and anterior semicircular canals into the utricle. Rarely, patients will develop a persistent vertigo and nystagmus after returning to the sitting posi- tion. This phenomenon might result from a jamming of the otolithic debris (a canalith jam) when migrating from a wider to narrower seg- ment, such as from the ampulla to the canal or
at the bifurcation of the common crus to the posterior and anterior canals.7 Repeating the particle repositioning with vibration applied to the skull (i.e., mastoid region of affected side) will usually break up the “canalith jam” and cure the BPV.
What happens to the particles once they enter the utricle? When the otoconia are sepa- rated from the gelatinous layer of the otolith membrane, they can dissolve in the endolymph fluid, be taken up into the membrane by the process of phagocytosis, or simply remain free floating within the endolymph. The calcium concentration of the endolymph seems to be critical for determining whether the otolithic debris will dissolve.50 Otolith debris may not be cleared in patients who have recurrent attacks of BPV. As noted earlier, osteopenia and osteo- porosis are more common in patients with recurrent BPV than in patients with de novo BPV.12
After performing a particle repositioning maneuver, we recommend that patients avoid all extreme head-back positions such as those mentioned earlier. Other restrictions, such as having the patient sleep propped up for a few nights or wearing a cervical neck collar, do not improve the outcome.51Antivertiginous medi- cations such as meclizine or promethazine have relatively little use in the management of patients with BPV because the acute attacks are not suppressed by these drugs; moreover, the particle repositioning maneuver is much more effective in controlling the condition. For the very rare patient with prolonged refractory BPV, one might still consider a surgical proce- dure such as the singular neurectomy or the canal plugging procedure.9,52,53 The main com- plication of these procedures is a sensorineural hearing loss, which may occur in as many as 10% of patients.
Patients who have multiple recurrences of BPV can be taught to perform the particle repositioning maneuver on their own.55,56 We regularly give our patients a diagram for per- forming the maneuver on a bed at home (e.g., Figs. 10–3 and 10–4). One study compared the efficacy of self-treatment with a modified Epley maneuver and a modified Semont maneuver and found that the response rate was signifi- cantly greater with the modified Epley maneu- ver.56 Although minor complications such as those described earlier can occur, none of them is serious and most are cured by simply
repeating the maneuver. Patients can pre- sedate themselves and often they feel more comfortable performing the maneuver on their own in the controlled environment of their bedroom. Vibration applied to the mastoid is rarely required,57 but a simple neck massage vibrator can be used if one is available. Of course, one key point that cannot be ignored is that the particle repositioning maneuver (i.e., Epley maneuver or Semont maneuver) only works for treating PC-BPV. It is of no use for treating other causes of vertigo.
OTHER VARIANTS OF BENIGN POSITIONAL VERTIGO
As noted earlier, the most common type of BPV occurs when otoconial debris are within the posterior semicircular canal, probably because this is the canal in which it is most easily trapped. However, the otoconia can enter either one of the other canals and patients do present clini- cally with BPV from these canals. After poste- rior canal BPV, the next most common variant is horizontal canal BPV. Anterior canal BPV is much less common. The clinician must also add a bit more caution when making the diagnosis of either the horizontal or anterior canal vari- ants of BPV because the patterns of nystagmus seen with these variants can be similar to pat- terns of positional nystagmus caused by lesions in the central nervous system.58,59 This is differ- ent than the clinical scenario with posterior canal BPV because the characteristic pattern of nystagmus in posterior canal BPV (i.e., a burst of upbeat, torsional nystagmus lasting <30 sec- onds, and then having the nystagmus convert to downbeat torsional nystagmus if the patient is brought directly back up to the sitting position) is not a pattern expected or ever reported to be caused by a central lesion. For the PC-BPV variant, further adding to the confidence in the bedside diagnosis is the fact it is readily cured at the bedside. On the other hand, horizontal canal BPV can at times be difficult to make an immediate impact at the bedside.
The horizontal and anterior canal variants have clinical syndromes that resolve on average more quickly than the posterior canal variant,29 likely because the debris in these canals more readily falls back out based on the anatomy. Although, in rare cases the otolithic debris in
these variants can become attached to the cupula, producing true cupulolithiasis.
It is also possible for otoconia to enter multiple canals at once, particularly after head trauma,60 which leads to more challenging approach to repositioning.
Lastly, these other variants of BPV can be produced after performing the particle reposi- tioning maneuver for the typical posterior canal variant. For example, as the debris is moved from the posterior semicircular canal, it can enter the anterior canal from the common crus or it can enter the horizontal canal after it falls into the utricle (Fig. 10–5).
Horizontal Canal Benign Positional Vertigo
The clinical history of horizontal canal BPV is similar to that of the posterior canal variant, although there are important differences.61–63 With both syndromes, positional vertigo com- monly occurs in bed, particularly when patients turn over from one side to the other. Patients with PC-BPV, however, develop vertigo when getting in and out of bed and when bending down and straightening up or extending the head backward to look up. In contrast, patients with horizontal canal BPV develop vertigo pri- marily when turning over in bed or when turn- ing the head to the side when lying back in an easy chair. Occasionally, patients with horizon- tal canal BPV experience episodes of vertigo when turning the head to the side while sitting or walking. Remissions and exacerbations com- monly occur with both types of BPV, but exacerbations are typically shorter in duration with the horizontal canal variant than with the posterior canal variant, 29,64 though recurrences may be more common in the horizontal canal variant.54 With horizontal canal BPV, patients can develop two patterns of positionally trig- gered nystagmus (i.e., “geotropic” or “apogeo- tropic”) depending on whether the canaliths are in the anterior segment or the posterior segment of the horizontal canal.
GEOTROPIC NYSTAGMUS
When the canaliths are in the posterior seg- ment (or “long arm”) of the horizontal canal, patients will develop a paroxysmal, horizontal direction–changing nystagmus that beats
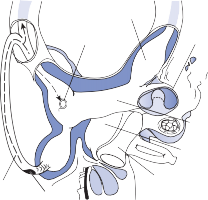
Horizontal canal
Horizontal canal ampulla
Utricle Saccule
clot moves to the bottom of the canal in the lateral position, returning to the supine posi- tion results in a movement of the clot back to its original position and a burst of nystagmus in the opposite direction. Furthermore, rotation of the head to the opposite side causes the clot to move in the opposite direction and produces ampullofugal displacement of the cupula and geotropic nystagmus.
Unlike PC-BPV, the horizontal canal variant has only minimal latency and no fatigability. With PC-BPV, latency is explained by a delay in move-
Posterior
canal Debris
Vestibule
ment of the calcium carbonate clot and fatigabil- ity by dispersion of the calcium carbonate particles with repeated positioning. There may
Figure 10–5. Drawing of the vestibular labyrinth showing
the proposed path of debris as it moves out of the posterior semicircular canal and into the anterior or horizontal canal (dashed line and arrows).
toward the ground (so-called geotropic nystag- mus) when the head is turned to the side while they are lying supine (the nystagmus will also be triggered by a body roll to the side). Thus, when the head is turned to the left side, a left- beating nystagmus is triggered. Then, when the head is turned toward the right side, a right-beating nystagmus is triggered. The nys- tagmus lasts about a minute. Though it occurs with the head to either side, it is nearly always stronger on one side (the abnormal side). The debris enters the canal side of the horizontal semicircular canal when the patient lies supine. When the head is rapidly turned to the abnor- mal side, the mass is accelerated downward in the canal. The deceleration that occurs once the lateral position is reached would normally rapidly return the cupula to the center position and there would be no post-positioning nystag- mus. Because of the continued effect of gravity on the freely floating clot, however, it contin- ues to move downward in the canal toward the ampulla until it reaches the bottom of the canal in that lateral position. Movement of the clot within the canal results in an ampullopetal deviation of the cupula and a burst of nystag- mus beating toward the ground. When the clot stops moving, the cupula returns to the primary position, with the normal time constant. The longer duration of horizontal canal variant BPV compared to the posterior canal variant is explained by the longer time constant of the horizontal vestibulo-ocular reflex (VOR) than that of the vertical VOR.65 Once the free-floating
be a viscous plug or gel in the horizontal canal, rather than a clot of calcium carbonate crystals, which could explain the lack of fatigability.61
APOGEOTROPIC NYSTAGMUS
The second pattern of nystagmus that occurs with otoconia in the horizontal canal is the “apogeotropic” (i.e., away from the ground) pattern that is caused by otoconia in the ante- rior segment of the canal.66 This again can result from otoconia adherent to the cupula (cupulolithiasis) or free floating in the anterior segment of the canal (canalithitiasis). When patients have otoconia in these regions of the horizontal canal, a head turn to the left side while lying supine triggers a right-beating (thus away from the ground, “apogeotropic”) nystag- mus. Turning the head to the right side will trigger a left-beating nystagmus. The dynamics of the buildup and decay of nystagmus depends on whether the debris is free floating or attached to the cupula and the dynamics of the horizontal VOR. When the debris is attached to the cupula, the stimulus is constant accelera- tion, which causes a gradual buildup of slow- phase velocity determined by the dominant time constant, defined as the time it requires for the response to reach 63% of maximum. An average time constant of the horizontal VOR in normal subjects is in the range of 12–20 sec. The gradual decay in slow-phase velocity after reaching a peak response can be explained on the basis of central VOR adaptation.
TREATMENT OF HORIZONTAL BPV
Unlike treatment for the posterior canal variant, there are no high-level randomized controlled

![]()
2
2





1
3
3
5 7
6

Figure 10–6. Maneuver for treating the horizontal canal variant of benign positional vertigo. While lying supine with the head turned toward the affected ear (right side in this case) the patient rolls across toward the unaffected side (1–3); the patient then continues the roll by moving through the prone position to the right lateral position (3–5); finally the patient returns to the sitting position (6–7). (From Fife TD et al. Quality Standards Subcommittee, American Academy of Neurology. Practice parameter: therapies for benign paroxysmal positional vertigo (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2008;70:2067 with permission).
trials of positional treatments for the horizontal canal variant.32 However, there are several different positional treatments that suggest efficacy in cohort and case series designs. In general, it is easier to treat HC-BPV when the otoconia are in the posterior segment (render- ing geotropic positional nystagmus) because the particles are closer to the exit of the canal to the utricle and because they are less likely to be adherent to the cupula. In fact, when the patient is identified as having apogeotropic nys- tagmus, the first goal in repositioning is gener- ally to move the otoconia from the anterior segment to the posterior segment, thus convert- ing the apogeotropic nystagmus to a geotropic nystagmus.
For patients with geotropic pattern of nys- tagmus, one commonly used maneuver is the “barbeque” maneuver (Fig. 10–6). With the
“barbeque” maneuver, the patient is rotated in the plane of the horizontal canal.67–69 The patient starts in the supine position and is rolled 90 degrees to the normal side (the side with lesser nystagmus), then in 90-degree steps to prone, to the abnormal side, and back to the supine. Another maneuver gaining acceptance is the Gufoni maneuver. With the Gufoni maneuver70 the patient is rapidly moved from the sitting position (feet forward) to a head lat- eral position (normal ear down). The head is held in this position for about 2 minutes until the geotropic nystagmus disappears. The head is then quickly turned to the side, nose down toward the ground. After 2 minutes the patient returns to the starting position. (See Video 10–2) Finally, the “forced prolonged position” is another option. With the “forced prolonged position” the patient is instructed
to sleep overnight with the normal ear down,71 which allows the otoconia to naturally fall out of the affected canal. The forced prolonged position is typically selected if the other maneu- vers are not effective or if the patient is not tolerating the other maneuvers.
When the pattern of nystagmus is apogeo- tropic, performing the barbeque maneuver toward the affected side can convert the nys- tagmus to geotropic.72 The Gufoni maneuvers can also do this.73 Once the nystagmus is con- verted, the typical treatments for geotropic can be used. More than half of patients with apo- geotropic BPV were cured by having them lie on the side of weakest nystagmus for 2 nights.74 Barbaque rotation and the Gufoni maneuver (toward the side with weaker nystagmus) can also get the debris to move from the anterior to posterior arm and out of the horizontal canal.72,73
Anterior Canal Benign Positional Vertigo
Rarely patients with a typical history of BPV, particularly after head trauma, exhibit a tor- sional downbeat nystagmus rather than the usual torsional upbeat nystagmus on the standard Dix-Hallpike positional test.28,75,76 The positioning nystagmus otherwise has all of the typical features of posterior canal BPV— the most important of these features is the brief duration, since persistent downbeat nystagmus is a central nervous system sign. With the stan- dard Dix-Hallpike test, turning the head to the right and lowering the patient into the head- hanging right position activates the right poste- rior semicircular canal and the left anterior semicircular canal. Since the ampullae are on the opposite ends of these two vertical canals, this positional change results in ampullofugal movement (excitation) of endolymph in the posterior semicircular canal and ampullopetal movement (inhibition) of the endolymph in the contralateral anterior canal. Thus, if there were debris in the anterior canal of the contralateral ear, it would be driven back toward the cupula rather than away from the cupula, as occurs in the posterior semicircular canal. Once the final position is reached, however, debris within the canal would move downward away from the cupula of the anterior canal since the
ampullary segments of both anterior canals point downward in the head-hanging positions. The direction of the torsional component of the positioning nystagmus indicates which ear is the origin of the nystagmus.76,77 If the upper pole of the eye beats away from the ground toward the uppermost ear, then it originates from the anterior canal of the uppermost ear. If it beats toward the ground, then it originates from the anterior canal of the undermost ear. The standard particle repositioning maneuver used for treating PC-BPV on the same side is also successful for treating the anterior canal variant of BPV.76
Mimics of Benign Positional Vertigo
BPV is by far the most common cause of brief recurrent attacks of positional vertigo. But it is important to know that any patient with con- stant or prolonged vertigo will report worsening of the symptom with certain position changes. For example, if a patient has vestibular neuritis and presents with severe and constant vertigo, the patient will typically report feeling much better lying still and much worse with any movement. This history is frequently misinter- preted as a positional vertigo syndrome.
It is also important to know that positionally triggered nystagmus can be a central nervous system finding. The most common pattern of a central positional nystagmus is a persistent downbeating nystagmus, which can be misin- terpreted as nystagmus from anterior canal BPV.58 The next most common central pattern of positional nystagmus is a horizontal direction changing nystagmus, which can be identical to the patterns of nystagmus seen with horizontal canal BPV.59 Thus, when man- aging a patient with findings suggestive of either anterior or horizontal canal BPV but when the patient is not able to be effectively treated over a reasonable time frame or if there are any important inconsistencies, then a cen- tral lesion should be considered. The most common central lesions that mimic anterior and horizontal canal BPV are Chiari malforma- tions, tumors or other structural lesions involv- ing the cerebellum or peri-forth ventricular regions, or neurodegenerative disorders involv- ing the cerebellum (i.e., spinocerebellar ataxia syndromes).
Uneri A. Migraine and benign paroxysmal positional vertigo: an outcome study of 476 patients. Ear Nose Throat J. 2004;83(12):814.
Gordon CR, Levite R, Joffe V, Gadoth N. Is post-
has been no convincing report of a central lesion causing all the characteristic features of PC-BPV (i.e., a burst of upbeat torsional nys- tagmus in the head-hanging position with the duration of nystagmus being less than 30 sec- onds). Any reports that suggest this occurrence are much more likely to be an example of a common disorder (i.e., PC-BPV) co-occurring with a rare central lesion.
REFERENCES
Barany R. Diagnose von Krankheitserschirnungen im Bereiche des Otolithenapparates [in Swedish]. Acta Otolaryngol (Stockh). 1921;2:434.
Dix MR, Hallpike CS. Pathology, symptomatology and diagnosis of certain disorders of the vestibular system. Proc R Soc Med. 1952;45:341.
Schuknecht HF. Positional vertigo: clinical and experi- mental observations. Trans Am Acad Ophthalmol Otol. 1962;66:319.
Schuknecht HF. Cupulolithiasis. Arch Otolaryngol. 1969;90:113.
Hall SF, Ruby RRF, McClure JA. The mechanics of benign positional vertigo. J Otolaryngol. 1979;8:151.
Epley JM. New dimensions of benign positional ver- tigo. Otolaryngol Head Neck Surg. 1980;88:599.
Epley JM. The canalith repositioning procedure: For treatment of benign paroxysmal positional vertigo. Otolaryngol Head Neck Surg. 1992;107:199.
Hilton M, Pinder D. The Epley (canalith reposition- ing) manieuvre for benign paroxysmal positional ver- tigo. Cochrane Database Sys Rev. 2004;2:CD003162.
Parnes LR, McClure JA. Free floating endolym- phatic particles: a new operative findings during pos- terior semi-circular canal occlusion. Laryngoscope. 1992;102:988.
Vilbert D, Kompis M, Häusler R. Benign paroxys- mal positional vertigo in older women may be related to osteoporosis and osteopenia. Ann Otol Rhinol Laryngol. 2003;112:885.
Jang YS, Kang MK. Relationship between bone mineral density and clinical features in women with idiopathic benign paroxysmal positional vertigo. Otol Neurotol. 2009;30(1):95.
Jeong SH, Choi SH, Kim JY, Koo JW, Kim HJ, Kim JS. Osteopenia and osteoporosis in idiopathic benign positional vertigo. Neurology. 2009;72(12):1069.
Vibert D, Sans A, Kompis M, et al. Ultrastructural changes in otoconia of osteoporotic rats. Audiol Neurootol. 2008;13(5):293.
Jang YS, Hwang CH, Shin JY, Bae WY, Kim LS. Age- related changes on the morphology of the otoconia. Laryngoscope. 2006;116(6):996.
Ishiyama A, Jacobson KM, Baloh RW. Migraine and benign positional vertigo. Ann Otol Rhinol Laryngol. 2000;109:377.
traumatic benign paroxysmal positional vertigo different from the idiopathic form? Arch Neurol. 2004;61(10):1590.
Giacomini PG, Ferraro S, Di Girolamo S, Villanova I, Ottaviani F. Benign paroxysmal positional vertigo after intense physical activity: a report of nine cases. Eur Arch Otorhinolaryngol. 2009;266(11):1831.
Vibert D, Redfield RC, Häusler R. Benign paroxysmal positional vertigo in mountain bikers. Ann Otol Rhinol Laryngol. 2007;116(12):887.
Aksoy S, Sennaro lu L. Benign paroxysmal positional vertigo in swimmers. Kulak Burun Bogaz Ihtis Derg. 2007;17(6):307.
Viccaro M, Mancini P, La Gamma R, De Seta E, Covelli E, Filipo R. Positional vertigo and cochlear implantation. Otol Neurotol. 2007;28(6):764.
Harada Y. Metabolic disorder, absorption area and for- mation area of the statoconia. J Clin Electron Microsc. 1982;15:1.
Chiarella G, Leopardi G, De Fazio L, Chiarella R, Cassandro C, Cassandro E. Iatrogenic benign par- oxysmal positional vertigo: review and personal experience in dental and maxillo-facial surgery. Acta Otorhinolaryngol Ital. 2007;27(3):126.
Aydin E, Akman K, Yerli H, Ozluoglu LN. Benign par- oxysmal positional vertigo after radiologic scanning: a case series. J Med Case Reports. 2008;2:92.
von Brevern M, Seelig T, Neuhauser H, Lempert
T. Benign paroxysmal positional vertigo predomi- nantly affects the right labyrinth. J Neurol Neurosurg Psychiatry. 2004;75(10):1487.
Korres SG, Papadakis CE, Riga MG, Balatsouras DG, Dikeos DG, Soldatos CR. Sleep position and lateral- ity of benign paroxysmal positional vertigo. J Laryngol Otol. 2008;122(12):1295.
Honrubia V, Baloh RW, Harris MR, Jacobson KM. Paroxysmal positional vertigo syndrome. Am J Otol. 1999;20:465.
Baloh RW, Honrubia V, Jacobson K. Benign positional vertigo: clinical and oculographic features in 240 cases. Neurology. 1987;37:371.
Imai T, Ito M, Takeda N, et al. Natural course of the remission of vertigo in patients with benign paroxysmal positional vertigo. Neurology. 2005;64: 920.
Neuhauser HK, von Brevern M, Radtke A, et al. Epidemiology of vestibular vertigo: a neurotologic survey of the general population. Neurology. 2005;65: 898.
von Brevern M, Radtke A, Lezius F, et al. Epidemiology of benign paroxysmal positional vertigo: a population based study. J Neurol Neurosurg Psychiatry. 2007;78:710.
Fife TD, Iverson DJ, Lempert T, et al. Practice param- eter: therapies for benign paroxysmal positional ver- tigo (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2008;70(22):2067.
Lopez-Escamez JA, Gamiz MJ, Fernandez-Perez A, Gomez-Finana M. Long term outcome and health related quality of life in benign paroxysmal positional vertigo. Eur Arch Otorhinolaryngol. 2005;262:507.
Fife TD, Fitzgerald JE. Do patients with benign par- oxysmal positional vertigo receive prompt treatment? Analysis of waiting times and human and financial costs associated with current practice. Int J Audiol. 2005;44:50.
Oghalai JS, Manolidis S, Barth JL, et al. Unrecongized benign paroxysmal postional vertigo in elderly patients. Otolaryngol Head Neck Surg. 2000;122:630.
Harrison MD, Ozsahinoglu C. Positional vertigo. Arch Otolaryngol. 1975;101:675.
Katsarkis A, Kirkham TH. Paroxysmal positional ver- tigo: a study of 255 cases. J Otolaryngol. 1978;7:320.
Brandt T, Steddin S. Current view of the mechanism of benign paroxysmal positional vertigo: cupulolithiasis or canalithiasis? J Vestib Res. 1993;3:373.
Lanska DJ, Render B. Benign paroxysmal positioning vertigo: classic descriptions, origins of the provocative positioning technique, and conceptual developments. Neurology. 1997;48:1167.
Harbert F. Benign paroxysmal positional nystagmus.
Arch Ophthalmol. 1970;84:298.
Baloh RW, Sakala SM, Honrubia V. Benign paroxysmal positional nystagmus. Am J Otolaryngol. 1979;1:1.
Dallan I, Bruschini L, Neri E, et al. The role of high- resolution magnetic resonance in atypical and intracta- ble benign paroxysmal positional vertigo: our prelimi- nary experience. ORL J Otorhinolaryngol Relat Spec. 2007;69(4):212.
Schratzenstaller B, Wagner-Manslau C, Strasser G, Arnold W. Canalolithiasis of the superior semicircular canal: an anomaly in benign paroxysmal vertigo. Acta Otolaryngol. 2005;125(10):1055.
Bhattacharyya N, Baugh RF, Orvidas L, et al. Clinical practice guideline: benign paroxysmal positional ver- tigo. Otolaryngol Head Neck Surg. 2008;139(5 suppl 4): S47.
Woodworth BA, Gillespie MB, Lambert PR. The canalith repositioning procedure for benign positional vertigo: a meta-analysis. Laryngoscope. 2004;114: 1143.
Teixeira LJ, Machado JN. Maneuvers for the treatment of benign positional paroxysmal vertigo: a systematic review. Braz J Otorhinolaryngol. 2006;72:130.
Polensek SH, Tusa R. Unnecessary diagnostic tests often obtained for benign paroxysmal positional ver- tigo. Med Sci Monit. 2009;15:MT89.
Oh HJ, Kim JS, Han BI, Lim JG. Predicting a suc- cessful treatment in posterior canal benign paroxysmal positional vertigo. Neurology. 2007;68(15):1219.
Uneri A. Falling sensation in patients who undergo the Epley maneuver: a retrospective study. Ear Nose Throat J. 2005;84(2):82, 84.
Zucca G, Valli S, Valli P, Perin P, Mira E. Why do benign paroxysmal positional vertigo episodes recover spontaneously? J Vestib Res. 1998;8:325.
Fyrmpas G, Demitrios R, Bettina HA, et al. Are pos- tural restrictions after an Epley maneuver unneces- sary? First results of a controlled study and review of the literature. Auris Nasus Larynx. 2009;36(6):637.
Pournaras I, Kos I, Guyot JP. Benign paroxysmal posi- tional vertigo: a series of eight singular neurectomies. Acta Otolaryngol. 2008;128(1):5.
Leveque M, Labrousse M, Seidermann L, Chays
Surgical therapy in intractable benign paroxys- mal positional vertigo. Otolaryngol Head Neck Surg. 2007;136(5):693.
Sakaida M, Takeuchi K, Ishinaga H, Adachi M, Majima Y. Long-term outcome of benign paroxysmal positional vertigo. Neurology. 2003;60(9):1532.
Tanimoto H, Doi K, Katata K, Nibu KI. Self-treatment for benign paroxysmal positional vertigo of the pos- terior semicircular canal. Neurology. 2005;65(8): 1299.
Radtke A, von Brevern M, Tiel-Wilck K, Mainz- Perchalla A, Neuhauser H, Lempert T. Self- treatment of benign paroxysmal positional vertigo: Semont maneuver vs Epley procedure. Neurology. 2004;63(1):150.
Ruckenstein MJ, Shepard NT. The canalith reposi- tioning procedure with and without mastoid oscillation for the treatment of benign paroxysmal positional ver- tigo. ORL J Otorhinolaryngol Relat Spec. 2007;69(5): 295.
Yabe I, Sasaki H, Takeichi N, et al. Positional vertigo and macroscopic downbeat positioning nystagmus in spinocerebellar ataxia type 6 (SCA6). J Neurol. 2003;250:440.
Johkura K. Central paroxysmal positional vertigo: iso- lated dizziness caused by small cerebellar hemorrhage. Stroke. 2007;38:e26; 2007;78:710.
Tomaz A, Ganança MM, Ganança CF, Ganança FF, Caovilla HH, Harker L . Benign paroxysmal posi- tional vertigo: concomitant involvement of differ- ent semicircular canals. Ann Otol Rhinol Laryngol. 2009;118(2):113.
McClure JA. Horizontal canal BPV. J Otolaryngol. 1985;14:30.
Pagnini P, Nuti D, Vannuchi P. Benign paroxys- mal positional vertigo in the horizontal canal. ORL J Otorhinolaryngol Relat Spec. 1989;51:161.
Baloh RW, Jacobson K, Honrubia V. Horizontal semi- circular canal variant of benign positional vertigo. Neurology. 1993;43:2542.
Chung KW, Park KN, Ko MH, et al. Incidence of hori- zontal canal benign paroxysmal positional vertigo as a function of the duration of symptoms. Otol Neurotol. 2009;30(2):202.
Baloh RW, Richman L, Yee RD, Honrubia V. The dynamics of vertical eye movements in normal human subjects. Aviat Space Environ Med. 1983;54:32.
Baloh RW, Yue Q, Jacobson KM, Honrubia V. Persistent direction-changing positional nystagmus: another variant of benign positional nystagmus? Neurology. 1995;45:1297.
Lempert T. Horizontal benign positional vertigo [let- ter]. Neurology. 1994;44:2213.
Baloh RW. Reply to the letter by Lempert: horizontal benign positional vertigo. Neurology. 1994;44:2214.
Escher A, Ruffieux C, Maire R. Efficacy of the bar- becue manoeuvre in benign paroxysmal vertigo of the horizontal canal. Eur Arch Otorhinolaryngol. 2007;264(10):1239.
Francesco R, Francesco D, Salvatore G, Gautham K, Rosalia G, Riccardo S. Management of benign paroxysmal positional vertigo of lateral semicircu- lar canal by Gufoni’s manoeuvre. Am J Otolaryngol. 2009;30(2):106.
Nuti D, Agus G, Barbieri M, Passali D. Management of horizontal–canal paroxysmal positional vertigo. Acta Otolaryngol (Stockh). 1998;118:455.
Appiani GC, Catania G, Gagliardi M, Cuiuli G. Repositioning maneuver for the treatment of the
apogeotropic variant of horizontal canal benign par- oxysmal positional vertigo. Otol Neurotol. 2005; 26(2):257.
Boleas-Aguirre MS, Perez N, Batuecas-Caletrio A. Bedside therapeutic experiences with horizontal canal benign paroxysmal positional vertigo (cupulolithia- sis). Acta Otolaryngol. ePub ahed of print, May 20, 2009:1.
Chiou WY, Lee HL, Tsai SC, Yu TH, Lee XX. A single therapy for all subtypes of horizontal canal positional vertigo. Laryngoscope. 2005;115(8):1432.
Jackson LE, Morgan B, Fletcher JC, Jr., Krueger WW. Anterior canal benign paroxysmal positional
vertigo: an underappreciated entity. Otol Neurotol. 2007;28(2):218.
Tusa R, Herdman S. Assessment and treatment of anterior canal benign paroxysmal positional vertigo using the canalith repositioning maneuver (CRM). Neurology. 1997;48:A384.
Imai T, Takeda N, Ito M, Nakamae K, Sakae H, Fujioka H, Kubo T. Three-dimensional analysis of benign paroxysmal positional nystagmus in a patient with anterior semicircular canal variant. Otol Neurotol. 2006;27(3):362.
This page intentionally left blank
![]()
BACKGROUND OCCURRENCE CLINICAL FEATURES PATHOPHYSIOLOGY ANIMAL MODELS ETIOLOGY
Genetics
Migraine and Meniere’s Syndrome
Infection/Autoimmune
DIAGNOSIS
Audiometric Testing Vestibular Testing Imaging MANAGEMENT
Medical Management Surgical Management
When Prosper Meniere described a series of patients with hearing loss and vertigo in 1861, he was not attempting to define a disease but rather to convince his medical colleagues that vertigo could be a symptom of damage to the inner ear.1,2 Prior to this time, vertigo was con- sidered to be a cerebral symptom and was lumped together with epileptic seizures and stroke under the rubric of “apoplectiform cere- bral congestion,” a condition thought to result from overfilling of blood vessels in the brain.2 To support his argument, Meniere described the autopsy findings in a young girl who died 5 days after experiencing the sudden onset of hearing loss and vertigo. Meniere observed a red plastic material, in the child’s inner ear, a bloody exudate filling the semicircular canals. In retrospect, this likely represented a case of acute leukemia with hemorrhage into the inner ear. It provided convincing evidence that dam- age to the semicircular canals of the inner ear could cause vertigo. It would take almost another century before English and Japanese
researchers independently reported hydrops of the inner ear in autopsy specimens from patients with the typical symptom triad of modern-day Meniere’s disease.3,4 However, because of Meniere’s description of the young girl’s autopsy in his classical paper, the concept that Meniere’s disease was caused by hemorrhage into the inner ear persisted well into the twen- tieth century. Meniere was not suggesting that the patients he described with episodic vertigo and fluctuating hearing loss had the same dis- ease as the young girl with sudden deafness and vertigo. He was simply trying to make the point that vertigo and hearing loss commonly occur together with inner ear disease.
The clinical profile of Meniere’s disease evolved over the years from a nonspecific description of just about any combination of hearing loss and vertigo to a characteristic clin- ical triad of fluctuating hearing loss, tinnitus, and vertigo. Once a characteristic pathology— endolymphatic hydrops—was discovered in the late 1930s,3,4 the next question was whether patients with the typical clinical profile consistently showed this pathology. As more
273
and more pathological specimens have been reported, the answer to this question is, usually but not always.5–8 Complicating matters fur- ther, endolymphatic hydrops can be found in ears from patients without the typical symp- toms of Meniere’s disease and, occasionally, even in patients without ear symptoms.8–10 Furthermore, it has become apparent that Meniere’s syndrome and the characteristic inner ear pathology can be produced by several different etiologies, including infections (syph- ilitic, bacterial, and viral) and after traumatic and metabolic injuries to the inner ear. Some suggest using the term Meniere’s syndrome to describe the broad spectrum of disorders with the characteristic clinical triad regardless of cause, while restricting the use of Meniere’s disease to describe only those cases without an identifiable cause (idiopathic). However, as we will see, there are undoubtedly multiple etiolo- gies for the idiopathic group so that the separa- tion between Meniere’s syndrome and Meniere’s disease is arbitrary and likely to change constantly. For this reason, we prefer the general term Meniere’s syndrome to describe the classical clinical profile of fluctuat- ing hearing loss, tinnitus, and vertigo. When a cause can be determined, it can be described as Meniere’s syndrome due to infection or autoimmune disease, for example, and if no cause can be found, Meniere’s syndrome of unknown cause.
It is difficult to obtain accurate measures of the prevalence and incidence of Meniere’s syn- drome because of problems with a definitive diagnosis and patient selection. In 1984, Wladislavosky-Wasserman et al.11 reviewed the data of the Mayo Clinic in Rochester, Minnesota, and estimated a prevalence of 218 per 100,000 and an incidence of 15 per 100,000. In 2005, Shojaku et al.12 reported an average annual prevalence and incidence of 34.5 and 5.0, respectively, per 100,000 population in the Nishikubiki district of Japan. Figures for the incidence of Meniere’s syndrome in European countries varies from a low of 4 per 100,000 in Finland13 to as high as 157 per 100,000 in Britain.14 A recent study from Germany found a population prevalence of 0.12%.15 It affects
all races and is recognized in all countries. Kitahara et al.16 found that black Americans were affected only about half as often as white Americans. Although some studies have found a female preponderance among Meniere’s patients, others have not, and the slight differ- ences that have been reported can probably be attributed to patient selection. A Swedish study of 356 patients with advanced Meniere’s syn- drome found a preponderance of males over females,17 whereas a Japanese study found a slight female preponderance.18
There is general agreement that Meniere’s syndrome is a disease of middle age.19 The mean age of onset is remarkably consistent around the age of 40, with the mean age at diagnosis being closer to 50. Meniere’s syn- drome can occur in children, but it is rare. Initial audiograms in children often show a high-frequency hearing loss rather than the low-frequency loss seen in adults.20 In the material from the Mayo Clinic study, no patients with Meniere’s syndrome were under the age of 15 years.11
There is a good deal of confusion and con- troversy regarding when and how often Meniere’s syndrome becomes bilateral. Thomas and Harrison21 followed 610 cases of Meniere’s syndrome and noted that those cases that were destined to become bilateral invari- ably developed within 5 years of presentation. In contrast, Friberg and colleagues22 monitored 161 patients with repeated examinations and noted an almost linear increase in the number of bilateral cases from only a few percent at onset to almost 50% in those who had been fol- lowed for as long as 30 years. In another study with long-term follow-up, Kitahara et al.23 reported development of bilateral involvement in 41% of cases. In the Mayo Clinic study, about a third of patients developed bilateral involvement.11 Complicating matters further, the contralateral ear may show audiometric changes without overt symptoms.24,25
A major problem in interpreting all of these studies on the epidemiology of Meniere’s syn- drome is the lack of uniform criteria for diag- nosis and the likelihood that many different etiologies are being included in the patient groups being studied. For example, it is possi- ble that certain causes of Meniere’s syndrome might be more prevalent in one sex than the other, have a different racial distribution, and be more likely to become bilateral. Until there
are diagnostic markers that identify specific categories of Meniere’s syndrome, it will be very difficult to obtain accurate statistics regarding the epidemiology of the disease.
Typically, an attack of Meniere’s syndrome begins with a sensation of fullness and pressure along with decreased hearing and tinnitus in one ear. Vertigo rapidly follows, reaching a maximum intensity within minutes and then slowly subsides over the next several hours. The patient is usually left with a sense of unsteadiness and dizziness for days after the acute vertiginous episode. In the early stages the hearing loss is completely reversible, but in later stages a residual hearing loss remains. Tinnitus may persist between episodes but usually increases in intensity immediately before or during the acute episode. It is typi- cally described as a roaring sound (the sound of the ocean or a hollow seashell sound). After vomiting the patient prefers to lie in bed without eating until the acute symptoms pass. Such episodes occur at irregular intervals for years, with periods of remission unpredictably intermixed.26,27 Eventually, severe permanent hearing loss develops and the episodic nature spontaneously disappears (“burnt-out phase”).7 However, some patients can continue to be bothered by severe dizziness even after a 20-year disease history.28
Variations from this classic picture are com- mon, particularly in the early stages of the disease process, but the diagnosis remains uncertain unless the combination of fluctuating hearing loss and vertigo occurs. Isolated epi- sodes of vertigo or hearing loss often precede the characteristic combination of symptoms by months or even years. Although so-called ves- tibular Meniere’s and cochlear Meniere’s have been proposed as variations of the classic syn- drome, clinical–pathologic correlation of iso- lated vestibular and auditory disorders with selective endolymphatic hydrops of the vestib- ular and auditory labyrinth is lacking. A small percentage of patients with sudden deafness— particularly if there is associated vertigo—will later develop typical symptoms of Meniere’s syndrome, so-called delayed endolymphatic hyrdrops.29 Some patients with well-documented
Meniere’s syndrome experience abrupt epi- sodes of falling to the ground without loss of consciousness or associated neurologic symp- toms. These episodes have been called otolithic catastrophes by Tumarkin30 because of his sus- picion that they represented acute stimulation of the otoliths from the hydrops. Patients often report feeling as though they were pushed to the ground by some external force.31 These epi- sodes can be confused with drop attacks seen with vertebrobasilar insufficiency and may suggest an inaccurate diagnosis in a patient with otherwise typical symptoms and signs of Meniere’s syndrome.32
With delayed endolymphatic hydrops, the patient reports a long history of hearing loss, often since early childhood, followed years later by typical symptoms and signs of endo- lymphatic hydrops.33,34 The hydrops can develop in either ear. When it develops in the ear with longstanding hearing loss, the vertigo attacks may not be accompanied by fluctuating hear- ing loss and tinnitus, confusing the diagnosis. Schuknecht34 speculated that the initial hear- ing loss results from a viral infection that also produces subclinical damage to the resorptive mechanism (in either ear). Hydrops develops years later as the balance between endolymph secretion and resorption is disrupted. Delayed hydrops might also result from an autoimmune process triggered by antigens released from the previously damaged inner ear or from genetic causes of unilateral hearing loss.35,36
As indicated earlier, the principal pathologic finding in patients with Meniere’s syndrome is an increase in the volume of endolymph associ- ated with distension of the entire endolym- phatic system (Fig. 11–1).3,4,7 The membranous labyrinth progressively dilates until the saccu- lar wall makes contact with the stapes footplate and the cochlear duct occupies the entire vestibular scala. The cochlear and vestibular end organs and nerves show modest pathologic changes. Interestingly, even the contralateral ears show significantly more damage than the inner ears of controls.37 Membranous labyrinth herniations and ruptures are common, the latter frequently involving Reissner’s mem- brane and the walls of the sacculus, utriculus,
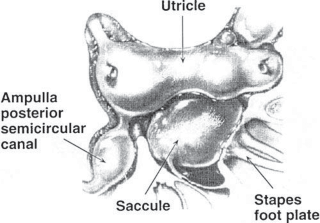
Figure 11–1. Dilated membranous labyrinth in Meniere’s syndrome. The drawing was made from a three-dimensional model developed from serial sections of the ear of a patient with Meniere’s syndrome. (Adapted from Schuknecht HF. Pathology of the Ear. Lea & Febiger, Philadelphia, 1993, with permission.)
and ampullae. Occasionally, a rupture is fol- lowed by complete collapse of the membra- nous labyrinth.
Althoughthepathologicchangesin Meniere’s syndrome have been well described, the mech- anism for its fluctuating symptoms and signs are still not completely understood. Some of the fluctuating symptoms with Meniere’s syn- drome probably result from mechanical deformation of the end organ, which is revers- ible as the endolymphatic pressure decreases. The characteristic hearing loss can be explained by increased pressure at the apex of the cochlear duct leading to displacement of the basilar membrane and altered auditory thresholds.38,39 The dramatic sudden falling attacks initially described by Tumarkin are likely due to sud- den deformation or displacement of one of the otolith organs.30,31 In addition to mechanical effects on the sensorineural elements of the inner ear, increases in endolymphatic pressure can cause symptoms by altering blood flow to the inner ear.39 Experimental and clinical data support the notion that pressure changes in the inner ear can induce ischemia that either tran- siently or permanently damages the sensory cells in the auditory and vestibular organs.40 Furthermore, hydrops can affect the neural regulation of inner ear blood flow. Hypoxic changes within the ear could lead to changes in chemistry and resting potentials, which could explain reversible episodes of auditory and ves- tibular dysfunction. Prolonged ischemia would lead to permanent damage to the sensorineural elements.40
Another possible explanation for the epi- sodes of hearing loss and vertigo is ruptures in the membranes separating endolymph from perilymph, producing a sudden increase in potassium concentration in the latter.7 If the perilymph space of animals is infused with a potassium solution, the bioelectric activity of the labyrinthine receptors is inhibited.41 When the artificial infusate is stopped, potassium is slowly cleared from the perilymph, and laby- rinthine function returns to normal in 2 to 3 hr (the typical duration of a Meniere’s attack). However, the stereotyped episodes of vertigo, tinnitus, and low-frequency hearing loss would be difficult to explain on the basis of random ruptures in the membranous labyrinth.
As discussed in Chapter 2, there are conflict- ing theories regarding the mechanisms in regu- lating endolymph volume.42 The longitudinal flow theory, initially proposed by Guild, holds that endolymph is secreted by the stria vascu- laris in the cochlea and dark cells in the ves- tibular labyrinth, and the endolymph gradually flows toward the endolymphatic duct and sac where it is resorbed. The radial theory assumes a local transverse and active diffusion process between endolymph and perilymph through- out the labyrinth. Finally, the dynamic theory is a combination of the two, whereby endo- lymph volume is determined by both a radial ionic diffusion and a slow longitudinal bulk process. Endolymphatic hydrops could result from an increase in production or a decrease in the resorption of endolymphatic fluid. An increased production of endolymph could
result from an increase in active transport of cations (sodium, potassium, and calcium) with their anionic pair (chloride) as well as water into the endolymph to maintain osmolality and electroneutrality.39 Since the stria vascularis of the cochlea and the dark cells of the vestibular labyrinth are the sites of maximum ion trans- port and maximum energy consumption, they are likely candidates for the site of overproduc- tion of endolymph resulting in hydrops. The adrenal hormone aldosterone controls the level of membrane-bound Na/K ATPase of the stria vascularis and dark cells.43–45 And, in turn, it controls secretion of potassium ions and endo- lymph production. Increased aldosterone lev- els induced by diet or stress could be a trigger for inducing Meniere’s attacks.42 The vasopres- sin–aquaporin 2 system is also important for water homeostasis in the inner ear just as it is in the kidney.46,47 Plasma vasopressin levels are elevated during Meniere’s attacks48 and vaso- pressin type 2 receptors are up-regulated in surgical specimens of inner ears from patients with Meniere’s syndrome.49 Whether this sys- tem is involved in the pathogenesis of Meniere’s syndrome is yet to be determined.
Movement of solutes along the scala media toward the endolymphatic duct and sac has been shown to be an extremely slow process.42 While the bulk flow of water and ions across the endolymphatic sac is minimal, the sac has been shown to be important in the extraction of debris and breakdown of macromolecules as well as secretion of macrophages and immune cells. Lesions that obstruct the endolymphatic duct or sac can cause typical symptoms of Meniere’s syndrome.50
Endolymphatic hydrops can be reliably pro- duced in animals by either decreasing endo- lymph absorption or increasing secretion. Since the initial studies in guinea pig by Kimura and Schuknecht,51 endolymphatic hydrops has been produced in several animal species by surgical obliteration of the endolymphatic duct and sac.52,53 Injecting foreign substances such as lipopolysaccharides into the middle ear can also produce hydrops possibly by inducing endolymphatic sac dysfunction.54 Over- production models include injecting cholera
toxin into the inner ear55 and long-term admin- istration of alderosterone or vasopressin.56,57 A problem with all of these animal models is that they produce hydrops and hearing loss but no attacks of vertigo. Thus, they may be producing the interictal phase of Meniere’s syndrome but not the ictal phase. Recently, Takumada et al. produced a mouse model with episodic vestib- ular dysfunction by using a combination of intratympanic injections of lipopolysaccharides and intraperitoneal injections of aldersterone.53 Intratympanic injections of epinephrine caused episodes of vestibular dysfunction in these ani- mals but not in controls or in animals that just received lipopolysaccharides or aldosterone. This suggests that an additional “stressor” is required to trigger episodes of vestibular dys- function with endolymphatic hydrops.
Genetics
As noted earlier, the incidence of Meniere’s syndrome varies between populations in differ- ent continents, suggesting that genetic factors may be important in the pathophysiology. Between 5% to 15% of patients with Meniere’s syndrome report a first-degree relative with Meniere’s syndrome.58,59 Morrison et al. reported 46 families from the United Kingdom with at least two affected family members (27 families with two affected members, 12 fami- lies with three affected members, 6 families with four affected members, and 1 family with five affected members).58 The mode of trans- mission was most consistent with autosomal dominant with reduced penetrance. Frykholm et al.60 reported a large Finnish family with Meniere’s syndrome in five generations. A genome-wide linkage scan of the family identi- fied five candidate regions with a lod score greater than one. Two additional Finnish fami- lies with autosomal dominant Meniere’s syn- drome were analyzed for linkage to these regions and a cumulative lod score of 3.5 was obtained for a single region on chromosome 12p.61 Two of the three families shared a
1.7 Mb haplotype in the region, suggesting a common ancestral origin. However, so far no mutations have been found on chromosome 12p. Since mutations in the COCH gene can
produce a syndrome that is similar to Meniere’s syndrome, two groups screened the COCH gene for mutations in patients with Meniere’s syndrome but found no mutations.62,63 Single nucleotide polymorphism (SNP) analysis of candidate genes found associations between Meniere’s syndrome and variants in the potassium channelgenes KCNE1 and KCNE364 and host cell factor C1 gene,65 but these find- ings have not been replicated in other patient populations.
Migraine and Meniere’s Syndrome
Speculation on an association between migraine and Meniere’s syndrome dates back to the ini- tial description by Meniere.1,2 Numerous stud- ies have found an increased prevalence of migraine in patients with Meniere’s syndrome compared to controls.66,67 Whether migraine mimics Meniere’s syndrome or causes Meniere’s syndrome is yet to be determined. Since there is a major genetic component to migraine, the small percentage of patients with familial Meniere’s syndrome could have an inherited migraine/Meniere’s syndrome.68 We compared the clinical features of patients with Meniere’s syndrome and migraine with those who had Meniere’s syndrome alone.69 Age of onset was earlier and bilateral symptoms and signs were significantly more common in migraine/Meniere’s syndrome compared with Meniere’s syndrome alone. Forty percent of patients with migraine/Meniere’s syndrome had a family history of episodic vertigo compared to 2% of patients with Meniere’s syndrome alone. Inherited metabolic abnormalities shared by brain and inner ear could explain the combina- tion of brain and ear symptoms in patients with migraine/Meniere’s syndrome.
Infection/Autoimmune
Bacterial, viral, and syphilitic infections can lead to endolymphatic hydrops and typical symptoms and signs of Meniere’s syndrome. The hydrops presumably results from damage to the fluid resorption mechanism due to inflammation and scarring of the endolym- phatic duct and sac. Paparella and Djalitian found that 75 of 194 temporal bones from patients with a history of otitis media showed
typical pathological features of endolymphatic hydrops.70 Bacterial endotoxins placed on the middle ear of chinchillas permeate the round window and cause inflammatory cell recruit- ment, strial swelling, and sensory cell degen- eration.71 Overt or subclinical viral infections could damage the resorptive mechanisms of the inner ear, leading to an eventual decom- pensation in the balance between secretion and absorption of endolymph.7,72 Infections in utero may lead to developmental hypoplasia of the endolymphatic duct and sac predisposing to later development of Meniere’s syndrome. Autoimmune injury to the endolymphatic sac may play an important role in the pathogenesis of Meniere’s syndrome.73 The endolymphatic sac is the only site that contains immuno- competent cells in the inner ear and is capable of mounting an active immune response if appropriately stimulated.
Audiometric Testing
The key to the diagnosis of Meniere’s syndrome is to document fluctuating hearing levels in a patient with the characteristic clinical history. A shift of more than 10 to 15 dB at two differ- ent frequencies is required.74 In the early stages, the sensorineural hearing loss typically affects only the low frequencies (low-frequency trough) (Fig. 11–2). This is followed by a “peaked pattern” with a prominent low- frequency hearing loss, best hearing at 2000 Hz, and a fall-off again in the higher frequencies.75,76 The fluctuation in hearing levels is usually most prominent in the low frequencies. Speech dis- crimination is relatively preserved and recruit- ment often occurs consistent with a cochlear site of dysfunction. Brainstem auditory-evoked response (BAER) and stapedius reflex mea- surements are typically normal. Otoacoustic emissions are often abnormal in patients with Meniere’s syndrome, particularly as the disease evolves.77 If the hearing loss is >30 or 40 dB at a given frequency, then the otoacoustic emis- sions are absent at that frequency, consistent with a cochlear origin for the hearing loss. Smaller amplitude otoacoustic emissions in the opposite “normal ear” may be an early indica- tor of bilateral involvement.78
125 250 500 1000 2000 4000 8000
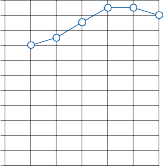
0
10
20
30
40
125 250 500 1000 2000 4000 8000
0
10
20
30
40
50
60
70
Hearing level in dB
Hearing level in dB
80
90
100
110
6/1976
SRT - 20 dB
Discrim - 100%
50
60
70
80
90
100
110
8/1978
SRT - 35 dB
Discrim - 85%
0
10
20
30
40
50
60
70
80
90
100
12/1980
SRT - 50 dB
Discrim - 58%
0
10
20
30
40
50
60
70
80
90
100
4/1984
SRT - 90 dB
Discrim - 0%
110
125 250 500 1000 2000 4000 8000
110
125 250 500 1000 2000 4000 8000
Frequency (Hz)
Figure 11–2. Series of audiograms from a patient with Meniere’s disease. This patient’s hearing loss was initially fluctuant. With time, the loss became permanent and progressive. SRT, speech reception threshold.
Electrocochleography (ECoG) is a clinical test that was developed to aid in the diagnosis of Meniere’s syndrome, and the test has also added insights about the underlying pathophys- iological process.79 With ECoG, the cochlear microphonic (CM), summating potential (SP), and the eighth nerve compound action poten- tial (AP) are measured in response to brief acoustic stimulation (clicks, tone pips, and tone bursts) (see Fig. 8–5 in Chapter 8). Summating potential is superimposed on CM during steady-state pure-tone stimulation such as with a tone burst. It can be positive or negative depending on the frequency and intensity of the pure tone. The AP is the synchronized response of the auditory nerve to the transient acoustic stimulation. In patients with Meniere’s syndrome, the SP/AP ratio (amplitude or area under the curve) is often increased, largely because of an increase in SP. Large SPs are rarely seen with other types of sensorineural hearing loss, so the finding is relatively specific for Meniere’s syndrome.75 On the other hand,
an abnormal ECoG is not very sensitive for Meniere’s syndrome. About a third of patients with definite Meniere’s syndrome have normal ECoGs.80 An elevated SP/AP ratio is helpful for identifying patients with sudden low frequency hearing loss who will go on to develop Meniere’s syndrome.29,81 Overall, ECoG is not widely used as a clinical tool because it has not proved to be a valid tool for discriminating endolym- phatic hydrops from other causes of recurrent dizziness at the time of clinical uncertainty.
Many patients with Meniere’s syndrome note that their symptoms fluctuate with the amount of fluid intake. Dehydration of the patient, most commonly achieved with the glycerol test, can transiently reverse the low-frequency hearing loss in many patients with Meniere’s syndrome (particularly those who are experiencing fluctuating hearing loss).75 This test has not received general acceptance, however, because some patients (<10%) will develop severe headaches with or without nausea and vomiting during the testing.
Other dehydrating agents such as urea, furo- semide, or mannitol have fewer adverse effects, but they are also less effective in elevating plasma osmolality. A minimum increase in osmolality of 10 millios-moles/kg is necessary for a valid test. The test is typically conducted with an oral dosage of glycerol of 1.5 ml/kg body weight in the fasting state together with an equal volume of water and a flavoring agent. The maximum dehydration occurs about 90 min after the glycerol is ingested, which is the best time to check for a change in hearing on audiometry. One must monitor blood osmo- lality since, even with this standard dosage, between 10% and 15% of patients will not reach the critical change in osmolality. A posi- tive glycerol test requires a 15 dB pure-tone change and/or a 15% discrimination gain. A small percentage of patients actually have a worsening of hearing after glycerol, possibly because of glycerol entering the scala media and attracting fluid and thereby increasing the degree of hydrops.
Vestibular Testing
Electronystagmography may reveal a periph- eral spontaneous nystagmus and either a ves- tibular paresis or directional preponderance on caloric testing. The degree of caloric paresis increases with the duration of disease, eventu- ally stabilizing at approximately 50% of the normal functional level. In one study, after 7 years of follow-up, 65% of patients had a reduced response, rising to 75% after 20 years.82 A complete loss of caloric responses is unusual with Meniere’s syndrome. Both VEMP and head-thrust tests can be abnormal with Meniere’s syndrome, but abnormalities on these tests are less frequent than caloric abnor- malities.83,84 It is more typical that the head- thrust test is normal with Meniere’s syndrome unless a destructive procedural has been per- formed.85 During an acute attack of Meniere’s syndrome, the nystagmus may be directed toward the involved ear, but this “wrong- direction nystagmus” may be a reversal phe- nomena due to central compensation, since if the episodes are monitored from the begin- ning, they typically begin with nystagmus in the opposite direction (i.e., fast component toward the good ear).86
Imaging
Radiologic studies of the temporal bones in patients with Meniere’s syndrome may show narrowing of the endolymphatic duct or decreased pneumatization of the temporal bone.87 However, these features are also seen in normal subjects and therefore are of limited value in the diagnosis of Meniere’s syndrome. On computed tomography (CT) scan, the endolymphatic duct and sac may be more dif- ficult to visualize in patients with Meniere’s syndrome than in controls, but this is not a reli- able enough finding to be of diagnostic value. Contrast enhancement of the endolymphatic duct and sac on magnetic resonance imaging (MRI) has been reported in a few patients with Meniere’s syndrome, but again, this is not a consistent finding.88 MRI (3T) of endolymph and perilymph after intratympanic administra- tion of gadolinium is a promising although invasive technique for identifying endolym- phatic hydrops.88–90
Medical Management
SYMPTOMATIC TREATMENT OF ACUTE SPELLS
Because the cause of Meniere’s syndrome is usually unknown, treatment is empiric. Medical management consists of symptomatic treatment of the acute spells (Table 11–1, also see Table 19–2 in Chapter 19) and long-term prophylaxis with salt restriction and diuretics. Phenergan at 25 to 50 mg, orally or via suppository, is usually effective for relieving the acute vertigo, nausea, and vomit- ing. It should be taken as soon as possible, pref- erably during the prodrome if there are reli- able warning symptoms. Intravenous diazepam or droperidol may be required for severe attacks with repeated vomiting. Meclizine is often adequate for milder attacks. Antiemetics such as prochlorperazine (Compazine) are sometimes useful if nausea and vomiting are severe. Hearing can be improved with aids that can be manually adjusted as hearing fluctuates.91
Table 11–1 Dosage and Effects of Commonly Used Antivertiginous Medications
Class | Drug | Dosage | Sedation | Anti- emetic Actions | Dryness of Mucous Membranes | Extra- pyramidal Symptoms |
Anticholinergic | Scopolamine | 0.6 mg orally | + | + | +++ | − |
q4–6 h or
0.5 mg transdermally q3d
Monoamingergic Amphetamine 5 or 10 mg
orally q4–6h
Ephedrine 25 mg orally q4–6h
− + + +
− + + –
Antihistamine Meclizine
(Antivert)
Dimenhydrinate (Dramamine)
Promethazine (Phenergan)
25 mg orally q4–6h
25 mg orally q4–6h
25–50 mg orally or IM q4–6h or 25–100 mg suppository q8h
+ + + −
+ + + −
++ ++ + −
Phenothiazine Prochlorperazine 5 or 10 mg orally + +++ + +++
(Compazine)
Benzodiazepine Diazepam
(Valium)
![]()
IM, intramuscularly; IV, intravenously.
PROPHYLAXIS
or IM q6h or 25 mg suppository q12h
5 or 10 mg orally, IM, or IV
q4–6h
+++ + − −
patients. We have seen patients who had had
No high-level evidence has demonstrated an average beneficial effect of medical interven- tions for Meniere’s disease. The main problem is that there is a lack of high-quality randomized trials rather than negative results from adequate trials. Importantly, there are several options for medical management that are very reasonable to try even though high-level evidence is lack- ing, because these treatments have a very low risk of harm to patients and also very low cost.
One of the first options to consider is an aggressive low-salt diet. The mechanism by which a low-salt diet may decrease the fre- quency and severity of attacks with Meniere’s syndrome is unclear, but there is some empiric evidence for its efficacy.92,93 Though no average beneficial effect has been demonstrated by randomized controlled trials,94 a low-salt diet seems to have a dramatic effect in some
severe disabling episodes on a weekly basis have prolonged remissions (years) on a low-salt diet. Other patients show little or no improve- ment, possibly reflecting the multifactorial pathogenesis of Meniere’s syndrome, though differences in adherence to the diet or placebo effects are also possible explanations. Since there are no common risks associated with a low-salt diet (other than inconvenience to the patient), we recommend salt restriction as the first step in any patient felt to have Meniere’s syndrome. An adequate trial of a low-salt diet is restricting the salt to about 1 to 2 g/day for a minimum of 2 to 3 months, which is not an easy task. If a good response is obtained, then the level of salt intake can be gradually increased while symptoms and signs are carefully moni- tored. Fluid and food intake should be regu- larly distributed throughout the day, and binges
(particularly foods with high sugar and/or salt content) should be avoided. Occasionally, patients will notice that certain foods (e.g., alcohol, coffee, chocolate) may precipitate attacks. Diuretics (e.g., hydrochlorothiazide, 50 mg two times a day) may provide additional benefit in some patients, although there also have been no adequately controlled studies to demonstrate an average beneficial effect.95 When attacks reliably occur at the time of the menstrual period, a diuretic can be started 4–5 days before menses and discontinued after menses. Acetazolamide is a carbonic anhydrase inhibitor that has long been used to lower ocu- lar pressure with glaucoma. Some patients on acetazolamide report an improvement in symp- toms. How it potentially works in Meniere’s syndrome is unknown, although it may decrease endolymph production. Acetazolamide is also known to decrease cerebrospinal fluid (CSF) secretion and was shown to decrease the osmotic pressure of the inner ear in experi- mental endolymphatic hydrops in guinea pigs.96 An average trial dose of acetazolamide for treating Meniere’s syndrome is 250 mg twice a day.97 Prophylactic use of betahistine has also been reported to decrease the frequency of Meniere’s attacks in an open, nonmasked trial,98 but as with other medical treatment options adequate trial data are lacking.99
Surgical Management
SHUNTS
Two different types of surgery have been used for treating Meniere’s syndrome: endolym- phatic shunts and destructive procedures. Although shunts are logical, based on the pre- sumed pathophysiology of Meniere’s syndrome, several factors limit the probability of achieving a functional shunt with this disorder.100 The most popular shunt procedure at the present time is used to drain the endolymphatic sac to the mastoid cavity. A major conceptual prob- lem with this procedure is that pathologic stud- ies of temporal bones in patients with Meniere’s syndrome show evidence of blockage of the endolymphatic pathways proximal to the endo- lymphatic sac. Furthermore, Schuknecht91 pointed out that any drain device that is implanted in the endolymphatic sac will almost certainly become rapidly encapsulated in
fibrous tissue. Revision operations and tempo- ral bone studies in patients who have had shunts implanted have shown fibrous encapsulation of shunt devices. Not surprisingly, there are con- flicting reports regarding the clinical efficacy of these surgical shunt procedures.101,102
PRESSURE PULSE TREATMENT
Recently a minimally invasive device was intro- duced for the treatment of Meniere’s disease. The device, called the Meniett device, uses a pulse generator to provide positive pressure into the ear canal. A tympanostomy tube is required so that the pressure is transferred into the middle ear. The idea for this sort of treat- ment was based on observation that changes in ambient pressure improve Meniere’s disease symptoms103; however, the mechanism by which the external pressure works to reduce vertigo attacks is not clear. One theory is that the intermittent pressure could decrease endo- lymphatic fluid volume by forcing outflow into the endolymphatic sac. A randomized con- trolled trial of the Meniett device found a ben- eficial effect in patients who had at least two vertigo attacks per month for the 2 months prior to enrollment.104 The results indicate that the control group had vertigo attacks on 13% of the days over the 4 months, whereas the treat- ment group had vertigo attacks on 7% of the days (p = 0.048). However, all of the benefit of the device was seen in the first 3 months because in the fourth month the frequency of vertigo attacks in the control group dropped down to that of the treatment group. The authors of a longer term follow-up study of the device argue that there is a beneficial effect out to 2 years, but this study was uncontrolled, had a high dropout rate, and also problems with compliance with the device.105 As a result, pres- sure pulse treatment may have an important impact in reducing the frequency of vertigo attacks, but the effect appears to be moderate and also limited to only several months. This is a common theme in Meniere’s disease treat- ment since the natural history of the disorder is that vertigo episodes decrease in frequency over time.
DESTRUCTIVE PROCEDURES
The rationale for destroying the labyrinth in treatment of Meniere’s syndrome is that the
nervous system is better able to compensate for complete loss of vestibular function than for partial loss that is fluctuating in degree. Ablative procedures can be a particularly good option in patients with unilateral involvement who have no functional hearing on the dam- aged side. This is because a unilateral severe hearing loss is the most valid indicator of the affected side and also because one of the most concerning risks of the procedure (i.e., hearing loss) is no longer a factor. Obviously, ablative procedures should not be considered if the abnormal side is not well defined or if an impor- tant level of uncertainty still exists about the cause of the vertigo attacks. Severe vertigo is expected during the immediate posttreatment period, but most patients who follow a struc- tured vestibular exercise program can return to normal activity within 1 to 3 months (see Chapter 20).106 Ablative procedures generally should be avoided in elderly patients because the elderly have great difficulty adjusting to the vestibular imbalance.
A chemical labyrinthectomy can be achieved by introducing an ototoxic drug into the middle ear, where it will be absorbed into the inner ear via the round window or the angular ligament of the round window.107 Initially, streptomycin was used, but more recently gentamicin has been the drug of choice. The advantages of this procedure are that it can be done as an outpa- tient with minimal discomfort and the ototoxic effects can be titrated over multiple treatments so that the patient may gradually compensate for the unilateral vestibular loss with only mild dizziness symptoms. The main disadvantage is the marked variability in the rate of entry of the ototoxic drug into the inner ear in different patients and thus more heterogeneity in the clinical response compared with surgical labyrinthectomy. Some patients receiving gentamicin injections may show a severe oto- toxic effect after one or two doses, whereas
with some deficiencies in the design or report- ing of the trial.
The two main types of destructive surgery are labyrinthectomy and vestibular nerve sec- tion. Labyrinthectomy is useful only when there is no functional hearing on the damaged side since any remaining hearing cannot be spared with this procedure. The purpose of a labyrinthectomy is to remove the neural epi- thelium of the vestibular end organs.110 Sectioning the vestibular nerve, or vestibular neurectomy, has the advantage of preserving hearing in patients with salvageable residual cochlear function, but the risks of complication are greater than with labyrinthectomy.111 Again, adequate randomized clinical trials of either of these procedures are lacking. Though dramatic and immediate reductions in the frequency of vertigo attacks have been demonstrated in case series reports,112 the patients selected for these procedures are generally at the severe stages of the disorder and thus may not be far off from the late stage of the disorder when the fre- quency of the vertigo attacks drops substantially based on the natural history of the disorder. As a result, it may be that destructive surgical pro- cedures improve the time to remission of ver- tigo attacks but not the long-term outcome.
REFERENCES
Atkinson M. Meniere’s original papers: Reprinted with an English translation together with commentaries and biographical sketch. Acta Otolaryngol Suppl (Stockh). 1961;162:14.
Baloh RW. Prosper Meniere and his disease. Arch Neurol. 2001;58:1151.
Hallpike CS, Cairns H. Observations on the pathol- ogy of Meniere’s syndrome. J Laryngol Otol. 1938; 53: 625.
Yamakawa K. Über die pathologische Verdäderung bei einem Meniere-Kranken [in German]. Z Otol Rhinol Laryngol. 1938;34:181.
Lindsay JR. Labyrinthine dropsy and Meniere’s disease.
others may show minimal or no effect after multiple doses. A wide range of protocols has been used to introduce the drug into the mid- dle ear; the dose of applied gentamicin varies from 30–40 mg/ml. Two recent prospective, double-blind placebo-controlled trials have shown large treatment effects of intratympanic gentamicin for relief of vertigo attacks with only a small increase in hearing loss (on aver-
Arch Otolaryngol. 1942;35:853.
Lindsay JR. Hydrops of the labyrinth. Arch Otolaryngol. 1960;71:500.
Schuknecht HF. Pathology of the Ear. Philadelphia: Lea & Febiger; 1993.
Merchant SN, Adams JC, Nadol JB, Jr. Pathophysiology of Meniere’s syndrome: are symptoms caused by endolymphatic hydrops? Otol Neurotol. 2005;26(1): 74.
Rauch SD, Merchant SN, Thedinger BA. Meniere’s
age about 10 dB).108,109 However, these were small trials (22 and 28 patients, respectively)
syndrome and endolymphatic hydrops: double-blind
temporal bone study. Ann Otol Rhinol Laryngol. 1989;98:873.
Vasama JP, Linthicum FH, Jr. Meniere’s disease and endolymphatic hydrops without Meniere’s symptoms: temporal bone histopathology. Acta Otolaryngol (Stockh). 1999;119:297.
Wladislavosky-Waserman P, Facer GW, Mokri B, Kurland LT. Meniere’s disease: a 30-year epidemio- logic and clinical study in Rochester, MN, 1951–1980. Laryngoscope. 1984;94:1098.
Shojaku H, Watanabe Y, Fujisaka M, et al. Epidemiologic characteristics of definite Ménière’s disease in Japan. ORL J Otorhinolaryngol Relat Spec. 2005;67(5):305.
KotimakiJ, Sorri M, Aantaa E, Nuutinen J. Prevalence of Meniere disease in Finland. Laryngoscope. 1999;109:748.
Cawthorne T, Hewlett AB. Meniere’s disease. Proc R Soc Med. 1954;47:663.
Radtke A, von Brevern M, Feldmann M, et al. Screening for Menière’s disease in the general popu- lation—the needle in the haystack. Acta Otolaryngol. 2008;128(3):272.
Kitahara M, Futaki T. Nakano K. Ethnic aspect of Meniere’s disease. Int J Equilib Res (Suppl). 1971;1:104.
Stahle J. Advanced Meniere’s disease: a study of 356 severely disabled patients. Acta Otolaryngol (Stockh). 1976;81:113.
Watanabe Y, Mizukoshi K, Sojaku H, et al. Epidemiological and clinical characteristics of Meniere’s disease in Japan. Acta Otolaryngol (Stockh). 1995;519:206.
Friberg U, Stahle J. The epidemiology of Meniere’s disease. In: Harris JP, ed. Meniere’s Disease. The Hague, Netherlands: Kugler Publications; 1999.
Choung YH, Park K, Kim CH, Kim HJ, Kim KJ. Rare cases of Ménière’s disease in children. Laryngol Otol. 2006;120(4):343.
Thomas K, Harrison MS. Long-term follow up of 610 cases of Meniere’s disease. Proc R Soc Med. 1971;64:853.
Friberg U, Stahle J, Svedberg A. The natural course of Meniere’s disease. Acta Otolaryngol Suppl (Stockh). 1984;406:72.
Kitahara M, Kitano H, Suzuku M. Meniere’s disease with bilateral fluctuant hearing loss. In: Kitahara M, ed. Meniere’s Disease. Tokyo, Japan: Springer-Verlag; 1990.
Perez R, Chen JM, Nedzelski JM. The status of the contralateral ear in established unilateral Menière’s disease. Laryngoscope. 2004;114(8):1373.
Takumida M, Kakigi A, Takeda T, Anniko M. Ménière’s disease: a long-term follow-up study of bilateral hearing levels. Acta Otolaryngol. 2006;126(9): 921.
Eggermont JJ, Schmidt PH. Meniere’s disease: a long- term follow-up study of hearing loss. Ann Otol Rhinol Laryngol. 1985;94:1.
Perez-Garrigues H, Lopez-Escamez JA, Perez P, et al. Time course of episodes of definitive vertigo in Meniere’s disease. Arch Otolaryngol Head Neck Surg. 2008;134(11):1149.
Havia M, Kentala E. Progression of symptoms of diz- ziness in Ménière’s disease. Arch Otolaryngol Head Neck Surg. 2004;130(4):431.
Junicho M, Aso S, Fujisaka M, Watanabe Y. Prognosis of low-tone sudden deafness—does it inevitably
progress to Meniere’s disease? Acta Otolaryngol. 2008;128(3):304.
Tumarkin I. Otolithic catastrophe: a new syndrome.
BMJ. 1936;2:175.
Baloh RW, Jacobson K, Winder AT. Drop attacks with Meniere’s syndrome. Ann Neurol. 1990;28:384.
Ishiyama G, Ishiyama A, Jacobson K, Baloh RW. Drop attacks in older patients secondary to an otologic cause. Neurology. 2001;57:1103.
Nadol JB, Weiss AD, Parker SW. Vertigo of delayed onset after sudden deafness. Ann Otol. 1975;84:841.
Schuknecht HF. Delayed endolymphatic hydrops.
Ann Otol. 1978;87:743.
Harris JP, Afranian D. Role of autoimmunity in con- tralateral delayed endolymphatic hydrops. Am J Otol. 1994;15:710.
Dodson KM, Kamei T, Sismanis A, Nance WE. Familial unilateral deafness and delayed endolym- phatic hydrops. Am J Med Genet A. 2007;143A(14): 1661.
Kariya S, Cureoglu S, Fukushima H, et al. Histopathologic changes of contralateral human temporal bone in unilateral Ménière’s disease. Otol Neurotol. 2007;28(8):1063.
Tonndolf J. Endolymphatic hydrops: mechanical causes of hearing loss. Arch Otorhinolaryngol. 1976;212:923.
Honrubia V. Pathophysiology of Meniere’s disease. In: Harris JP, ed. Meniere’s Disease. The Hague, Netherlands: Kugler Publications; 1999.
Perlman HB, Kimura R. Experiments on tempo- rary obstruction of the internal auditory artery. Laryngoscope. 1959;69:591.
Silverstein H. The effects of perfusing the perilym- phatic space with artificial endolymph. Ann Otol Rhinol Laryngol. 1970;79:754.
Salt AN. Fluid homeostasis in the inner ear. In: Harris JP, ed. Meniere’s Disease. The Hague, Netherlands: Kugler Publications; 1999.
Rarey KE, Tyneway D, Patterson K. Decreased ade- nosine triphosphatase activity in the absence of adre- nocorticosteroids. Arch Otolaryngol Head Neck Surg. 1989;115:817.
Juhn SK, Ikeda K, Morizono T, Murphy M. Pathophysiology of inner ear fluid imbalance. Acta Otolaryngol Suppl. 1991;485:9.
ten Cate WJ, Curtis LM, Rarey KE. Effects of low-so- dium, high-potassium dietary intake on cochlear lateral wall Na+, K+-ATPase. Eur Arch Otorhinolaryngol. 1994;251:6.
Ishiyama G, López IA, Ishiyama A. Aquaporins and Meniere’s disease. Curr Opin Otolaryngol Head Neck Surg. 2006;14(5):332.
Takeda T, Taguchi D. Aquaporins as potential drug targets for Meniere’s disease and its related diseases. Handb Exp Pharmacol. 2009;(190):171.
Aoki M, Asai M, Nishihori T, Mizuta K, Ito Y, Ando
K. The relevance of an elevation in the plasma vaso- pressin levels to the pathogenesis of Meniere’s attack. J Neuroendocrinol. 2007;19(11):901.
Kitahara T, Doi K, Maekawa C, et al. Meniere’s attacks occur in the inner ear with excessive vasopressin type-2 receptors. J Neuroendocrinol. 2008;20(12):1295.
Cmejrek RC, Megerian CA. Obstructing lesions of the endolymphatic sac and duct mimicking Ménière’s dis- ease. Ear Nose Throat J. 2004;83(11):753.
Kimura RS, Schuknecht H. Membranous hydrops in the inner ear of the guinea pig after the obliteration of the endolymphatic sac. Pract Otorhinolaryngol. 1965;27:343.
Kimura RS. Animal models of endolymphatic hydrops.
Am J Otolaryngol. 1982;3:447.
Takumida M, Akagi N, Anniko M. A new animal model for Ménière’s disease. Acta Otolaryngol. 2008;128(3):263.
Takumida M, Anniko M, Popa R. Possible involvement of free radicals in lipopolysaccharide-induced labyrin- thitis in the guinea pig: a morphological and functional investigation. ORL J Otorhinolaryngol Relat Spec. 1998;60:246.
Feldman AM, Brusilow SW. Effects of cholera toxin on cochlear endolymph production: model for endolymphatic hydrops. Proc Natl Acad Sci USA. 1976;73:1761.
Dunnebier EA, Segenhout JM, Wit HP, Albers FWJ. Two-phase endolymphatic hydrops: a new dynamic guinea pig model. Acta Otolaryngol (Stockh). 1997;117:13.
Takeda T, Takeda S, Kitano H, Okada T, Kakigi A. Endolymphatic hydrops induced by chronic adminis- tration of vasopressin. Hear Res. 2000;140:1.
Morrison AW, Bailey ME, Morrison GA. Familial Ménière’s disease: clinical and genetic aspects. J Laryngol Otol. 2009;123(1):29.
Klockars T, Kentala E. Inheritance of Meniere’s dis- ease in the Finnish population. Arch Otolaryngol Head Neck Surg. 2007;133(1):73.
Frykholm C, Larsen HC, Dahl N, Klar J, Rask- Andersen H, Friberg U. Familial Ménière’s disease in five generations. Otol Neurotol. 2006;27(5):681.
Klar J, Frykholm C, Friberg U, Dahl N. A Meniere’s disease gene linked to chromosome 12p12.3. Am J Med Genet B Neuropsychiatr Genet. 2006;141B(5): 463.
Usami S, Takahashi K, Yuge I, et al. Mutations in the COCH gene are a frequent cause of autosomal dominant progressive cochleo-vestibular dysfunc- tion, but not of Meniere’s disease. Eur J Hum Genet. 2003;11(10):744.
Sanchez E, López-Escámez JA, López-Nevot MA, López-Nevot A, Cortes R, Martin J. Absence of COCH mutations in patients with Meniere disease. Eur J Hum Genet. 2004;12(1):75.
Doi K, Sato T, Kuramasu T, et al. Ménière’s disease is associated with single nucleotide polymorphisms in the human potassium channel genes, KCNE1 and KCNE3. ORL J Otorhinolaryngol Relat Spec. 2005;67(5):289.
Vrabec JT, Liu L, Li B, Leal SM. Sequence variants in host cell factor C1 are associated with Ménière’s dis- ease. Otol Neurotol. 2008;29(4):561.
Baloh RW, Andrews JC. Migraine and Meniere’s dis- ease. In: Harris JP, ed. Meniere’s Disease. The Hague, Netherlands: Kugler Publications; 1998.
Ibekwe TS, Fasunla JA, Ibekwe PU, Obasikene GC, Onakoya PA, Nwaorgu OG. Migraine and Meniere’s disease: two different phenomena with frequently observed concomitant occurrences. J Natl Med Assoc. 2008;100(3):334.
Cha YH, Kane MJ, Baloh RW. Familial clustering of migraine, episodic vertigo, and Ménière’s disease. Otol Neurotol. 2008;29(1):93.
Cha YH, Brodsky J, Ishiyama G, Sabatti C, Baloh RW. The relevance of migraine in patients with Men´ ière’s disease. Acta Otolaryngol. 2007;127(12):1241.
Paparella MM, Djalilian HR. Etiology, pathophysiol- ogy of symptoms, and pathogenesis of Meniere’s dis- ease. Otolaryngol Clin N Am. 2002;35:529.
Lim DJ, Kawauchi H, DeMaria TF. Role of middle ear endotoxin in inner ear inflammatory response and hydrops: long-term study. Ann Otol Rhinol Laryngol. 1990;99:33.
Welling DB, Daniels RL. Viral etiology in Meniere’s disease. In: Harris JP, ed. Meniere’s Disease. The Hague, Netherlands: Kugler Publications; 1999.
Tomiyama S. Development of endolymphatic hydrops following immune response in the endolymphatic sac of the guinea pig. Acta Otolaryngol (Stockh). 2004;124:1145.
Academy of Otolaryngology Head and Neck Surgery. Meniere’s disease: criteria for diagnosis and evalua- tion of therapy for reporting. AAO-HNS Bulletin. July 1985.
Morrison AW. Diagnostic and laboratory evaluation of Meniere’s disease. In: Harris JP, ed. Meniere’s Disease. The Hague, Netherlands: Kugler Publications; 1999.
Savastano M, Guerrieri V, Marioni G. Evolution of audiometric pattern in Meniere’s disease: long-term survey of 380 cases evaluated according to the 1995 guidelinesofthe American Academyof Otolaryngology- Head and Neck Surgery. J Otolaryngol. 2006;35(1): 26.
Lonsbury-Martin BL, Martin GK. Clinical utility of distortion product otoacoustic emissions. Ear Hear. 1990;11:90.
Harris FP, Probst R. Transiently evoked otoacoustic emissions in patients with Meniere’s disease. Acta Otolaryngol (Stockh). 1992;112:36.
Ferraro JA, Ruth RA. Clinical electrocochleography.
Hear J. 1985;38:51.
Kim HH, Kumar A, Battista RA, Wiet RJ. Electrocochleography in patients with Meniere’s dis- ease. Am J Otolaryngol. 2005;26(2):128.
Fushiki H, Junicho M, Aso S, Watanabe Y. Recurrence rate of idiopathic sudden low-tone sensorineural hear- ing loss without vertigo: a long-term follow-up study. Otol Neurotol. 2009;30(3):295.
Hulshof JH, Baarsma EA. Follow-up vestibular examinations in Meniere’s disease. Acta Otolaryngol (Stockh). 1981;92:397.
Osei-Lah V, Ceranic B, Luxon LM. Clinical value of tone burst vestibular evoked myogenic potentials at threshold in acute and stable Ménière’s disease. J Laryngol Otol. 2008;122(5):452.
Park HJ, Migliaccio AA, Della Santina CC, Minor LB, Carey JP. Search-coil head-thrust and caloric tests in Ménière’s disease. Acta Otolaryngol. 2005;125(8): 852.
Minor LB. Intratympanic gentamicin for control of vertigo in Meniere’s disease: vestibular signs that spec- ify completion of therapy. Am J Otol. 1999;20:209.
McClure JA, Copp JC, Lycett P. Recovery nystag- mus in Meniere’s disease. Laryngoscope. 1981;91: 1727.
Valvassori CE, Dobben GD. Multidirectional and computerized tomography of the vestibular aque- duct in Meniere’s disease. Ann Otol Rhinol Laryngol. 1984;93:547.
Fitzgerald DC, Mark AS. MR imaging in Meniere’s disease. In: Harris JP, ed. Meniere’s Disease. The Hague, Netherlands: Kugler Publications; 1999.
Naganawa S, Sugiura M, Kawamura M, Fukatsu H, Sone M, Nakashima T. Imaging of endolymphatic and perilymphatic fluid at 3T after intratympanic administration of gadolinium-diethylene-triamine pentaacetic acid. AJNR Am J Neuroradiol. 2008;29(4): 724.
Carfrae MJ, Holtzman A, Eames F, Parnes SM, Lupinetti A. 3 Tesla delayed contrast magnetic resonance imaging evaluation of Ménière’s disease. Laryngoscope. 2008;118(3):501.
McNeill C, McMahon CM, Newall P, Kalantzis
M. Hearing aids for Ménière’s syndrome: impli- cations of hearing fluctuation. J Am Acad Audiol. 2008;19(5):430.
Boles R, Rice DH, Hybels R, Work WP. Conservative management of Meniere’s disease: Furstenberg regimen revisited. Ann Otol Rhinol Laryngol. 1975;84:513.
Jackson CG, Glasscock ME, Davis WE, et al. Medical management of Meniere’s disease. Ann Otol. 1981;90:142.
James AL, Thorp M. Meniere’s Disease. BMJ Clinical Evidence. 2006;15:797.
Thirlwall AS, Kundu S. Diuretics for Ménière’s disease or syndrome. Cochrane Database Sys Rev. 2006;3:CD003599.
Shinkawa H, Kimura RS. Effect of diuretics on endolymphatic hydrops. Acta Otolaryngol (Stockh). 1986;101:43.
Brookes GB, Booth JB. Oral acetazolamide in Meniere’s disease. J Laryngol Otol. 1984;98:1087.
Strupp M, Hupert D, Frenzel C, et al. Long-term pro- phylactic treatment of attacks of vertigo in Menière’s disease—comparison of a high with a low dosage of betahistine in an open trial. Acta Otolaryngol. 2008;128(5):520.
James AL, Burton MJ. Betahistone for Meniere’s disease or syndrome. Cochrane Database Sys Rev. 2001;1:CD001873.
Schuknecht HF. Endolymphatic hydrops: can it be controlled? Ann Otol Rhinol Laryngol. 1986;95:36.
Thomsen J, Brettan P, Tos M, Johnsen NJ. Placebo effect of surgery for Meniere’s disease. Arch Otolaryngol. 1981;107:271.
Ress BD, Harris JP. Endolymphatic sac surgery. In: Harris JP, ed. Meniere’s Disease. The Hague, Netherlands: Kugler Publications; 1999.
Densert B, Densert O. Overpressure in treat- ment of Meniere’s disease. Laryngoscope. 1982;92: 1285.
Gates GA, Green JD, Tucci DL, Telian SA. The effects of transtympanic micropressure treatment in people with unilateral Meniere’s disease. Arch Otolaryngol Head Neck Surg. 2004;130:718.
Gates GA, Verrall A, Green JD, Tucci DL, Telian SA. Meniett clinical trial: long-term follow-up. Arch Otolaryngol Head Neck Surg. 2006;132:1311.
Takemori S, Ida M, Umezu H. Vestibular train- ing after sudden loss of vestibular functions. ORL J Otorhinolaryngol Relat Spec. 1985;47:76.
Hone SW, Nedelski JM. Selective chemical ablation as treatment for Meniere’s disease. In: Harris JP, ed. Meniere’s Disease. The Hague, Netherlands: Kugler Publications; 1999.
Stokroos R, Kingma H. Selective vestibular ablation by intratympanic gentamicin in patients with unilat- eral active Ménière’s disease: a prospective, double- blind, placebo-controlled, randomized clinical trial. Acta Otolaryngol. 2004;124(2):172.
Postema RJ, Kingma CM, Wit HP, Albers FW, Van Der Laan BF. Intratympanic gentamicin therapy for control of vertigo in unilateral Menire’s disease: a prospective, double-blind, randomized, placebo- controlled trial. Acta Otolaryngol. 2008;128(8): 876.
Glasscock ME, Davis ME, Hughes GB, Jackson GG. Labyrinthectomy versus middle fossa vestibular nerve section in Meniere’s disease. Ann Otol Rhinol Laryngol. 1980;89:318.
Silverstein H, Arruda J, Rosenberg S. Vestibular neurectomy. In: Harris JP, ed. Meniere’s Disease. The Hague, Netherlands: Kugler Publications; 1999.
Glasscock ME, Thedinger BA, Cueva RA, Jackson CG. An analysis of the retrolabyrinthine vs. the ret- rosigmoid vestibular nerve section. Otolaryngol Head Neck Surg. 1991;104:88.
![]()
BACKGROUND CLINICAL FEATURES
Migraine without Aura Migraine with Aura Migrainous Vertigo Basilar Migraine
Migraine and Meniere’s Syndrome Migraine Equivalents PATHOPHYSIOLOGY
Genetics
Spreading Wave of Depression
Vasomotor Abnormalities
DIAGNOSIS
Migraine without Aura Migraine with Aura
Migraine Aura without Headache Basilar Migraine
Migrainous Vertigo
MANAGEMENT
Symptomatic and Abortive Treatment Prophylactic Treatment
Migraine is a complex multigenetic disorder that renders one susceptible to vascular and chemical changes in the brain that can lead to many different symptoms.1 The hallmark fea- ture of the disorder is severe attacks of head- aches. However, migraine is also characterized by many other features, including dizziness, visual phenomenon, hypersensitivities to sen- sory stimuli, nausea, paresthesias, and even frank focal weakness in some cases.
Dizziness symptoms are among the most common symptoms reported by migraineurs. In some cases, the dizziness symptom is best characterized as a sensitivity to motion, a symp- tom that most patients with migraine are sus- ceptible to throughout their lives. Other times, patients use “dizziness” to label a vague and nonspecific symptom such as disorientation or difficulty focusing on a task. However, the diz- ziness of migraine can also be true and severe room-spinning vertigo. Migraine, in fact, is the great mimicker of all causes of vertigo because symptoms of it can closely resemble the attacks
of Meniere’s syndrome, benign positional ver- tigo, and even vestibular neuritis.
The association of migraine and dizziness extends back to the nineteenth century when Livening noted their connection in his classic book On Megrim: Sick Headaches and Some Allied Health Disorders.2 However, only in recent years has vertigo been recognized as a common migraine symptom. So-called migrain- ous vertigo or migraine-associated vertigo (ver- tigo caused by migraine) affects about 1% of the general population and about 10% of patients seen in dizziness clinics and in migraine clinics.3 The vertigo attacks can occur during the head- ache, but most often the attacks occur during headache-free intervals.4 Although the clinical association between migraine and dizziness is well documented, it is difficult to prove a causal relationship between migraine and dizziness or, for that matter, between migraine and any of the other transient symptoms that accompany it. For example, scintillating scotoma is a well- known accompaniment of migraine and is uni- versally accepted as a migrainous phenomenon, yet its pathophysiologic link with headache is
287
still unknown after hundreds of years of clinical association. The nonspecific, highly varied nature of dizziness makes the relationship between migraine and dizziness even more dif- ficult to characterize. Recent advances in our understanding of the genetics and pathophysiol- ogy of migraine provide new hope for improved understanding of the link between patients with periodic headaches and dizziness.5
Migraine without Aura
Migraine without aura can probably best be described as a “sick headache.” Vague pro- dromal symptoms precede it, but aura phe- nomena are absent. The headache, unilateral or bilateral, builds slowly in intensity and may go on for several days. Nausea, vomiting, diar- rhea, chills, and prostration can all accompany the headache. Nonspecific dizziness is a com- mon complaint, and patients frequently report visual blurring and a sense of unsteadiness dur- ing the entire headache phase. Vertigo can occur before, during, or entirely separate from the episodes of headache.4,6
There is still debate as to whether migraine without aura and migraine with aura are dis- tinct syndromes, different manifestations of the same disorder, or part of a continuum. Patients can have both types of headaches (with and without aura), and not infrequently, both types of migraine run in the same family (see later discussion). The headache phases of both types of migraine are almost identical, and the same treatments are usually effective in controlling both types of migraine. Conversely, certain epi- demiological characteristics, overall familial aggregation, and varying pathophysiologic find- ings suggest the possibility that these two types of migraine represent separate entities.7,8
Migraine with Aura
The aura with migraine typically precedes the onset of a severe, throbbing, unilateral head- ache. The aura symptoms slowly progress over several minutes, last 15 to 60 minutes, and then gradually abate. In a small percentage of patients, however, onset is more abrupt.
The headache begins as the aura diminishes, usually reaching its peak in about an hour and then gradually subsiding over the next 4 to 8 hours. Nausea and vomiting typically accom- pany the onset of the head pain.
The migraine aura consists of transient neu- rologic dysfunction, often with visual distur- bances, but also commonly includes prominent vertigo or somatosensory symptoms. Both posi- tive and negative visual phenomena occur. The latter include complete blindness, hemianopsia or quadrantanopsia, tunnel vision, altitudinal defects, monocular blindness, or one or more scotomata. Positive phenomena are more com- mon and may consist of stars, sparkling lights, unformed flashes of light (photopsia), geomet- ric patterns, or a jagged, sparkling zig-zag (teichopsia or fortification spectra). Although usually black and white, the visual phenomena can be in color. The positive and negative visual phenomena are combined in the so-called scin- tillating scotoma. Patients describe a hole in their vision with a sparkling border. The sco- toma will begin in one hemifield but gradually enlarge and move across to involve the other hemifield, with the leading edge being a zig- zag of sparkling lights.9
Somatosensory symptoms, particularly par- esthesias, are another common aura manifesta- tion. Numbness, tingling, or both may affect the hands, lower face, or half of the body. A characteristic feature of migrainous paresthe- sias is a migration of the numbness as it gradu- ally spreads over the face or extremity, some- times migrating from face to extremity on the same side or sometimes crossing over to the face and extremity on the opposite side. In a small percentage of patients, the paresthesias will simultaneously appear in multiple sites or be localized to a small area such as a single digit, lip, or cheek. When focal neurologic symptoms such as hemianopsia or unilateral paresthesias occur in an aura, they usually occur on the side opposite that of the headache. Only about 12% of patients with migraine regularly experience aura with their headache, but as many as two- thirds have occasional attacks with aura.13
Migrainous Vertigo
Overall, episodic vertigo occurs in about 25% of unselected migraine patients.11 The vertigo attacks can occur during the headache, but
most often the attacks occur during the head- ache-free interval. Only about a quarter of patients reliably experience headaches with their vertigo.10 Migrainous vertigo can be spon- taneous or positional and attacks typically last from minutes to days.10,12 If examined during an attack patients may show spontaneous or positional nystagmus (either in isolation or in combination) and the nystagmus can have either central or peripheral features.13 Complicating matters further, benign posi- tional vertigo is more common in patients with migraine than in the general population (see Chapter 10).14 Migrainous positional vertigo can be differentiated from benign positional vertigo by (1) short-duration symptomatic epi- sodes, (2) migrainous symptoms during epi- sodes, and (3) atypical positional nystagmus.15
Auditory symptoms are generally considered to be less common than the vestibular symp- toms of migraine. Phonophobia is probably the most common auditory symptom associated with migraine, occurring at some time in more than two-thirds of patients, usually in associa- tion with headache.16 Most patients with migrainous vertigo have normal audiometric findings.17 Several investigators have identified fluctuating low-frequency hearing levels in patients with migraine, which are typical of those seen in patients with Meniere’s syndrome.16,18,19 These episodes are most com- monly seen in young women around the time of their menstrual period and are usually com- pletely reversible. In the case of Meniere’s syn- drome, a progressive loss of hearing is required. Migraine has also been identified as the cause of sudden hearing loss that persists.20,21 Such patients report the abrupt onset of a profound hearing loss and may show some gradual improvement, but they are often left with a severe unilateral sensorineural hearing loss. Some patients with sudden hearing loss report a prior history of fluctuating hearing in the same ear and many develop persistent tinnitus. Hearing loss and tinnitus can also be symptoms of migrainous infarction.22
Basilar Migraine
Basilar migraine is a subtype of migraine with aura characterized by recurrent headaches, usu- ally localized to the occipital region, preceded by multiple neurological symptoms localized to
Table 12–1 Most Common Symptoms in 49 Patients with Basilar Migraine
![]()
Symptom Cases (%)
Headache (usually occipital) | 96 |
Nausea | 83 |
Vomiting | 71 |
Vertigo | 63 |
Gait ataxia | 63 |
Paresthesias (usually bilateral) | 61 |
Dysarthria | 57 |
Weakness (usually bilateral) | 55 |
Tinnitus | 26 |
Impaired hearing | 20 |
Double vision | 16 |
the posterior fossa (Table 12–1).23,24 The aura consists of posterior fossa symptoms such as vertigo, ataxia, dysarthria, and tinnitus along with visual phenomena consistent with ischemia in the distribution of the posterior cerebral arteries. Motor and sensory symptoms, such as circumoral or extremity paresthesias, weakness, and drop attacks, are occasionally seen as well. When vertigo occurs it usually has an abrupt onset and lasts 5 to 60 minutes. The headache following the aura is usually unilateral occipital or frontal, but it can occur anywhere, especially in children.
Basilar migraine affects about 10% of patients suffering from migraine with aura.24 One must be alert for the possibility of basilar migraine in any patient presenting with transient vertigo and other posterior fossa symptoms.25 In some individuals, the headache is not severe and is adequately managed by aspirin, sleep, or mild analgesics and sedatives. Some of these patients are unaware that migraine is the cause of their headaches and are much more concerned about the aura. If vertigo is prominent, the patient may not mention the headache, think- ing it is unimportant. Similarly, other transient manifestations may be given less importance than that given the vertigo. Such patients may be misdiagnosed as having a peripheral labyrin- thine disease if the physician is not alert to the possibility of basilar migraine.
Migraine and Meniere’s Syndrome
Speculation on a relationship between migraine and Meniere’s syndrome dates back to the initial
description of the syndrome by Prosper Meniere in 1861.26 He noted that both conditions com- monly manifested episodes of vertigo, fluctuat- ing hearing levels, and recurrent vomiting. Although many subsequent authors have also speculated on the relation between migraine and Meniere’s disease, there is still no generally accepted mechanism to explain the connec- tion.27–29 Diagnostic criteria have been estab- lished for both migraine and Meniere’s disease, but some of the criteria overlap. For example, the key diagnostic feature for Meniere’s syndrome is a fluctuating, low-frequency sensorineural hearing loss that many clinicians consider pathognomonic for Meniere’s syn- drome. As noted earlier, however, fluctuating low-frequency hearing loss can occur in patients with migraine. Although headache is the most common symptom of migraine, visual aura or episodes of vertigo can occur without headache. Therefore, even when using strict diagnostic cri- teria, it may not always be possible to separate the two conditions on clinical grounds alone.
Although most authors agree that there is an increased prevalence of migraine in patients with Meniere’s syndrome compared with the general population, the overall percentage of Meniere’s patients with migraine varies from study to study.30 A recent study that used cur- rent criteria for the diagnosis of migraine and Meniere’s syndrome found a lifetime preva- lence of migraine (with or without aura) of 56% in patients with Meniere’s syndrome compared to 25% in controls.31 Furthermore, 45% of patients with Meniere’s syndrome experienced at least one migrainous symptom (headache, photophobia, aura) with their Meniere’s attacks. Genetic factors are likely important for both disorders since migraine and Meniere’s syndrome tend to cluster in families.32 In three sets of twins in these families one twin had migraine and Meniere’s syndrome while the other twin had migraine and migrainous ver- tigo (without auditory symptoms).
Numerous studies have documented that migraine can lead to permanent auditory and vestibular deficits.4,33–35 Since Meniere’s syndrome can develop in an ear previously damaged by infection or trauma, another explanation for the association between migraine and Meniere’s syndrome is the devel- opment of endolymphatic hydrops in an ear previously damaged by migraine. For example, the vasospasm could lead to ischemic damage
to the endolymphatic duct and/or sac, resulting in impaired fluid circulation and the eventual development of hydrops. One would then expect a gradual progression of the disease typ- ical of Meniere’s syndrome of any cause.
Migraine Equivalents
BENIGN PAROXYSMAL VERTIGO OF CHILDHOOD
Basser36 described an episodic disorder in chil- dren under the age of 4 years that he called benign paroxysmal vertigo. A completely nor- mal child suddenly becomes frightened, cries out, clings to the parent, or staggers as though drunk, and exhibits pallor, diaphoresis, and often vomiting. Symptoms are accentuated by head movements, and sometimes nystagmus and torticollis are observed. Some children report a true spinning sensation, but most have difficulty describing what they are experienc- ing. The spells typically last for several minutes. Afterward, the child is immediately normal and can resume playing as though nothing has happened.
Vertigo spells typically begin before the age of 4 years and occur up to several times a month. After several years, they decrease in number and often gradually disappear. Many children have no further spells after the age of 7 or 8. The cause of benign paroxysmal vertigo of childhood is unknown, although most have a positive family history of vertigo or migraine.37 Follow-up studies of patients with typical benign paroxysmal vertigo during childhood show that nearly all patients eventually develop other features typical of migraine.38,39
VESTIBULAR MENIERE’S SYNDROME
In their initial recommendations on criteria for the diagnosis of Meniere’s disease, the American Academy of Ophthalmology and Otolaryngology Committee on Hearing and Equilibrium defined vestibular Meniere’s syn- drome as recurrent attacks of vertigo without associated auditory symptoms. It was assumed that most of these patients would progress to manifest all of the symptoms of classical Meniere’s syndrome. However, because there are so many causes for recurrent episodic ver- tigo other than Meniere’s syndrome, more
recently the American Academy of Otolaryn- gology Head and Neck Surgery Committee on Hearing and Equilibrium recommended dis- carding the term vestibular Meniere’s disease. Rassekh and Harker28 followed up 38 patients with a diagnosis of vestibular Meniere’s syn- drome using a standard questionnaire. Seventeen of the 38 no longer fulfilled the cri- teria for the diagnosis of vestibular Meniere’s syndrome at follow-up. Of these, eight went on to develop unilateral Meniere’s syndrome and seven became asymptomatic. Of the 21 patients who continued to meet the criteria for vestibu- lar Meniere’s syndrome, 17 (81%) had migraine headaches. Overall, this study suggests that only a small percentage of patients who present with recurrent episodes of vertigo typical of vestibular Meniere’s syndrome go on to develop the classical symptom triad of Meniere’s syn- drome, whereas the majority are associated with migraine.
BENIGN RECURRENT VERTIGO OF ADULTHOOD
Slater40 and Moretti et al.41 described patients who, between the ages of 7 and 55 years, began to experience repeated episodes of vertigo, nausea, vomiting, and diaphoresis. The attacks often occurred on awakening in the morning, being particularly common around menses in women. Duration varied from a few minutes to as long as 3 to 4 days, with the vertigo becom- ing primarily positional toward the end of the spell. Nearly all patients were asymptomatic between spells. During episodes, there were no auditory symptoms, specifically no hearing loss, tinnitus, or ear pressure or fullness. Most patients either had migraine themselves or a strong family history of migraine. Furthermore, the episodes of vertigo had several features in
common with migraine, including precipitation by alcohol, lack of sleep, emotional stress, and a female preponderance.
We studied the families of 24 adults who presented to our clinic with benign recurrent vertigo and who reported a family history of similar attacks of vertigo.42 All probands under- went diagnostic evaluation to exclude identifi- able causes of recurrent vertigo and they completed a standardized medical question- naire pertaining to episodic vertigo and the fea- tures of migraine. This questionnaire was also sent to all relatives of the probands who agreed to participate. Of 220 relatives who returned questionnaires, 37% reported BRV and 50% met the diagnostic criteria for migraine (Table 12–2). By contrast, only one of 43 (2%) unre-
lated spouses reported BRV and 10 of 43 (23%) met the diagnostic criteria for migraine. More than two-thirds of relatives with BRV met the diagnostic criteria for migraine and the majority reported that they had a typical migraine headache with at least some of their episodes of vertigo. Both benign recurrent vertigo and migraine showed a female prepon- derance (more than 2 to 1). Familial benign recurrent vertigo appears to be a migraine syn- drome, probably inherited in an autosomal dominant fashion with decreased penetrance in men.
OTHER MIGRAINOUS PHENOMENA
In young children the manifestations of migraine are protean, and headache is not always present.43 Migraine equivalents may appear as cyclic vomiting, attacks of abdominal pain, or even ophthalmoplegia. As the child matures, these nonspecific and other puzzling symptoms may cease and be supplanted by more typical paroxysmal head pain.
Table 12–2 Incidence of Vertigo and Migraine in Probands, First Degree Relatives (Parents, Siblings, Children), All Relatives, and Unrelated Spouses
Totala | Benign recurrent vertigo | Migraine | Both | |
Probands | 24 | 24 (100%) | 20 (83%) | 20 (83%) |
First degree relatives | 111 | 44 (40%) | 51 (46%) | 31 (28%) |
All relatives | 220 | 82 (37%) | 110 (50%) | 59 (27%) |
Unrelated spouses | 43 | 1 (2%) | 10 (23%) | 0 (0%) |
a Those who returned questionnaires. (From Oh AK, Lee H, Jen JC, Corona S, Jacobson KM, Baloh RW. Familial benign recurrent vertigo. Am J Med Genet. 2001;100:287 with permission.)
Symptoms of migraine equivalents can also begin in adulthood. Isolated episodes of scintil- lating scotomas are not uncommon after the age of 40. Fisher44 reported 60 patients with what he called “transient migrainous accompa- niments,” which were attacks of paresthesias, aphasia, dysarthria, paresis, and diplopia with or without the visual manifestations of migraine. None of these patients had headache. Normal angiography, long-term follow-up, and, in a few cases, necropsy, suggested a migrainous syndrome despite the absence of associated headaches.
Genetics
Although it is clear that some varieties of migraine are inherited, it is not clear whether migraine is a single syndrome or a variety of syndromes, some of which are inherited and some of which are not.45 The situation is analo- gous to the epilepsy syndromes. Some seizure syndromes are inherited and genetic factors are important for most types of epilepsy, but anyone can have a seizure with adequate prov- ocation. Similarly, several inherited syndromes are associated with migraine headache, and genetic factors are important in determining the threshold for migraine headaches, but structural lesions such as vascular malforma- tions and vasculopathies can trigger migraine headaches in anyone. Genotype heterogeneity has already been established for several migraine syndromes. These syndromes are defined based on the presence of hemiplegic episodes or other aura symptoms that accom- pany migraine headaches. Within such fami- lies, however, some members may have only migraine headaches, indicating that there is also phenotypic heterogeneity. Not surpris- ingly, since headache is the least specific symp- tom of the different migraine syndromes, the genetic basis for migraine without aura has been more difficult to establish.
Numerous studies over the years have docu- mented familial aggregation of migraine, and some neurologists have suggested that a posi- tive family history should be part of the diag- nostic criteria.46,47 In nearly all studies, the inci- dence of a positive family history in patients
with migraine headaches was significantly greater than in controls. The incidence of a positive family history varied from approxi- mately 40% to 90% compared with approxi- mately 5% to 20% in controls. The percentage of positive family histories tended to be greater in those studies in which family members were individually interviewed than in studies that relied on questionnaires or on the recall of the proband. Studies in monozygotic and dizygotic twins have also supported a strong genetic component for migraine, particularly for migraine with aura. Also, the fact that the prev- alence of migraine in African and Asian popu- lations is lower than in European and North American populations favors a major genetic component. Studies of migrainous vertigo show the same familial aggregation seen in other migraine syndromes.48–50
RARE MIGRAINE SYNDROMES WITH KNOWN GENETIC CAUSES
The recent identification of the genes for sev- eral rare subtypes of migraine may provide the first true molecular insight into migraine pathophysiology. Within families with these relatively rare syndromes, some members will have only MO or MA, suggesting that these more common migraine syndromes may have a similar pathophysiologic mechanism.
Familial hemiplegic migraine (FHM) is an autosomal dominant disease characterized by headache attacks preceded by or accompanied by episodes of hemiplegia, sometimes lasting days.51 Within reported families with FHM, some affected members have interictal nystag- mus, ataxia, essential tremor, and seizures. MA and episodes of hemiplegia may alternate within individuals and co-occur within families. Based on this observation, Russell and Olesen52 concluded that the pathophysiology and etiol- ogy of FHM and other more common uncom- plicated migraine variants may be similar. By contrast, after comparing clinical features of patients with MA and patients with FHM, Thomsen et al.53 concluded that hemiplegic migraine is a separate entity from MA. Haan et al.54 suggested the FHM may be a hereditary form of basilar migraine. They studied aura symptoms in 83 patients from six unrelated families with FHM and found that 55 of the patients reported symptoms that met the International Headache Society (IHS) criteria
So far, mutations in two genes have been identified in FHM: CACNA1A55 and ATP1A2.56 Both of these genes code for the transmem- brane component of a neuronal ion channel. Although there is overlap in clinical features, most families with FHM due to mutations in CACNA1A have progressive ataxia and interic- tal nystagmus,55,57 while the families reported with mutations in ATP1A2 had associated epi- leptic seizures.56 All of the mutations in CACNA1A associated with FHM have been missense mutations.57 Nonsense mutations in the same gene produce a related disorder, epi- sodic ataxia type 2 (EA-2), which is also associ- ated with migraine headaches.55,58 In some families there can be an overlap between epi- sodes of hemiplegia and episodes of ataxia.59 In families with mutations in either FHM gene, some members can have just MO or MA as the only manifestation.56,57
Some families with FHM are not linked to either the CACNA1A locus on chromosome 19p or the ATP1A2 locus on chromosome 1q.60 Terwindt et al.61 screened the CACNA1A gene for mutations in 27 patients with sporadic hemiplegic migraine and found only two muta- tions (one patient had ataxia and interictal nys- tagmus, while the other had no cerebellar signs). Jen et al.62 screened 19 patients with hemiplegic migraine (8 familial and 11 spo- radic) for mutations in CACNA1A and ATP1A2 and found only a single mutation in CACNA1A in one of the sporadic cases (who also had cer- ebellar ataxia). Of the approximately 40 FHM familieswithidentifiedmutationsin CACNA1A, about half had the T666M mutation. Most of the patients with the T666M mutation also have symptoms and signs of cerebellar ataxia, although there is a broad clinical spectrum.63 Likely mutations in several other genes will be identified, particularly in cases of sporadic hemiplegic migraine.
CANDIDATE GENES FOR COMMON MIGRAINE SYNDROMES
Within families with known mutations in CACNA1A or ATP1A2, some members have just MO or MA. Furthermore, linkage studies performed in a few large families with MO or
MA inherited in an autosomal dominant fashion found linkage to the 19p and 1q loci of these two genes.64,65 However, Kim et al.66 and Wieser et al.67 could not find mutations in CACNA1A in patients with these common migraine syndromes. Jen et al.62 screened
50 probands from families with migraine (7 basilar migraine, 25 MO, 18 MA) and did not find any mutation in either CACNA1A or ATP1A2. Based on this preliminary data, it does not appear that CACNA1A or ATP1A2 is important for the common migraine syndromes.
One way to explain the heterogeneity of migraine syndromes is to postulate a group of defects in genes that code for a family of pro- teins with similar properties and functions. The family of ion channels is appealing because many of the migraine syndromes share the clinical features of known inherited ion channel disorders (Table 12–3). A defective ion channel could explain the local buildup of extracellular potassium that initiates the spreading wave of depression in migraine (see later discussion). Since ion channels in the inner ear are critical for maintaining the potassium-rich endolymph and neuronal excitability, a defective ion channel shared by the brain and inner ear could lead to a reversible hair cell depolarization and auditory and vestibular symptoms. Further- more, many of the well-known triggers for migraine symptoms, including stress and menstruation, could result from hormonal influences on the defective ion channels. Finally prophylactic drugs such as beta blockers, calcium channel blockers, aceta- zolamide, and tricyclic amines might work by stabilizing abnormal ion channels. Acetazol- amide may work in episodic ataxia type 2 by changing cerebellar pH and stabilizing calcium channels.68
Table 12–3 Clinical features shared by known inherited ion channel disorders
![]()
Autosomal dominant inheritance Reduced penetrance
Periodic symptoms
Episodes triggered by stress, exercise May have no interictal findings Response to acetazolamide
![]()
Probably the most characteristic of migraine symptoms is the classic visual aura. It typically begins with a small scintillating scotoma that gradually enlarges over 20 to 30 minutes. There is convincing evidence that the visual aura is secondary to a spreading wave of cortical depression beginning at the occipital pole, which gradually spreads across the cortex before stopping at the central sulcus.69 Although decreased cerebral perfusion is associated with the spreading wave of depression, it is probably a secondary phenomenon rather than a primary process.70 The spreading wave of cortical depression is associated with a marked accu- mulation of extracellular potassium that must be cleared before neural activity can return to normal. Although the exact mechanism for the spreading wave of depression is not known, most agree that the initial event is local buildup of potassium in the extracellular space.
Nearby synaptic terminals then depolarize in response to the high extracellular potassium, and both excitatory and inhibitory neurotrans- mitters are released. This release in turn leads to the opening of subsynaptic channels, result- ing in further ionic exchange between the intracellular and extracellular fluids. During the spreading wave of depression, neurons are completely silent for approximately 1 minute and they then slowly recover their predepres- sion level of firing. The rate of movement of the spreading wave of depression across the visual cortex (measured with functional mag- netic resonance imaging [MRI]) nicely corre- lated with the rate of enlargement of the scintillating scotoma observed by patients.71 A spreading wave of cortical depression was observed on positron emission tomography (PET) in a patient having a typical migraine headache without aura.72
Consistent with a basic neuronal defect, patients with migraine have cortical abnormali- ties not only during their attacks but also in the interictal period. Paroxysmal slowing on interictal electroencephalographs (EEG) is commonly seen in migraine, and the classical association of migraine and epilepsy is well documented.73 Interestingly, identical paroxys- mal slowing on EEG is seen in patients with EA-2. Magnetic resonance spectroscopy shows decreased interictal energy metabolism in the cortex of patients with migraine with or
without aura.74,75 Single-photon emission com- puted tomography (SPECT) and other tech- niques for measuring cerebral blood flow have shown areas of regional hypoperfusion, not only during migraine attacks but also in the interictal phase.76 These changes were found in migraine without visual aura, with visual aura, and with episodes of hemiplegia. Acetazolamide reversed these interictal areas of hypoperfu- sion in all three classes of migraine patients.
How does the spreading wave of depression and associated increase in extracellular potas- sium lead to a typical migraine headache? Trigeminal nerve fibers surrounding pial arter- ies on the ventral surface of the brain could be depolarized by the high potassium concentra- tion.77 This in turn would lead to the release of neurotransmitters such as substance P and cal- citonin gene–related peptide (CGRP) by both orthodromic and antidromic conduction. The result is an increase in vascular permeability, dilatation of cerebral vessels, and a local inflam- matory response further activating pain- provoking fibers of the trigeminal vascular sys- tem. Thus, the headache of migraine could be a secondary phenomenon, the end result of a local increase in extracellular potassium con- centration. Many of the drugs used for prophy- lactic treatment of migraine have been shown to suppress the cortical spreading wave of depression in an animal model of migraine.78
Vasomotor Abnormalities
Vasomotor abnormalities have long been con- sidered in the pathophysiology of migraine symptoms. Vasodilatation of extracranial ves- sels accompanies the typical migraine head- ache. Vasospasm occurs in some intracranial vessels with migraine, although there is contro- versy regarding its role in the production of symptoms. Although vasospasm is associated with the classical migraine visual aura, most likely, vasospasm results from a metabolic defect slowly spreading across the cerebral cortex and is secondary to hypometabolism. Vasospasm is more likely a cause of retinal migraine.79 Some patients experience episodes of monocular blindness, and when examined during these episodes there is vasospasm of retinal arteries. Furthermore, such patients respond to antispasmodic agents.80,81 Sudden episodes of hearing loss and/or vertigo
associated with migraine could be explained by the vasospasm of the cochlear and/or vestibular branches of internal auditory artery.
It is interesting to note that MRI studies in migraine patients found an increased preva- lence of small white matter hyperintensities, particularly involving the cerebellum and brain stem, compared to non-migraine control sub- jects.82–84 (Fig. 12–1).Though these are nonspe- cific findings, a leading hypothesis is that these
lesions reflect ischemia which could be related to vasospasm.
The diagnosis of migraine is relatively easy when headaches are the major feature and there is a strong family history. In patients in
Figure 12–1. Cerebellar lesions in patients with migraine from the CAMERA study. Corresponding magnetic resonance T2-weighted (left) and fluid-attenuated inversion-recovery images (right) showing cerebellar infarct-like lesions (arrows) in four representative cases. (Kruit et al. Brain. 2005;128:2068-2077, with permission)
Migraine without Aura
The criteria for the diagnosis of migraine with- out aura, established by the IHS, are summa- rized in Table 12–4.85 The patient must have had at least five headache attacks that meet the criteria and other causes of headache must be ruled out. If they meet all but one criteria they are designated as probable migraine. No labo- ratory or radiological findings are specific for migraine. The physical and neurological exami- nations are normal and serve primarily to exclude other causes of headache. A classic migraine attack typically has five phases: a prodrome (e.g, depression, cognitive dysfunc- tion, food craving), aura (e.g, visual, sensory, or motor phenomena), headache (usually unilat- eral and throbbing), resolution (when pain wanes), and recovery.86 None of these phases is obligatory for the migraine diagnosis, however. Symptoms (severity, duration, nature of pro- drome or aura) also vary substantially between individuals. Thus, the diagnosis of migraine is based on a combination of sequentially occur- ring symptoms in paroxysmal attacks. Motion sickness is often the first symptom of migraine
Table 12–4 Diagnostic Criteria for Migraine without Aura
![]()
At least five attacks fulfilling B–D
Headache lasts 4–72 hr (untreated)
Headache has at least two of the following features:
Unilateral Pulsating
Moderate or severe (inhibits or prohibits daily activities)
Aggravated by walking, stairs, or similar physical activities
During headache at least one of the follow- ing:
Nausea and vomiting Photophobia and phonophobia
Other causes of headache have been ruled out
![]()
Source: Adapted from International Headache Society.85
in children and has been recommended for inclusion as another minor criterion for the diagnosis.87
Migraine with Aura
The IHS Headache Classification Committee defined migraine with aura as “an idiopathic recurring disorder manifesting with attacks of neurological symptoms unequivocally localiz- able to cerebral cortex or brain stem, usually gradually developed over 5 to 20 min and usu- ally lasting less than 60 min.”85 Visual symp- toms, including the classic scintillating scotoma and fortification spectra, are the most com- monly recognized aura phenomena, but somatosensory and vestibular symptoms are probably equally common.
Migraine Aura without Headache
Although less common, migraine aura without headache is of particular interest from a neurotologic point of view. The occurrence of migraine aura without headache has been rec- ognized for many years, although this contin- ues to be a difficult concept for both patients and physicians to deal with. Some patients will have isolated aura symptoms at one point in their life and typical migraine headaches at another point. In some patients the aura symp- toms occur alone on occasion and with typical headaches on other occasions. Some have only aura symptoms and never experience a head- ache. Terms such as migraine equivalent and migraine accompaniment have been used to describe these isolated aura symptoms, but a diagnosis is difficult to arrive at without the presence of typical headaches at some time during the course.
Basilar Migraine
In 1961, Bickerstaff described a subset of migraine with aura characterized by a combi- nation of symptoms that he felt reflected isch- emia within the distribution of the basilar artery.88 Most of Bickerstaff’s patients were adolescent girls, but subsequent reports have confirmed that basilar migraine can occur in both sexes at any age. Vertigo, tinnitus, and
decreased hearing are common symptoms with basilar migraine and could confound the dif- ferential diagnosis between basilar migraine and Meniere’s syndrome since these same symptoms are characteristic of the latter dis- ease. The IHS criteria for the diagnosis of basi- lar migraine require an aura that contains two or more of the symptoms listed in Table 12–1.85 Patients with isolated episodes of vertigo do not meet the criteria for basilar migraine.
Migrainous Vertigo
The IHS currently does not have criteria for the diagnosis of migrainous vertigo. However, Neuhauser and Lempert have suggested crite- ria that are becoming generally accepted.3,6 The criteria include the following: (1) recur- rent vertigo attacks, (2) migraine according to IHS criteria, (3) migrainous symptoms during at least two vertiginous attacks (headache, pho- tophobia, phonophobia, or aura symptoms), and (4) vertigo cannot be attributed to another disorder. Patients who meet criteria 1, 2, and 4, but not 3 are considered probable migrainous vertigo. Using these criteria in a screen of the general population in Germany, Neuhauser et al.10 concluded that migrainous vertigo is underdiagnosed and has considerable personal and health-care impact.
Treatment of migraine can be divided into three general categories: symptomatic, abor- tive, and prophylactic. Some drugs are useful in ameliorating the symptoms of the acute attack, others can abort an attack if taken just after the onset, and still others are effective in reducing the frequency and severity of attacks or eliminating their occurrence entirely.
Before embarking on drug treatment of migraine, it is important to recognize that there are many common triggers for migraine symp- toms (Table 12–5). It is often helpful to have the patient keep a log of his or her migraine attacks, noting any possible triggers that might be regularly associated with the attacks. Though rigorous clinical trials are lacking, knowledge of triggers and focusing on general lifestyle fac- tors can go a long way in reducing the burden
Table 12–5 Common Factors that Trigger Migraine Symptoms
![]()
Stress, emotional upset
Hormones: menstruation, oral contraceptives, pregnancy
Sleep deprivation
Food: red wines, fermented cheeses, chocolate, coffee
Eating disorders: fasting, binges
![]()
of migraine symptoms in many patients. The general lifestyle factors that are important include stress management, good quality sleep, awareness of food-related factors, and cardio- vascular exercise. As a general rule, migraineurs have fewer symptoms when following a regi- mented schedule to ensure all of these factors are adequately addressed. These factors are perhaps more important for the management of migrainous vertigo than for migraine head- aches because there are no good trials of the effect of abortive or prophylactic medication treatment for migrainous vertigo, whereas there are adequate trials demonstrating the efficacy of medicines to manage migraine headaches. Although we presume similar mechanisms cause headaches and vertigo, we cannot presume the same benefit of these medications.
Symptomatic and Abortive Treatment
Symptomatic and abortive treatment for migrainous symptoms largely depends on which symptoms the patient is most bothered by. Headache symptoms, by far, have the most research to support the use of medicines, and society guidelines have been published. 89 The treatments available for acute attacks of migraine headaches include serontonin recep- tor agonsists (i.e, triptans), antiemetics, analge- sics, and combination preparations.
Clinical trials of abortive therapies for head- aches have also demonstrated that the medi- cines can reduce other symptoms of migraine attacks, including hypersensitivities (photopho- bia and phonophobia) and nausea. 90 However, no good clinical trial data exist to support the effect of abortive therapies for migraine vertigo. Only one randomized controlled trial
has been published on the treatment of migrainous vertigo with a triptan medication, but this trial had substantial limitations mainly stemming from the very few subjects enrolled.91 In this trial, 3 out of 8 patients randomized to zolmitriptan met the endpoint of vertigo symp- tom improvement at 2 hours, compared with 2 out of 9 patients taking placebo. Thus, the number of participants was much too small to make any conclusions about the effect of zol- mitriptan.
Symptomatic treatment of the vertigo attacks has also not been well studied specifically in migraine vertigo. As a result, migraine vertigo is managed symptomatically the same way that vertigo in general is managed. Antivertiginous and antiemetic medications are useful in reduc- ing the severity of the symptom (see Tables 19–2 and 19–3 in Chapter 19). Promethazine at 25 or 50 mg, orally or via suppository, is par- ticularly effective for the relief of both vertigo and nausea. Drugs in this class also have a sed- ative effect, which is probably one of the main reasons for a positive effect in patients with severe vertigo. However, the sedating effect typically limits the use of these medicines for less severe attacks. The decrease in gastric motility that occurs during migraine attacks can decrease the absorption of oral drugs as well as contribute to the nausea and vomiting. Metoclopramide (Reglan) promotes normal gastric motility and may improve absorption of oral drugs.
Prophylactic Treatment
Many studies of prophylactic treatments have demonstrated an average improvement in headache severity and frequency in patients with migraine headaches89 (Table 12–6). But, as with abortive and symptomatic treatments, no good trials of prophylactic agents have been conducted to measure the effect on vertigo attacks in a population of patients with migraine vertigo. As a result, we do not know whether migraine prophylactic medications are benefi- cial in patients with migraine vertigo.
A general sentiment exists in clinical care that migraine prophylactic medicines can be effective in reducing the frequency and severity of migraine vertigo attacks, but this sentiment is based on anecdotes and observa- tional studies. In the absence of data from rig- orous clinical trials, it is a large—and poten- tially dangerous—leap to assume that a therapy is effective.
All of the prophylactic treatment studies in migraine vertigo have profound shortcomings. In fact, no control groups were even used in any of the prophylactic treatment studies iden- tified in a recent review on migraine vertigo.92 Not using control groups is a major shortcom- ing because of the natural tendency for symp- toms such as vertigo attacks to improve over time in the absence of any treatment. In fact, close to 50% (9 out of 19) of the patients expe- rienced spontaneous remission (i.e, no attack
Table 12–6 Common Drugs for Treating Migraine
![]()
Class Actions Sample Drugs
![]()
Serotonergic 5HT1 agonist Sumatriptan
5HT2 antagonist Dihydroxyergotamine
5HT reuptake inhibitor Methysergide, amitriptyline, fluoxetine
Dopaminergic DRD2 antagonist, ion channel stabilization Prochlorperazine,a metoclopramidea Beta blockers 5HT2 antagonist Propranolol, atenolol
Calcium channel blockers
Non-steroidal
anti-inflammatory
Ion channel stabilization, neuronal protection, DRD2 antagonist
Prostaglandin suppression, neurogenic
inflammation
Flunarizine, nimodipine Aspirin, naproxen, ibuprofen
Anticonvulsants Ion channel stabilization, neuronal
inhibition
Valproate, gabapentine, topiramate
Carbonic anhydrase inhibitors
Ion channel stabilization Acetazolamide
![]()
aCan be used for both symptomatic and prophylactic treatment.
or very mild attacks) in the trial of zolmitriptan for migraine vertigo attacks.91 The authors spe- cifically point out that three of these patients had 10 or more attacks of vertigo within 12
Lempert T, Neuhauser H. Epidemiology of ver- tigo, migraine and vestibular migraine. J Neurol. 2009;256(3):333.
Cutrer FM, Baloh RW. Migraine-associated dizziness.
months prior to enrollment but then did not have even a single attack during the following 15 to 24 months. If no control group is used, then any “intervention” could appear to have a
Headache. 1992;32:300.
Baloh RW, Jen JC. Genetics of familial episodic ver- tigo and ataxia. Ann NY Acad Sci. 2002;956:338.
Neuhauser H, Lempert T. Vestibular migraine. Neurol Clin. 2009;27(2):379.
Rasmussen BK. Epidemiology of migraine. In:
treatment effect, which of course is the reason placebo-controlled trials are necessary.
When an important level of uncertainty exists about the potential benefits of a therapy, then
Olesen J, Tfelt-Hansen P, Welch KMA, et al. eds. The Headaches. 3rd ed. Philadelphia: Lippincott, Williams and Wilkins; 2006.
Rasmussen BK, Olesen J. Migraine with aura and
the impact of the potential harms naturally increases. The main potential harms of migraine prophylactic agents include side effects and the associated costs of the medicines.
Despite the evidence void, physicians must
migraine without aura: an epidemiological study.
Cephalalgia. 1992;12:221,186.
Manzoni GC, Farina S, Lafranchi M, Solari A. Classic migraine—clinical findings in 164 patients. Eur Neurol. 1985;24:163.
Neuhauser HK, Radtke A, von Brevern M, et al.
still make treatment decisions in current patients presenting with migraine vertigo. Prophylactic migraine medications are reason- able options when patients are suffering from recurrent migraine vertigo attacks. But the first
Migrainous vertigo: prevalence and impact on quality of life. Neurology. 2006;67(6):1028.
Kayan A, Hood JD. Neuro-otological manifestations of migraine. Brain. 1984;107:1123.
Brantberg K, Trees N, Baloh RW. Migraine-associated vertigo. Acta Otolaryngol. 2005 Mar;125(3):276.
von Brevern M, Zeise D, Neuhauser H, Clarke AH,
area of emphasis should be for patients to search for and reduce triggers, and also to focus on the general lifestyle measures that can impact symptoms. It is when attacks persist despite these general measures that trials of prophylactic medicines are reasonable, though patients should be clearly informed about the limitations of these medicines and also about
Lempert T. Acute migrainous vertigo: clinical and oculographic findings. Brain. 2005;128(pt 2):365.
Ishiyama A, Jacobson KM, Baloh RW. Migraine and benign positional vertigo. Ann Otol Rhinol Laryngol. 2000;109(4):377.
von Brevern M, Radtke A, Clarke AH, Lempert T. Migrainous vertigo presenting as episodic positional vertigo. Neurology. 2004;62(3):469.
Harker LA. Migriane-associated vertigo. In: Baloh
the optimal use of them.
For the same reasons already mentioned, there is no strong evidence to support the use of one typical prophylactic agent over another. One option for selecting prophylactic medica-
RW, Halmagyi GM, eds. Disorders of the Vestibular
System. New York: Oxford University Press; 1996.
Battista RA. Audiometric findings of patients with migraine-associated dizziness. Otol Neurotol. 2004;25(6):987.
Parker W. Migraine and the vestibular system in
tions in individual patients is to base the choice of agent on the presence of comorbidities. Thus, patients with hypertension could be started on a beta-blocker or verapamil; patients with depression or anxiety could be started on a
adults. Am J Otol. 1991;12:25.
Olsson JE. Neurotologic findings in basilar migraine.
Laryngoscope. 1991;101(suppl 52):1.
Lipkin AF, Jenkins HA, Coker NJ. Migraine and sud- den sensorineural hearing loss. Arch Otolaryngol Head Neck Surg. 1987;113:325.
Viirre ES, Baloh RW. Migraine as a cause of sudden
tricyclic amine or serotonin reuptake inhibitor; and patients with epilepsy could be started on one of the antiseizure agents (i.e, valproic acide, topiramate). In patients with benign recurrent vertigo, acetazolamide is also an option.
REFERENCES
Goadsby PJ, Lipton RB, Ferrari MD. Migraine— current understanding and treatment. N Engl J Med. 2002;346:257.
Liveing E. On Megrim: Sick Headache and Some Allied Health Disorders: A Contribution to the Pathology of Nerve Storms. London: Churchill; 1873.
hearing loss. Headache. 1996;36:24.
Lee H, Whitman GT, Lim JG, et al. Hearing symptoms in migrainous infarction. Arch Neurol. 2003;60(1): 113.
Sturzenegger MH, Meienberg O. Basilar artery migraine: a follow-up study of 82 cases. Headache. 1985;25:408.
Kirchmann M, Thomsen LL, Olesen J. Basilar-type migraine: clinical, epidemiologic, and genetic features. Neurology. 2006;66(6):880.
Harker LA, Rassekh HC. Episodic vertigo in basi- lar migraine. Otolaryngol Head Neck Surg. 1987;96: 239.
Meniere P. Memoir sur des lesions de l’orreile interne donnant lieu a des symptomes de congestion cerebral apoplectiforme [in Frenc]. Gaz Med Paris. 1861;16:597.
Atkinson M. Migraine and Meniere’s disease. Arch Otolaryngol. 1962;75:220.
Rassekh CH, Harker LA. The prevalence of migraine in Meniere’s disease. Laryngoscope. 1992;102:135.
Parker W. Meniere’s disease. Etiologic considerations.
Arch Otolaryngol Head Neck Surg. 1995;121:377.
Baloh RW. Neurotology of migraine. Headache. 1997;37:615.
Radtke A, Lempert T, Gresty MA, Brookes GB, Bronstein AM, Neuhauser H. Migraine and Ménière’s disease: is there a link? Neurology. 2002;59(11): 1700.
Cha YH, Kane MJ, Baloh RW. Familial clustering of migraine, episodic vertigo, and Ménière’s disease. Otol Neurotol. 2008;29(1):93.
Harno H, Hirvonen T, Kaunisto MA, et al. Subclinical vestibulocerebellar dysfunction in migraine with and without aura. Neurology. 2003;61(12):1748.
Baier B, Stieber N, Dieterich M. Vestibular-evoked myogenic potentials in vestibular migraine. J Neurol. ePub ahead of print, Apr 18, 2009.
Marcelli V, Furia T, Marciano E. Vestibular path- ways involvement in children with migraine: a neuro- otological study. Headache. ePub ahead of print, May 27, 2009.
Basser LS. Benign paroxysmal vertigo of childhood: a variety of vestibular neuronitis. Brain. 1964;87:141.
Marcelli V, Piazza F, Pisani F, Marciano E. Neuro- otological features of benign paroxysmal vertigo and benign paroxysmal positioning vertigo in children: a follow-up study. Brain Dev. 2006;28(2):80.
Mira E, Piacentino G, Lanzi G, Ballotin U, Fazzi E. Benign paroxysmal vertigo in childhood: a migraine equivalent. ORL J Otorhinolaryngol Relat Spec. 1984;46:97.
Lanzi G, Ballontin U, Fazzi E, Tagliasacchi M, Manfrin M, Mira E. Benign paroxysmal vertigo of childhood: a long-term follow-up. Cephalalgia. 1994;14:458.
Slater R. Benign recurrent vertigo. J Neurol Neurosurg Psychiatry. 1979;42:363.
Moretti G, Manzoni GC, Caffara P, Parma M. “Benign recurrent vertigo” and its connection with migraine. Headache. 1980;20:344.
Oh AK, Lee H, Jen JC, Corona S, Jacobson KM, Baloh RW. Familial benign recurrent vertigo. Am J Med Genet. 2001;100(4):287.
Lewis DW, Pearlman E. The migraine variants.
Pediatr Ann. 2005;34(6):486, 490, 494.
Fisher CM. Late-life migraine accompaniments as a cause of unexplained transient ischemic attack. J Can Sci Neurol. 1980;7:9.
Baloh RW. Genes and migraine. Drugs Today. 2004;40:577.
Baloh RW. The genetics of migraine. In: Pulst SM, ed. Neurogenetics. New York: Oxford University Press; 2000.
Palotie A, Baloh RW, Wessman M. Genetics of migraine. In: King RE, Rotter JI, Motulsky A, eds. The Genetic Basis of Common Diseases. New York: Oxford University Press; 2001.
Cha YH, Baloh RW. Migraine associated vertigo.
J Clin Neurol. 2007;3(3):121.
Eggers SD. Migraine-related vertigo: diagnosis and treatment. Curr Pain Headache Rep. 2007;11(3): 217.
Lee H, Jen JC, Cha YH, Nelson SF, Baloh RW. Phenotypic and genetic analysis of a large fam- ily with migraine-associated vertigo. Headache. 2008;48(10):1460.
Ducros A, Thomsen LL. Sporadic and familial hemi- plegic migraines. In: Olesen J, Tfelt-Hansen P, Welch KMA, et al. (eds.) The Headaches. 3rd ed. Philadelphia: Lippincott, Williams and Wilkins; 2006.
Russell, M.B, Olesen, J. The genetics of migraine without aura and migraine with aura. Cephalalgia. 1993;13:245.
Thomsen LL, Ostergaard E, Olesen J, Russell MB. Evidence for a separate type of migraine with aura: sporadic hemiplegic migraine. Neurology. 2003;60: 595.
Haan J, Terwindt GM, Ophoff RA, et al. Is famil- ial hemiplegic migraine a form of basilar migraine? Cephalalgia. 1995;15:477.
Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hemiplegic migraine and episodic ataxia Type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543.
De Fusco M, Marconi R, Silvestri L, et al. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump on 2 subunit associated with familial hemiple- gic migraine type 2. Nature Genet. 2003;33:192.
Ducros A, Denier C, Joutel A, et al. The clinical spec- trum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N Engl J Med. 2001;345:17.
Denier C, Ducros A, Vehdi K, et al. High prevalence of CACNA1A truncations and broader clinical spec- trum in episodic ataxia type 2. Neurology. 1999;52: 1816.
Jen J, Yue Q, Nelson SF, et al. A novel nonsense muta- tion in CACNA1A causes episodic ataxia and hemiple- gia. Neurology. 1999;53:34.
Ducros A, Joutel A, Vahedi K, et al. Mapping of a sec- ond locus for familial hemiplegic migraine to 1q21-q23 and evidence for further heterogeneity. Ann Neurol. 1997;42:885.
Terwindt G, Kors E, Haan J, et al. Mutation analysis of the CACNA1A calcium channel subunit gene in 27 patients with sporadic hemiplegic migraine. Headache. 2003;43:303.
Jen J, Kim GW, Dudding KA, Baloh RW. No mutations in CACNA1A and ATP1A2 in probands with common types of migraine. Arch Neurol. 2004;61:926.
Kors EE, Haan J, Giffin NJ, et al. Expanding the phenotype spectrum of the CACNA1A gene T666M mutation. A description of 5 families with familial hemiplegic migraine. Arch Neurol. 2003;60:684.
Terwindt GM, Ophoff RA, van Eijk R, et al. Involvement of the CACNA1A gene containing region on 19p13 in migraine with and without aura. Neurology. 2001;56:1028.
Lea RA, Shepard AG, Curtain RP, et al. A typical migraine susceptibility region localizes to chromosome 1q31. Neurogenetics. 2002;4:17.
Kim JS, Yue Q, Jen JC, Nelson SF, Baloh RW. Familial migraine with vertigo: no mutations found in CACNA1A. Am J Med Genet. 1998;79:148.
Weiser T, Mueller C, Evers S, Zierz S, Deufel T. Absence of known familial hemiplegic migraine (FHM) mutations in the CACNA1A gene in patients
with common migraine: implications for genetic test- ing. Clin Chem Lab Med. 2003;41:272.
Bain PG, O’Brien MD, Keevil SF, Porter DA. Familial periodic cerebellar ataxia: a problem of cerebellar intra- cellular pH homeostasis. Ann Neurol. 1992;31:147.
Lauritzen M. Pathophysiology ofthe migraine aura. The spreading depression theory. Brain. 1994;117:199.
Farkkila M. The pathophysiology of migraine. Ann Med. 1994;26:7.
Hadjikhani N, Sanchez Del Rio M, Wu O, et al. Mechanisms of migraine aure revealed by functional MRI in human visual cortx. Proc Natl Acad Sci USA. 2001;98:4687.
Woods RP, Iacoboni M, Mazziotta JC. Brief report: bilateral spreading cerebral hypoperfusion dur- ing spontaneous migraine headache. N Engl J Med. 1994;331:1689.
Lipton RB, Ottman R, Ehrenberg BL, Hauser WA. Comorbidity of migraine: the connection between migraine and epilepsy. Neurology. 1994;44:S28.
Montagna P, Cortelli P, Barbiroli B. Magnetic reso- nance spectroscopy studies in migraine. Cephalalgia. 1994;14:184.
Welch KM, Levine SR, D’Andréa G, Schultz LR, Helpern JA. Preliminary observations on brain energy metabolism in migraine studied by in vivo phospho- rous 31 NMR spectroscopy. Neurology. 1989;39:538.
Schlake HP, Bottger IG, Grotemeyer KH, Husstedt IW, Oberwittler C, Schober O. The influence of aceta- zolamide on cerebral low-flow regions in migraine—an interictal 99mTc-HMPAO SPECT study. Cephalalgia. 1992;12:267, 284.
Moskowitz M, Bolay H, Dalkara T. Deciphering migraine mechanisms: clues from familial hemiplegic migraine. Ann Neuro. 2004;55:276.
Ayata C, Jin H, Kudo C, Dalkara T, Moskowitz M. Suppression of cortical spreading depression in migraine prophylaxis. Ann Neurol. 2006;59:652.
Hupp SL, Kline LB, Corbett JJ. Visual disturbances of migraine. Surv Ophthalmol. 1989;33:221.
Winterkorn JM, Kupersmith MJ, Virtschfter JD, Forman S. Brief report: treatment of vasospas- tic amaurosis fugax with calcium-channel blockers. N Engl J Med. 1993;329:396.
Grosberg BM, Solomon S, Friedman DI, Lipton RB. Retinal migraine reappraised. Cephalalgia. 2006;26:1275.
Kruit MC, van Buchern MA, Launer LJ, Terwindt GM, Ferrari MD. Migraine is associated with an increased risk of deep white matter lesions, subclinical posterior circulation infarcts and brain iron accumulation: the population-based MRI CAMERA study. Cephalalgia. ePub ahead of print, June 8, 2009.
Kruit MC, Launer LJ, Ferrari MD, van Buchem MA. Brain stem and cerebellar hyperintense lesions in migraine. Stroke. 2006;37:1109.
Kruit MC, Launer LJ, Ferrari MD, van Buchem MA. Infarcts in the posterior circulation territory in migraine. The population-based MRI CAMERA study. Brain. 2005;128:2068.
Headache Classification Committee of the International Headache Society. The international clas- sification of headache disorders, 2nd ed. Cephalalgia. 2004;24(suppl 1):1.
Blau JN. Migraine: theories of pathogenesis. Lancet. 1992;339:1202.
Aromaa M, Sillanpää ML, Rautava P, Helenius H. Childhood headache at school entry. A controlled clinical study. Neurology. 1998;50:1729.
Bickerstaff ER. Basilar artery migraine. Lancet. 1961;1:15.
Silberstein S. Practice parameter: Evidence based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommitte of the American Academy of Neurology. Neurology. 2000;55:754.
Brandes JL, Kudrow D, Stark SR. Sumatriptan- naproxen for acute treatment of migraine: a random- ized trial. JAMA. 2007;297:1443.
Neuhauser H, Radtke A, von Brevern M, Lempert T. Zolmitriptan for treatment of migrainous vertigo: a pilot randomized placebo-controlled trial. Neurology. 2003;60:882.
Fotuhi M, Glaun B, Quan SY, Sofare T. Vestibular migraine: a critical review of treatment trials. J Neurol. 2009;256(5):711.
This page intentionally left blank
![]()
AUTOIMMUNE INNER EAR DISEASE
Background Pathophysiology Clinical Features Diagnosis Management
PARANEOPLASTIC IMMUNE DISORDERS
Background Pathophysiology Clinical Features
Diagnosis Management MULTIPLE SCLEROSIS
Background Pathophysiology Clinical Features Diagnosis Management
The immune system is implicated in many neu- rologic diseases. There are some primary immune-based neurologic diseases, such as multiple sclerosis and myasthenia gravis, and also many secondary immune-based diseases, including paraneoplastic syndromes. In pri- mary immune-mediated diseases the cause is typically not known, but genetic and environ- mental factors undoubtedly play a role. On the other hand, in secondary immune-based dis- eases a known inciting event occurs, typically an infection, cancer, or environmental stimu- lus, in the setting of genetic susceptibility. Immune-mediated disorders are important to identify because in many cases effective thera- pies are available. In the absence of adequate therapeutic trials, identifying an immune response can at least direct therapeutic approaches for what can otherwise be severely disabling deficits.
In this chapter, we focus on the immune- mediated syndromes that can involve the peripheral audio-vestibular system and also the central vestibular system.
Background
The endolymphatic sac is the key structure for immunoregulation within the inner ear.1 Ultra- structural studies have identified lymphocytes and macrophages within the endolymphatic sac and a surrounding network of lymphatic vessels containing these mononuclear cells.2 Injection of exogenous antigens into the inner ear of a guinea pig is as effective for initiating systemic immunity as intraperitoneal injection. When an antigen is placed into the inner ear of animals that have been immunized against the antigen, immuno- competent cells infiltrate the ear and there is local production of antibody.3,4 The inner ear is dam- aged during the immune response, even though the antigen is not expressed in the inner ear. Surgical ablation of the endolymphatic sac or blockade of the endolymphatic duct markedly curtails the immunological response and inner
303
ear damage.5 Lymphocytes engaged in the immune response within the inner ear are recruited from the circulation, presumably via the spiral modiolar vein. With this secondary immune response, there is activation of spiral modiolar vein endothelium, release of cytokines such as interleukin-2, and up-regulation of adhesion molecule expression.6
The inner ear is also capable of mounting an immune response against one of its own anti- gens. Several groups have immunized mice, rats, and guinea pigs with specific inner ear antigens and produced inner ear damage.7–11 The histopathology of the inner ear in these ani- mals shows degenerative changes with mono- nuclear cell infiltration. Antibodies directed against several different inner ear proteins have been identified in these animals, but so far, pas- sive transfer of injury with these antibodies has not been produced. Sensitized lymphocytes, however, can produce inner ear damage and sensorineural hearing loss after passive transfer to other animals.11–13 Although there are still many unanswered questions regarding these animal models of immune-mediated inner ear damage and how they translate to humans, they clearly show that the inner ear can be damaged either directly by attack on inner ear proteins or indirectly by a general immune response against an irrelevant antigen.
In 1958 Lehnhardt14 was the first to suggest that bilateral deafness might be caused by anti- cochlear antibodies. In 1979, McCabe described the clinical syndrome of autoimmune inner ear disease.15 He reported a series of patients with rapidly progressive bilateral sensorineural hear- ing loss often associated with vertigo or imbal- ance. There was histopathologic evidence com- patible with vasculitis in a temporal bone biopsy from one patient and evidence of cell-mediated immune responses to inner ear membranous tissue from several other patients. The most convincing evidence of immune-mediated damage, however, was the response to corticos- teroids and cytotoxic drug therapy. Subsequently, there have been numerous simi- lar case reports of patients with this clinical profile who often show good response to immu- nosuppression with corticosteroids and other cytotoxic agents. However, the effect of corti- costeroids was variable in an open-label study (about 58% [67/116] of those enrolled met the criteria for improved hearing after initiating corticosteroid treatment), and there was no
average beneficial effect of a cytotoxic agent (methotrexate) in one large randomized clinical trial.16 Autoantibodies have been identified in some patients, but, as in the case of the animal models, it is unclear whether the antibodies are involved in the pathogenesis or are simply an epiphenomenon (see later discussion).
Pathophysiology
A wide range of multisystem autoimmune dis- orders can involve the inner ear (Table 13–1). With these disorders, there appears to be an episodic breakdown in immunologic tolerance to self-molecules. These autoimmune diseases are thought to result from a defect in a cell sur- face protein on T cells required for the intra- thymic death of autoreactive T lymphocytes. The defective protein prevents negative selec- tion of autoreactive T cells so that there are excessive numbers of circulating autoreactive T cells. In a mouse model with this T cell surface protein defect (the MRL mouse), cochlear damage occurs in the early stages of the systemic illness.17,18
There are three major mechanisms by which an immune-mediated disease can damage the inner ear: (1) deposition of antigen antibody immune complexes in the tissue; (2) autoanti- bodies directed against inner ear antigens; and
(3) infiltration and destruction of inner ear tissue by specific cytotoxic T cells. A wide range of non-organ-specific autoimmune disorders presumably damages the inner ear via the first mechanism.16 Temporal bone specimens from patients with polyarteritis nodosa, systemic
Table 13–1 Systemic Immunologic Disorders that Involve the Inner Ear15
![]()
Vasculitis Syndromes Polyarteritis nodosa Wegener’s granulomatosis Bechet’s syndrome Cogan’s syndrome
Connective Tissue Diseases Systemic lupus erythematosus Sjögren’s syndrome Rheumatoid arthritis
Vogt-Koyanagi-Harada syndrome Relapsing polychondritis Ulcerative Colitis
![]()
lupus erythematosus, and Wegener’s granulo- matosis show evidence of small vessel vasculitis with inflammation and ischemic changes. Endolymphatic hydrops has been found in some specimens, possibly secondary to the inflammatory changes. Cogan’s syndrome,19 which is characterized by interstitial keratitis and inner ear involvement, may blur the dis- tinction between organ-specific and non– organ-specific inner ear immune-mediated dis- ease.20,21 Lunardi et al.22 used pooled IgG immunoglobulins from patients with Cogan’s syndrome to screen a random peptide library to identify disease-relevant autoantigens. They found an immunodominant peptide with a sequence similar to the cell-density enhanced protein tyrosine phosphatase-1 (DEP-1/ CD148) that is expressed on the sensory epi- thelia of the inner ear and on endothelial cells. Antibodies directed against the “Cogan pep- tide” were able to transfer the disease to other animals and mice or rabbits immunized with the Cogan’s peptide or a different peptide from DEP-1/CD148; the animals developed hearing loss and interstitial keratitis. Furthermore, antibodies against the Cogan peptide cross- reacted with a structural protein of the reovirus type III, suggesting the possibility of molecular mimicry as a disease mechanism. About 50% of patients with Cogan’s syndrome report an upper respiratory infection prior to the onset of disease.
Central to the notion of an organ-specific immune disease is that antibodies or immune cells are directed at a specific inner ear antigen. Numerous clinical studies have examined cel- lular and humoral immune reactivity to a vari- ety of inner ear tissues from humans and ani- mals with immune-mediated inner ear disease.23 Cellular immune assays using lymphocyte migration inhibition and lymphocyte transfor- mation have produced some positive findings, but these assays are technically demanding and are not very sensitive.24 Detection of serum antibodies directed at inner ear antigens is a much simpler procedure and thus has potential to lead to a clinically useful laboratory test. Many patients with the typical syndrome of idiopathic bilateral progressive sensorineural hearing loss first described by McCabe have antibodies in their serum that bind a 68 kDa protein on Western blot analysis.25–27 This 68 kDa protein is not species specific, since it is detectable using human, bovine, and guinea
pig inner ear tissue, and it is not even organ specific, since it is present in many other tis- sues. There is now convincing evidence that the antigen recognized by these antibodies is heat-shock protein 70 (HSP-70), a highly con- served molecule that is up-regulated during times of stress.28,29 Whether HSP-70 located in the inner ear is the target of these antibodies or simply shares an epitope with the true target antigen is unknown. However, the level of these anti-HSP-70 antibodies correlates both with the activity of disease and the steroid responsiveness of the disease.29 The HSPs are ubiquitous in all normal cells and are desig- nated as chaperone proteins because of their role in aiding protein folding. Since they are up-regulated in response to injury or stress, antibodies directed against them may be a sec- ondary phenomenon unrelated to the underly- ing cause of injury.30
Numerous investigators have shown that serum from some patients with immune-medi- ated inner ear disease contains antibodies directed against specific inner ear structures, using immunofluorescence.31 However, there has been no consistent pattern of staining and no specific cellular or subcellular structure consistently stained. Disher and colleagues30 noted that patient sera containing antibodies directed against the 68 kDa protein produced a characteristic punctate “wine glass” staining pattern of the supporting cells in the guinea pig organ of Corti. Such a staining pattern was never seen with control sera. These authors concluded that, because of the localized stain- ing pattern, it was unlikely that HSP-70 was the primary target of these autoantibodies. Baek et al.32 found that patients with autoim- mune inner ear disease had a high frequency of circulating T cells producing interferon gamma or IL-5 in response to recombinant cochlin, the most abundant inner ear protein. Patients also showed significantly elevated cochlin-specific serum antibody titers compared to controls or patients with other causes of hearing loss. Cochlin expression is largely confined to the inner ear and mutations in the COCH gene that codes for cochlin causes progressive cochlear and vestibular pathology.
So far there have been only a few histopatho- logic studies of temporal bone specimens from patients with inner ear-specific immunologic disease. Schuknecht33 reported infiltration of lymphocytes, plasma cells, and macrophages in

B
Figure 13–1. Spiral modiolar vein of patient with auto- immune inner ear disease showing perivascular round cell infiltration (A) and new bone formation (B) in an area of intense inflammation (Courtesy, Harold F Schuknecht MD).
the endolymphatic and perilymphatic spaces and within the tympanic lamellae and spiral ligaments (Fig. 13–1). Osteoneogenesis and fibrous tissue were present in the scala tympani adjacent to the round window and dense bone formation surrounded the spiral modiolar vein and modiolus. There were also focal mononu- clear infiltrates in the cochlear and vestibular nerve trunks. Although hydrops was also pres- ent, the pathology was distinct from that of typ- ical Meniere’s disease. These findings are also typical of those observed in experimental ani- mals undergoing chronic antigen stimulation.
Clinical Features
SYSTEMIC IMMUNE-MEDIATED DISEASES WITH INVOLVEMENT OF THE INNER EAR
Systemic lupus erythematosus (SLE) is the pro- totypical immune complex disease. Both sudden
unilateral and progressive bilateral sensorineu- ral hearing loss have been well documented with SLE.34,35 In a case series of female SLE patients, 57.5% (23/40) had impaired hearing, particularly in the low frequencies.36 In some cases, fluctuating low-frequency hearing loss along with episodic vertigo suggests the diagnosis of endolymphatic hydrops. The most consistent inner ear pathology is a vasculitis, presumably caused by deposition of circulating immune complexes in the walls of blood ves- sels.37 In theory at least, the inner ear—just like the kidney—is a prime target in SLE because there are antigenic similarities between the stria vascularis and the glomerular basement mem- brane and both structures show deposits of immune complexes.38
Polyarteritis nodosa (PN) is a necrotizing vasculitis of small muscular arteries that com- monly involves the inner ear; hearing loss is occasionally the initial symptom.24,39 Patients can have hearing loss for several years before the systemic features of polyarteritis become evident. Obliterative vasculitis of the labyrin- thine artery can result in ischemic necrosis of the entire labyrinth. Wegener’s granulomatosis is another vasculitic syndrome characterized by necrotizing granulomas primarily within the upper respiratory tract, lung parenchyma, and kidneys. Although both acute and chronic sen- sorineural hearing loss occur with Wegener’s granulomatosis, a conductive hearing loss due to otitis media is a more common finding.40 Behçet’s disease is a rare vasculitic syndrome characterized by aphthous ulcers, genital ulcers, and iritis or uveitis. Auditory and ves- tibular symptoms due to inner ear involvement are common at any time during the course.41 A small percentage of patients with Sjogren’s syndrome have a progressive bilateral hearing loss presumably secondary to infiltration of the inner ear by autoreactive T cells.42
As noted earlier, Cogan’s syndrome is typi- fied by audiovestibular symptoms and recur- rent nonsyphilitic interstitial keratitis.19–21 Initially, the clinical picture is indistinguishable from Meniere’s syndrome with episodic vertigo and fluctuating hearing loss, but the course is more fulminant, typically rapidly leading to deafness if untreated. Although the disorder can begin with either eye or ear involvement, both organs are usually involved within 1 year of onset. Cogan’s syndrome has features of both non–organ-specific and organ-specific immune-mediated disease. In most cases, there
Inner ear involvement occurs in about 50% of patients with relapsing polychondritis.24 This rare condition is characterized by recurrent episodes of inflammatory necrosis of cartilage.43 Conductive hearing loss is also common because of swelling and degeneration of carti- lage in the middle ear.44 Autoantibodies directed against collagen type 2 and type 9 and other cartilage components have been identi- fied with this disorder, but it is unclear whether the inner ear damage is due to vasculitis or to these collagen autoantibodies.
AUTOIMMUNE INNER EAR DISEASE
The typical presentation of immune-mediated inner ear disease is a rapidly progressive bilat- eral sensorineural hearing loss that extends beyond the arbitrary 72 hr typical of sudden sensorineural hearing loss.15,45 Moscicki and colleagues26 defined the following criteria for the disorder: (1) bilateral sensorineural hearing threshold of 30 dB at any frequency, and
(2) evidence of progression in at least one ear on two serial audiograms performed 3 months apart (progression being defined as a threshold loss of 15 dB at one frequency or 10 dB at two or more consecutive frequencies). Fluctuating hearing loss qualifies if there is also progression as defined by the criteria. Most investigators have found a female preponderance, although others have not.29,46 Immune-mediated inner ear disease typically begins in the 30s and 40s, although there is a wide range of age of onset from childhood into the eighth decade. Vestibular symptoms typically occur in about 50% of patients. Most commonly, there are episodes of vertigo with or without episodes of fluctuating hearing loss. Occasionally, patients show just a deterioration in balance due to bilateral progressive vestibular loss, which can be identified at the bedside with the head-thrust test. Whether a subset of patients has just bilateral vestibular loss without hearing loss is unclear, although to date, no convincing patients with this syndrome have been reported. Arbusow et al.47 found autoanti- bodies directed at inner ear antigens in 8 of 12 patients with idiopathic bilateral vestibul- opathy and normal hearing, but autoantibody
levels did not correlate with clinical activity and 3 of these patients treated with immunosup- pressive therapy showed no benefit. Deutschlander et al.48 found antilabyrinthine or nervous tissue-specific serum antibodies in 12 patients with bilateral vestibulopathy and normal hearing, but only a few showed any response to immunosuppression treatment and it was unclear whether the response was spon- taneous or due to medication.
About 20% of patients with idiopathic pro- gressive bilateral sensorineural hearing loss who meet the diagnostic criteria for immune- mediated inner ear disease have a combination of fluctuating and progressive sensorineural hearing loss and episodic vertigo that meets the strict diagnostic criteria for Meniere’s syn- drome.29 About a third of patients with typical Meniere’s syndrome have been shown to have the anti–HSP-70 antibodies that are character- istic of immune-mediated inner ear disease.49 This suggests the possibility that a subset of patients in both diagnostic categories have a common pathophysiology.
Diagnosis
AUDIOVESTIBULAR TESTING
Serial audiograms performed every few months are necessary to confirm the presence of, and response to treatment for, bilateral progressive sensorineural hearing loss.50 There is no char- acteristic pattern of hearing loss with immune- mediated inner ear disease in which both up-sloping and down-sloping or even flat pat- terns occur. Occasionally, speech discrimina- tion is affected to a greater extent than hearing thresholds, although, in this case, one should consider a search for retrocochlear disease, such as that associated with multiple sclerosis (MS) or a vestibular schwannoma. Unilateral or bilateral decrease in caloric function will often correlate with the severity of hearing loss. A subset of patients will have severe auditory involvement without any evidence of vestibular dysfunction.
Routine serologic tests are recommended in patients with idiopathic bilateral progressive sensorineural hearing loss to evaluate for find- ings suggestive of systemic immune-mediated disease. The typical regimen would include a complete blood count with differential white count, erythrocyte sedimentation rate (ESR),
A wide range of antigen-specific laboratory tests have been recommended for the diagno- sis of immune-mediated inner ear disease, but to date, none of these tests are universally avail- able or have been demonstrated to be useful clinically. In his original description in 1979, McCabe15 reported that 6 of 18 patients with immune-mediated inner ear disease had a pos- itive migration inhibition test, using inner ear proteins for the antigenic stimulus. Hughes and colleagues52 at the Cleveland Clinic devel- oped a lymphocyte transformation test, also using inner ear proteins, and found abnormal results in about a quarter of patents with immune-mediated inner ear disease. Berger and colleagues53 used type 2 collagen in a lym- phocyte transformation test and reported a 50% positive rate in patients with immune- mediated inner ear disease versus a 6% posi- tive rate in controls. These cellular immune assays are probably too technically demanding for routine use in the clinical laboratory.54
Between one-third and two-thirds of patients with immune-mediated inner ear disease have anti–HSP-70 antibodies,23,28,29 but these anti- bodies can also be seen in normal subjects.55 The presence of anti-HSP-70 antibodies seems to be correlated with disease activity and with steroid responsiveness.25 Rauch estimated that a positive test indicated as much as a 75% chance of steroid responsiveness, whereas a negative test indicated a chance of response of
<20%.25 However, controlled studies are needed to confirm these observations, and it is unlikely that clinicians would withhold a trial of corticosteroids unless the test could identify patients with a probability of improved recov- ery very close to zero.
Management
The mainstay of treatment of immune- mediated inner ear disease is immune suppres- sion with high-dose corticosteroids.24,29,56 Unfortunately, there have been no prospective
randomized clinical trials of steroid treatment of immune-mediated inner ear disease. Because a short course of corticosteroids is associated with relatively few adverse effects and the medicine is inexpensive and widely available,57,58 most practitioners recommend a therapeutic trial of prednisone in the range of 1 mg/kg/day for 4 weeks, followed by tapering. Some patients will show an immediate response within hours to days of treatment, although many do not show improvement until later in the course of treatment. Shorter courses of treatment might lead to relapse.29 Hearing is typically tested at the initiation of therapy and at the end of the 4 weeks of high-dose steroids. If the threshold has improved by 15 dB at one frequency or 10 dB at two or more frequencies, the diagnosis of immune-mediated inner ear disease is confirmed and the patient is consid- ered a steroid responder. Patients who do not show a response after a month are tapered off the steroid over the next 12 days. Patients who respond to the initial month of therapy con- tinue on the full dosage until their audiogram shows that hearing levels have reached a pla- teau. The steroids are then slowly tapered over about 8 weeks and a smaller maintenance dose is continued for at least 6 months. Relapses are more common in patients who are treated for less than 6 months compared to those treated for 6 months or longer.29 Up to two-thirds of patients who meet the diagnostic criteria for autoimmune inner ear disease respond initially to steroids, but only about 15% continue to respond after 3 years. 16,50,57
Long-term corticosteroid treatment can lead to major side effects, including gastritis and ulcers, labile blood pressures, altered blood sugar metabolism and diabetes, mood changes, sleep disorders, accelerated cataract formation, cushingoid habitus, and ischemic bone necro- sis. All of these side effects tend to be more pronounced with long-term use; the risk dur- ing the initial 30-day high-dose steroids with rapid taper is relatively low.58 Despite these major long-term risks, corticosteroid therapy continues to be the mainstay for empiric treat- ment of immune-mediated inner ear disease.
Methotrexate and cyclophosphamide have been used as an alternative to systemic corti- costeroids, in combination with corticosteroids, or as an alternative to steroids in patients with unacceptable side effects.24,29 Most experience
has been with methotrexate using a low-dose protocol. An initial dosage of 7.5 mg/week (3 doses of 2.5 mg) is given for 2 weeks and, if there is not major toxicity, the dosage is dou- bled to 15 mg/week.29 This dosage is continued for 6 to 8 weeks and then, as with steroids, dis- continued in nonresponders and carried on for about 6 months in responders. The main toxic- ity associated with methotrexate includes myelosuppression, oral ulceration, gastrointes- tinal ulcers, and hepatic fibrosis. This latter severe complication is usually only seen when methotrexate is given for more than a year. In a randomized placebo-controlled trial, metho- trexate did not demonstrate an average benefi- cial effect compared to placebo for maintaining hearing improvement achieved with predni- sone (60 mg/day for 1 month followed by 18-week taper).16 In a pilot placebo-controlled study, the immunomodulatory drug etanercept was no better than placebo for treating autoim- mune inner ear disease.59
PARANEOPLASTIC IMMUNE DISORDERS
Background
Remote effects of cancer on the brain were first described at the end of the nineteenth century, but case reports were largely anec- dotal and, because similar syndromes occur without cancer, many investigators questioned the cause-and-effect relationship. Cancer can have remote effects on the nervous system, from the cortex to cranial and peripheral nerves, including the audiovestibular nerves. Whether there is a selective cochleovestibular paraneoplastic syndrome is unclear, but involvement of audiovestibular structures is common with paraneoplastic encephalomyeli- tis. Many cancers have been associated with paraneoplastic effects, but small-cell carcinoma of the lung is most common. In the majority of cases the underlying cancer is undetectable at the time the neurological symptoms present, so the diagnosis can be difficult.60 The discovery of antineuronal antibodies not only provides a diagnosis for the neurotologic symptoms but also may direct the search for a specific cancer type.
Pathophysiology
Paraneoplastic syndromes result from an immune-based attack on the nervous system, usually against neurons.61 Presumably, the tumor expresses antigens that are also expressed by neurons. The immune system recognizes the proteins expressed by the tumor as foreign and mounts an immune attack to control tumor growth. In many cases, the immune response is so effective that the tumor remains small and may not be detectable. The immune response consists of both an antibody and cytotoxic T cell response. In paraneoplastic syndromes affecting the brain, inflammatory infiltrates of T cells and plasma cells occur in both the ner- vous system and in the tumor. The same anti- bodies are found in the brain and the tumor. Different antibodies occur with different tumors and clinical syndromes (Table 13–2).61 These antibodies are directed against a num- ber of onconeural antigens, all important in neuronal function. In many cases the antibod- ies associated with paraneoplastic syndromes have been used as probes to immunohis- tochemically localize the antigen in the nervous system and to clone the gene from complimen- tary DNA expression libraries of genes that code for the onconeural antigens.61 Many of the genes associated with these antigens appear to be important in early neuronal development and in the regulation of gene expression.
The characteristic pathologic findings with paraneoplastic syndromes are perivascular cuffing by lymphocytes, activation of microglia with the formation of microglial nodules, and selective loss of neurons. The process is pre- dominantly, although not exclusively, confined to gray matter. Immunoglobulin G (IgG), nor- mally absent from the brain, can be found not only within the neuropil but also within neu- rons. These antibodies found in the brain are directed against specific onconeural antigens that are expressed within the tumor in the same patient.
Clinical Features
CEREBELLAR DEGENERATION
The clinical symptoms and signs of patients with paraneoplastic cerebellar degeneration are similar to those of patients with other diffuse
Table 13–2 Antineuronal Antibody Paraneoplastic Disorders
Antibody | Associated Cancer | Syndrome | Brain Antigen | Onconeuronal Antigen |
Anti-Hu | SCLC and | Encephalomyelitis, | All neuronal nuclei, | HuD, HuC, |
neuroblastoma | sensory neuropathy | 35–40 kDa | Hel-Nl | |
Anti-Yo | Gynecologic and | Cerebellar ataxia | Cytoplasm Purkinje | CDR34, CDR62-1, |
breast | cells, 34 and 62 kDa | CDR62-2 | ||
Anti-Ri | Breast, | Cerebellar ataxia, | Neuronal nuclei CNS, | NOVA1, NOVA2 |
gynecologic, | opsoclonus | 55 and 80 kDa | ||
and SCLC | ||||
Antiamphiphysin | Breast | Stiff-person, | Synaptic vesicles, | Amphiphysin |
encephalomyelitis | 128 kDa | |||
Anti-Ma | Multiple | Cerebellar ataxia, | Neuronal nuclei and | Mai, Ma2 |
brain stem | cytoplasm, 37 and |
dysfunction
Anti-Ta Testicular Limbic encephalitis,
brain stem dysfunction
40 kDa
Neuronal nuclei and cytoplasm, 40 kDa
Ma2
Anti-Tr Hodgkin’s lymphoma
Cerebellar ataxia Cytoplasm neurons,
Purkinje cells, spiny dendrites
Unknown
Anti-CV2 SCLC and others Encephalomyelitis,
cerebellar ataxia
Glia (subset), 66 kDa POP66
Anti-GAD Multiple Stiff-person, Cerebellar ataxia
Glutamic acid decarboxylase
Unknown
Anti-VGKC Multiple Encephalomyelitis Voltage gated potassium
![]()
channels
Unknown
CNS, central nervous system; SCLC, small cell lung cancer.
Source: Data modified from Dalmau and Posner.61
cerebellar lesions. The trunk and extremities are ataxic and the speech is usually slurred. The course is subacute but can be fulminant. Often intractable nausea and vomiting occur early. Most patients are nonambulatory within 2-4 months from the onset of symptoms.62 Less commonly patients can remain ambulatory throughout the course.62 After about 6 months the symptoms generally stabilize. 63 All types of spontaneous nystagmus are seen, but downbeat nystagmus is most specific for cerebeller involve- ment.64,65 Gaze-evoked and rebound nystagmus are also common. Eye movement recordings document impairment of smooth pursuit, opto- kinetic nystagmus, and fixation-suppression of vestibular nystagmus. Saccades may be dysmet- ric, but peak velocity remains normal. Vestibular responses are normal or hyperactive.
Patients with anti-Ri antibodies may have a less severe course (less likely to have nystag- mus and more likely to be ambulatory) and also a better outcome when compared to patients with paraneoplastic cerebellar degeneration
associated with anti-Yo, anti-Hu, and anti-Tr antibodies. 63
OPSOCLONUS
A dramatic neurophthalmologic paraneoplastic syndrome is the opsoclonus-myoclonus syn- drome.66 This disorder is characterized by cha- otic conjugate eye movements in all directions, along with myoclonus of the trunk and limbs. It is usually seen in children with neuroblastoma, although it also occurs in adults with occult malignancies in the lung, ovaries, uterus, and breast. With opsoclonus, conjugate saccades occur continuously in all directions, often with torsional components.65 Eye movement record- ings document that the saccades have normal peak velocity for their amplitude.
ENCEPHALOMYELITIS
Paraneoplastic encephalomyelitis can present with a wide range of central and peripheral
nervous system symptoms and signs.67 Involvement of the limbic system in the medial temporal lobes results in a combination of per- sonality change and memory deficits. Brain stem involvement can lead to prominent ver- tigo, nausea, and vomiting, with involvement of oculomotor pathways causing strabismus and nystagmus. Cranial and peripheral nerves are also commonly involved, leading to a combina- tion of central and peripheral signs. Often peripheral sensory nerves are involved to a much greater extent than motor nerves. Cases have been reported with a combination of encephalomyelitis and inner ear involvement, also possibly secondary to a paraneoplastic immune-based mechanism, but so far no anti- bodies directed at the specific inner ear anti- gens have been identified.68,69
Diagnosis
Although the role of the antibodies listed in Table 13–2 in the pathogenesis of the different paraneoplastic syndromes is still unclear, iden- tification of these specific antibodies in the clinical laboratory can help lead to a diagno- sis.61 Any patient with subacute cerebellar degeneration should be examined carefully for an occult malignancy. Antineuronal antibodies are identified in about 50% of patients with paraneoplastic cerebellar degeneration. The most specific antibody is the anti–Purkinje cell antibody found in women with gynecological tumors (the anti-Yo antibody).70 With immuno- histochemical staining, this antibody binds to the Purkinje cell cytoplasm, producing a char- acteristic “ring of pearls” on microscopic exam- ination of the cerebellum. It is so specific for gynecological tumors that surgical exploration of the pelvis is indicated in any woman with the antibody, even without an obvious malignancy, regardless of the outcome of other diagnostic studies. A subgroup of adults with opsoclonus and cerebellar ataxia (primarily truncal) has an antineuronal antibody called anti-Ri. Nearly all of these patients have cancer of the breast. The antineuronal nuclear antibody (anti-Hu) is most commonly associated with small-cell lung cancer and either a sensory neuropathy and/or encephalomyelitis.
If an antibody is detected, then the next step is to search for the primary malignancy.
The optimal method to search for a primary malignancy is not clearly defined and can vary from patient to patient depending on the clini- cal scenario and available resources. In addi- tion, the ultimate value of the tests used to search for a malignancy (i.e., how having the tests impacts meaningful patient outcomes) has not been defined. In most cases, the initial imaging test is a whole-body computed tomog- raphy (CT) scan. If no mass is identified or if uncertainty about the etiology of an identified mass exists (e.g., benign pulmonary nodule ver- sus lung cancer), then fluoro-2-deoxy-glucose (FDG) positron emission tomography (PET) scan is typically the next step. In some cases, it may be reasonable to order a CT scan and PET scan simultaneously.71 The studies of body imaging to search for cancers in paraneoplastic presentations are all small cases series (10–80 patients) and have demonstrated concerning false-positive rates (typically infection or inflammation on PET scans, benign nodules on CT scans) and also false-negative rates (partic- ularly for small gynecologic cancers on CT scans, and slow-growing or very small tumors on PET scans).71 False-positive and false-nega- tive results may stem from suboptimal validity of the tests for malignancy in these presenta- tions or from suboptimal reliability (e.g., inter- observer agreement) of the interpretation of test results.
Even if an antibody is not detected in the serum, imaging studies of the body may still identify an occult neoplasm in patients with the symptom complex suggestive of a paraneoplas- tic disorder.72 However, the false-positive rate of imaging studies is likely to be higher in the absence of a positive antibody test.
Neuroimaging studies in patients with paraneoplastic cerebellar degeneration are usually normal early in the course, although occasionally there can be mild cerebellar atro- phy. Cerebrospinal fluid (CSF) examination may show a mild pleocytosis and elevated gamma globulin synthesis or may be completely normal.60
Management
The first goal is to identify and remove the under- lying cancer. Although infrequent, there are case reports of dramatic clinical improvement after
resection of the tumor. Regardless, the best chance to stabilize symptoms is when the cancer can be effectively treated.63,73 Most patients reach a “burnt-out” or plateau stage within 6 months where they remain with a moderate to severe neurologic deficit.63 We have followed a woman with paraneoplastic cerebellar degeneration for more than 25 years after a fallopian tube cancer was removed. Exploratory surgery was initiated after anti-Purkinje antibodies (anti-Yo) were iden- tified in her serum. Despite complete removal of the tumor and tumor markers returning to nor- mal, her clinical examinations remain relatively unchanged. She continues to have severe ataxia of the trunk and extremities, although her nausea and vomiting have markedly diminished.
Plasmapheresis and immunosuppression are frequently tried but only rarely associated with an improvement.62,63 In occasional cases, par- ticularly in patients with opsoclonus, dramatic improvement has been reported.65 However, unlike the cerebellar symptoms and signs associated with paraneoplastic cerebellar degeneration, spontaneous remissions are more common with paraneoplastic opsoclonus. Exacerbations can occur months after remis- sion. Clonazepam has been reported to improve opsoclonus in some adults,74 but many do not respond to this medication.
Rituximab has increased interest as a poten- tial treatment for the paraneoplastic neurologic symptoms because beneficial effects have been presumed in individual patients.75 However, any effect has generally been small. Furthermore, one must also consider that the natural history of paraneoplastic disorders is a symptomatic plateau which could easily be misinterpreted as a treatment effect. There are ongoing early-phase trials of medication treat- ments in paraneoplastic syndromes, including tacrolimus (Clinicaltrials.gov identifier NCT00378326) and rituximab in pediatric patients with opsoclonus-myoclonus syndrome (Clinicaltrials.gov identifier NCT00244361).
Background
Multiple sclerosis (MS) is a demyelinating dis- ease of the central nervous system (CNS) with
onset usually in the third and fourth decades of life.76 The key to the diagnosis is finding dis- seminated signs of CNS dysfunction mani- fested in an alternating remitting and exacer- bating course. Although the cause is still not definitely known, the characteristic findings at pathology, the similarity with known autoim- mune animal models, and the response to immunomodulatory and immunosuppressive therapies all suggest that the disorder is auto- immune in nature. Auditory and vestibular symptoms are common in patients with MS and not infrequently they are the presenting symptoms.77–79
Pathophysiology
The demyelination in MS is confined to CNS myelin—the myelin produced by oligodendro- gliocytes. Peripheral nerve myelin produced by Schwann cells is minimally affected. Because both peripheral and cranial nerves contain CNS myelin at their root entry zones, a demy- elinated plaque involving the root entry zone may produce signs of peripheral nerve dysfunc- tion. Next to the first and second cranial nerves, the eighth cranial nerve has the largest propor- tion of CNS myelin, perhaps making it particu- larly susceptible to the effects of MS.80 Plaques involving the vestibular and auditory root entry zones can explain the frequent finding of uni- lateral caloric hypoexcitability and hearing loss in patients with MS.79,81,82 In typical demyeli- nated plaques, most of the myelin sheaths are destroyed and those that remain become swol- len and fragmented. The axis cylinders and neurons are relatively spared so that conduc- tion of nerve impulses still occurs but at a decreased frequency and rate. Whether the remissions and exacerbations of symptoms and signs are related to repair of demyelinated regions or changes in the physiology of nerve conduction unrelated to demyelination is cur- rently debated. It has been repeatedly shown, however, that there is a poor correlation between the clinical symptoms experienced during life and the pathologic findings at necropsy.
The initial areas of demyelination seem to be dependent on T cell infiltration into the CNS.83 However, as the lesions expand, other mecha- nisms may come into play, including activation
of CNS glial and microglial cells. In the animal model for MS, autoimmune allergic encephalo- myelitis sensitization to myelin proteins such as myelin basic protein (MBP) leads to an infiltra- tion of the CNS by activated T cells that start the demyelinating process. There is a clear genetic component to developing MS that may be related both to factors in the immune system and in target CNS antigens.84 In vitro, oligoden- drogliocytes express major histocompatibility complex (MHC) class 1 molecules and are sus- ceptible to CD8 (positive plus) T cell–mediated cytotoxicity. Oligodendrogliocyte-specific anti- bodies can be identified in MS lesions and could induce injury by a complement-dependent mechanism. Oligodendrogliocytes are also sus- ceptible to injury mediated by nonspecific immune cell mechanisms, such as through the cytokines.
Clinical Features
Although many symptoms occur with MS, cer- tain ones deserve emphasis because of their consistent appearance. Blurring or loss of vision caused by demyelination of the optic nerve (retrobulbar or optic neuritis) is an initial symptom of MS in about 20% of patients. Diplopia, weakness, numbness, and ataxia also occur early in the disease process. Vertigo is the initial symptom in about 5% of patients and is reported some time during the disease in as many as 50%.76 Hearing loss occurs in about 10% of patients. No apparent relation- ship exists between the severity or duration of MS and the hearing loss; auditory impairment may be part of the initial episode or may occur more than 10 years after the onset.79,85,86 The hearing loss can be acute (hours to a few days), subacute (over months), or insidious in onset. Partial or complete remission after onset of hearing loss is common. A typical bout of ver- tigo associated with MS lasts from hours to days, although positional vertigo lasting seconds is also common. Multiple sclerosis can mimic vestibular neuritis, benign positional vertigo, and sudden deafness.82,86,87 Benign positional vertigo is common even in patients with MS, so this treatable cause of positional vertigo should always be considered even in patients with a clear central disorder such as MS.88
Diagnosis
The findings on examination in a patient with MS are as diverse as the symptoms. In most long-standing cases, there are signs of involve- ment of the pyramidal tracts (hyperreflexia, extensor plantar responses), cerebellum (inten- tion tremor, ataxia, slurred speech), sensory tracts (impaired vibratory and position sense), and visual pathways (decreased visual acuity and pallor of the optic disc). The finding of dis- sociated nystagmus on lateral gaze or acquired spontaneous pendular nystagmus is particularly helpful in the diagnosis of MS because these are common with MS and are relatively unusual in other disease processes.89,90 All varieties of positional nystagmus can be seen with MS and caloric examination is abnormal in about a quarter of patients.91 Pure-tone hearing levels, when abnormal, have no characteristic pattern with MS. Special audiometric studies (speech discrimination, tone decay, acoustic reflex) indicate a neural site for the lesion. Brainstem auditory-evoked responses (BAER) can be used to detect subclinical lesions in MS, even when hearing is normal (see Chapter 8).92 Vestibular evoked myogenic potentials (VEMPs) are abnormal in about a third of patients with MS.93,94 This is the only vestibular test able to detect dysfunction within central vestibular pathways (see Chapter 7).
No specific laboratory test for MS exists, but abnormalities in the CSF can be identified in about 80% to 90% of patients at some time in the disease course. These findings include elevated gamma globulin, increased gamma globulin synthesis, oligoclonal banding of gamma globulin, and elevated MBP. Unfortunately, none of these findings are spe- cific for MS. Magnetic resonance imaging is currently the gold standard for identifying demyelinating lesions with MS (Fig. 13–2). T2-
weighted and FLAIR images will show charac-
teristic white matter plaques in about 95% of patients with MS, although similar lesions are sometimes seen in patients without the clinical criteria for the diagnosis of MS.95,96
Management
Steroids and adrenocorticotropic hormone may hasten the remission of symptoms and signs
Figure 13–2. Magnetic resonance image demonstrates typical multiple sclerosis white matter lesions including ovoid peri-ventricular and juxta-cortical lesions.
after an acute exacerbation with MS, but no evidence indicates that these drugs alter the natural history.97,98 Immune modulation with interferon-beta (IFN-) and glatiramer acetate have been shown to decrease the frequency and severity of exacerbations with MS, although it is still unclear which of these drugs is best.99,100 Side effects tend to be more prominent with IFN- and include flu-like syndromes, tachy- cardia, headache, myalgias, and gastrointestinal disorders. Neurological toxicity includes gen- eral fatigue along with behavioral and cognitive changes. These latter symptoms may impair quality of life and result in treatment discon- tinuation. Autoantibodies often develop in patients on chronic IFN- treatment and these antibodies may not only interfere with treat- ment but may also be associated with systemic autoimmune symptoms in rare cases.101
OTHER IMMUNE-MEDIATED DISORDERS
If a patient presents with subacute or fulminate progressive cerebellar ataxia, then an immune- mediated disorder should be considered even
if the presentation does not suggest a paraneo- plastic disorder or MS. The case for an immune- based attack can be supported by serum autoantibodies that have been associated with subacute ataxia presentations or by CSF stud- ies indicating an immune-based response.
Serum autoantibodies not associated with a cancer have been found in patients with subacute cerebellar ataxia presentations. In these situa- tions, it is presumed that the antibodies developed spontaneously or in response to a noncancerous antigen and are attacking the cer- ebellum. Though the causal relationship is tenu- ous, the case that these antibodies are involved in the immune-based attack is typically supported by CSF pleocytosis, oligoclonal bands, increased IgG index, and at times even immunohistochem- istry. The main problem with the argument that the specific autoantibodies are causing the ataxia is that most of the identified antibodies also occur in the general population. This is different than the paraneoplastic autoantibodies, which are extremely rare to find outside of the specific neu- rologic presentations.
Probably the most widely reported, biologi- cally plausible, and thus convincing evidence of a
specific antibody association with a nonparaneo- plastic cerebellar ataxia syndrome is with anti- bodies directed at glutamic acid decarboxylase (anti-GAD). Purkinje cells contain high levels of GAD, and immunohistochemistry has found positive immunostaining of axons of Purkinje cells.102–104 Anti-GAD antibodies also have a strong association with other presumed immune- mediated disorders, including Stiffman syn- drome and insulin-dependent diabetes mellitus. Antigliadin antibodies have an association with cerebellar ataxia, but these antibodies are very common in the general population. Furthermore, these antibodies were elevated at higher rates in patients with a known genetic mutation causing the cerebellar ataxia com-
REFERENCES
Bovo R, Aimoni C, Martini A. Immune-mediated inner ear disease. Acta Otolaryngol. 2006;126(10): 1012.
Rask-Anderson H, Stahle J. Immunodefense of the inner ear? Lymphocyte–macrophage interaction in the endolymphatic sac. Acta Otolaryngol (Stockh). 1980;89:283.
Harris JP. Immunology of the inner ear. Otolaryngol Head Neck Surg. 1983;91:17.
Harris JP, Woolf NK, Ryan AF. A reexamination of experimental type II collagen autoimmunity: middle and inner ear morphology and function. Ann Otol Rhinol Laryngol. 1986;95:176.
Tomiyama S, Harris JP. The role of the endolymphatic sac in inner ear immunity. Acta Otolaryngol (Stockh). 1987;103:182.
Suzuki M, Harris JP. Expression of intercellular
pared to the general population,105–107 so it may be that antigliadin antibodies are a marker of cerebellar disturbance rather than a cause of it. Antigliadin antibodies are typically associated with Celiac disease, but some tests have a stronger association with Celiac disease, includ- ing anti-TTG antibodies, anti-endomysial anti- bodies, and small-bowel biopsy. Abnormalities in these tests have also been demonstrated in
adhesion molecule-1 in the inner ear during experi- mental labyrinthitis in rat. Ann Otol Rhinol Laryngol. 1995;104:69.
Terayama Y, Sasaki Y. Studies in experimental aller- gic (isoimmune) labyrinthitis in guinea pigs. Acta Otolaryngol (Stockh). 1964;58:49.
Harris JP. Experimental autoimmune sensorineural hearing loss. Laryngoscope. 1987;97:63.
Soliman AM. Experimental autoimmune inner ear dis- ease. Laryngoscope. 1989;99:188.
Cruz OLM, Miniti A, Cossermelli W, et al. Autoimmune
patients with subacute ataxia.105–108
Other antibodies associated with subacute cerebellar ataxia include antithyroid antibodies (Hashimoto’sencephalitis),109anti-GluR2,110,111 anti-zinc finger protein antibodies,112 anti-
sensorineural hearing loss: a preliminary experimental study. Am J Otol. 1990;11:342.
Solares CA, Hughes GB, Tuohy VK. Autoimmune sensorineural hearing loss: an immunologic perspec- tive. J Neuroimmunol. 2003;138(1-2):1.
Gloddek B, Rogowski M, Reiss G, et al. Adoptive
Homer 3 antibodies,113 and anti-GQ1b IgG antibodies (Miller-Fisher syndrome).114
Collectively, these disorders either show a favorable prognosis based on the natural history of the disorder (anti-GQ1b antibodies), or very little to no convincing or sustained improvement with immune-based therapies, including high-dose corticosteroids, IVIG, or plasmapheresis. If antigliadin antibodies are identified, then a gluten-free diet is generally recommended.115 Though adequate trials dem- onstrating a beneficial effect of the gluten-free diet in cerebellar ataxia presentation are lack- ing, the risks and cost of this diet modification are very low. Because evidence to support aggressive specific therapies are lacking, one should use caution when considering invasive (small-bowel biopsy) or expensive diagnostic testing, or aggressive and potentially harmful therapies, since the potential harms (adverse effects and extreme cost) are likely to substan- tially outweigh the potential for meaningful
transfer of an autoimmunological labyrinthitis in the guinea pig: animal model for a sympathetic cochleo- labyrinthitis. Clin Exp Immunol. 1994; 97:133.
Ikezono T, Tomiyama S, Pawankar R, Jinnouchi K, Suzuki Y, Yagi T. Passive transfer of experimen- tal autoimmune labyrinthitis. Audiol Neurootol. 2000;5(5):292.
Lenhardt E. Plotzliche Horstorungen, auf beiden Seiten gleichzeitig oder nacheinander aufgetreten [in German]. Z Laryngol Rhinol Otol Ihre Grenzgebiete. 1958;31:1.
McCabe BE. Autoimmune sensorineural hearing loss.
Ann Otol Rhinol Laryngol. 1979;88:585.
Harris JP, Weisman MH, Derebery JM, et al. Treatment of corticosteroid-responsive autoimmune inner ear disease with methotrexate: a randomized controlled trial. JAMA. 2003;290(14):1875.
Ruckenstein MJ, Mount RJ, Harrison RV. The MRL-1pr/1pr mouse: a potential model of autoim- mune inner ear disease. Acta Otolaryngol (Stockh). 1993;113:160.
Hefeneider SH, McCoy SL, Hausman FA, Trune DR. Autoimmune mouse antibodies recognize multiple antigens proposed in human immune-mediated hear- ing loss. Otol Neurotol. 2004;25(3):250.
Cogan DG. Syndrome of nonsyphilitic intersti-
benefit.
tial keratitis and vestibuloauditory symptoms. Arch
Ophthalmol. 1945;33:144.
Haynes BF, Kaiser Kupfer MI, Mason P, Fauci AS. Cogan syndrome: studies in thirteen patients, long-term follow-up, and a review of the literature. Medicine. 1980;56:426.
Mazlumzadeh M, Matteson EL. Cogan’s syndrome: an audiovestibular, ocular, and systemic autoimmune disease. Rheum Dis Clin North Am. 2007;33(4):855, vii.
Lunardi C, Bason C, Leandri M, et al. Autoantibodies to inner ear and endothelial antigens in Cogan’s syn- drome. Lancet. 2002;360(9337):915.
Agrup C, Luxon LM. Immune-mediated inner- ear disorders in neuro-otology. Curr Opin Neurol. 2006;19(1):26.
Barna BP, Hughes GB. Autoimmune inner ear disease—a real entity? Clin Lab Med. 1997;17:581.
Harris JP, Sharp PA. Inner ear autoantibodies in patients with rapidly progressive sensorineural hear- ing loss. Laryngoscope. 1990;100:516.
Moscicki RA, San Martin JE, Quintero CH, et al. Specificity of serum antibodies to a 68 kD inner ear antigen in disease associated with hearing loss and responsivitiy to corticosteroid therapy. JAMA. 1994;272:611.
Loveman DM, de Comarmond C, Cepero R, Baldwin DM. Autoimmune sensorineural hearing loss: clini- cal course and treatment outcome. Semin Arthritis Rheum. 2004;34(2):538.
Bloch DB, San Martin JE, Rauch SD, Moscicki RA, Bloch KJ. Serum antibodies to heat shock protein 70 in sensorineural hearing loss. Arch Otolaryngol. 1995;121:1167.
Rauch SD. Clinical management of immune-medi- ated inner-ear disease. In: Bernstein JM, Faden HS, Henderson D, Ryan AE, Barbara M, Quaranta A, eds. Immunologic Diseases of the Ear. New York: New York Academy of Sciences; 1997.
Gong SS, Yan Z. Expression of heat shock protein 70 in the cochlea in experimental autoimmune inner ear disease. Ann Otol Rhinol Laryngol. 2002;111 (3 pt 1):275.
Disher MJ, Ramakrishnan A, Nair TS, et al. Human autoantibodies and monoclonal antibody KHRI-3 bind to a phylogenetically conserved inner-ear supporting cell antigen. In: Bernstein JM, Faden HS, Henderson D, Ryan AF, Barbara M, Quaranta A, eds. Immunologic Diseases of the Ear. New York: New York Academy of Sciences; 1997.
Baek M-J, Park H-M, Johnson JM, et al. Increased frequencies of cochlin-specific T cells in patients with autoimmune sensorineural hearing loss. Immunology. 2006;177:4203.
Schuknecht HE. Ear pathology in autoimmune dis- ease. Adv Otorhinolaryngol. 1991;46:50.
Bowman CA, Linthicum FH, Nelson RA, et al. Sensorineural hearing loss associated with systemic lupus erythematosus. Otolaryngol Head Neck Surg. 1986;94:197.
Caldarelli DD, Rejowski JE, Corey JP. Sensorineural hearing loss in lupus erythematosus. Am J Otol. 1986;7:210.
Andonopoulos AP, Naxakis S, Coumas P, Lygatsikas C. Sensorineural hearing disorders in systemic lupus ery- thematosus: a controlled study. Clin Exp Rheumatol. 1995;13:1469.
Sone M, Schachern PA, Paparella MM, Morizono N. Study of systemic lupus erythematosus in temporal bones. Ann Otol Rhinol Laryngol. 1999;108:338.
Arnold W. Inner ear and renal diseases. Ann Otol Rhinol Laryngol. 1984;93(suppl 112):119.
Wolf M, KronenburgJ, Engelberg S, et al. Rapidly progressive hearing loss as a symptom of polyarteritis nodosa. Am J Otolaryngol. 1987;8:105.
Kempf HG. Ear involvement in Wegener’s granulo- matosis. Clin Otolaryngol. 1989;14:451.
Brama I, Fainaru M. Inner ear involvement in Behçet’s. Arch Otolaryngol. 1980;106:215.
Tucci M, Quatraro C, Silvestris F. Sjögren’s syndrome: anautoimmunedisorderwithotolaryngologicalinvolve- ment. Acta Otorhinolaryngol Ital. 2005;25(3):139.
McAdam LP, O’Hanlan MA, Bluestone CD, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193.
Cody DT, Sones DA. Relapsing polychondri- tis: audiovestibular manifestations. Laryngoscope. 1971;81:1208.
Bovo R, Ciorba A, Martini A. The diagnosis of autoim- mune inner ear disease: evidence and critical pitfalls. Eur Arch Otorhinolaryngol. 2009;266(1):37.
Dayal VS, Ellman M, Sweiss N. Autoimmune inner ear disease: clinical and laboratory findings and treatment outcome. J Otolaryngol Head Neck Surg. 2008;37(4):591.
Arbusow V, Strupp M, Dieterich M, Stocker W, Naumann, Schultz P, Brandt T. Serum antibod- ies against membranous labyrinth in patients with “idiopathic” bilateral vestibulopathy. J Neurol. 1998;245:132.
Deutschländer A, Glaser M, Strupp M, Dieterich M, Brandt T. Immunosuppressive treatment in bilat- eral vestibulopathy with inner ear antibodies. Acta Otolaryngol. 2005;125(8):848.
Shin S-O, Billings PB, Keithley EM, Harris JP. Comparison of anti-heat shock protein 70 (anti-hsp70) and anti-68-kDa inner ear protein in the sera of patients with Meniere’s disease. Laryngoscope. 1997;107: 222.
Niparko JK, Wang NY, Rauch SD, et al. Serial audi- ometry in a clinical trial of AIED treatment. Otol Neurotol. 2005;26(5):908.
Helmchen C, Jäger L, Büttner U, Reiser M, Brandt T. Cogan’s syndrome. High resolution MRI indicators of activity. J Vestib Res. 1998;8:155.
Hughes GB, Barna BP, Calabrese LH, et al. Clinical diagnosis of immune inner ear disease. Laryngoscope. 1988;98:251.
Berger P, Hillman M, Tabak M, et al. The lymphocyte transformation test with type II collagen as a diag- nostic tool of autoimmune sensorineural haring loss. Laryngoscope. 1991;101:895.
Harris JP, Heydt J, EM Keithley, Chen M-C. Immunopathology of the inner ear: an update. In: Bernstein JM, Faden HS, Henderson D, Ryan AF, Barbara M, Quaranta A, eds. Immunologic Diseases of the Ear. New York: New York Academy of Sciences; 1997.
Teom K, Gray J, Nair TS, et al. Antibodies to HSP-70 in normal donors and autoimmune hearing loss patients. Laryngoscope. 2003;113:1770.
Ruckenstein MJ. Autoimmune inner ear disease. Curr Opin Otolaryngol Head Neck Surg. 2004;12(5): 426.
Broughton SS, Meyerhoff WE, Cohen SB. Immune- mediated inner ear disease: 10-year experience. Semin Arthritis Rheum. 2004;34(2):544.
Alexander TH, Weisman MH, Derebery JM, et al. Safety of high-dose corticosteroids for the treatment of autoimmune inner ear disease. Otol Neurotol. 2009;30(4):443.
Cohen S, Shoup A, Weisman MH, Harris J. Etanercept treatment for autoimmune inner ear disease: results of a pilot placebo-controlled study. Otol Neurotol. 2005;26(5):903.
Vitaliani R, Zoccarato M, Giometto B. Diagnosis and treatment of paraneoplastic neurological syndromes. Curr Clin Pharmacol. 2008;3(1):46.
Dalmau JO, Posner JB. Paraneoplastic syndromes.
Arch Neurol. 1999;56:405.
Rojas I, Graus F, Keime-Guibert, et al. Long-term clinical outcome of paraneoplastic cerebellar degen- eration and anti-Yo antibodies. Neurology. 2000;55: 713.
Shams’ili S, Grefkens J, de Leeuw B, et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain. 2003;126:1409.
Baloh RW. Paraneoplastic cerebellar disorders.
Otolaryngol Head Neck Surg. 1995;112:125.
Ko MW, Dalmau J, Galetta SL. Neuro-ophthalmologi manifestations of paraneoplastic syndromes. J Neuro- Ophthalmol. 2008;28:58.
Bataller L, Graus F, Saiz A, et al. Clinical outcome in adult onset idiopathic or paraneoplastic opsoclonus- myoclonus. Brain. 2001;124:437.
Graus F, Keime-Guibert F, Rene I, et al. Anti-Hu- associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain. 2001;124:1138.
McGill T. Carcinomatous encephalomyelitis with aud- iroty and vestibular manifestations. Ann Otol Rhinol Laryngol. 1976;85:120.
Gulya AJ. Neurologic paraneoplastic syndromes with neurotologic manifestations. Laryngoscope. 1993;103:745.
Greenlee JE, Brashear HR. Antibodies to cerebellar Purkinje cells in patient with paraneoplastic cerebel- lar degeneration and ovarian carcinoma. Ann Neurol. 1983;14:609.
Linke R, Schroeder M, Helmberger T, Voltz R. Antibody-positive paraneoplastic neurologic syn- dromes: value of CT and PET for tumor diagnosis. Neurology. 2004;63:282.
Hadjivassiliou M, Alder SJ, Van Beek EJ, et al. PET scan in clinically suspected paraneoplastic neurologi- cal syndromes: a 6-year prospective study in a regional neuroscience unit. Acta Neurol Scand. 2009;119: 186.
Vedeler CA, Antoine JC, Giometto B. Management of paraneoplastic neurological syndromes: report of an EFNS Task Force. Eur J Neurol. 2006;13:682.
Bartos A. Effective high-dose clonazapam treatment in two patients with opsoclonus-myoclonus syndrome: GABAergic hypothesis. Eur Neurol. 2006;56:240.
Shams’ili S, de Beukelaar J, Gratama JW, et al. An uncontrolled trial of rituximab for antibody
associated paraneoplastic neurological syndromes. J Neurol. 2006;253:16.
Courtney AM, Treadaway K, Remington G, Frohman E. Multiple sclerosis. Med Clin North Am. 2009;93(2):451, ix.
Grenman R. Involvement of the audiovestibular system in multiple sclerosis: an otoneurologic and audiologic study. Acta Otolaryngol Suppl (Stockh). 1985;420:9.
Rae-Grant AD, Eckert NJ, Bartz S, Reed JE. Sensory symptoms of multiple sclerosis; a hidden reservoir of morbidity. Mult Scler. 1999;5:179.
Oh YM, Oh DH, Jeong SH, Koo JW, Kim JS. Sequential bilateral hearing loss in multiple sclerosis. Ann Otol Rhinol Laryngol. 2008;117(3):186.
Tarlov IM. Structure of the nerve root. I. Nature of the junction between the central and peripheral nervous system. Arch Neurol Psychiatry. 1937;37:555.
Sasaki O, Ootsuka K, Taguchi K, Kikukawa M. Multiple sclerosis presented acute hearing loss and vertigo. ORL J Otorhinolaryngol Relat Spec. 1994;56:55.
Thomke F, Hopf HC. Pontine lesions mimicking acute peripheral vestibulopathy. J Neurol Neurosurg Psychiatry. 1999;66:340.
Guo MF, Ji N, Ma CG. Immunologic pathogenesis of multiple sclerosis. Neurosci Bull. 2008;24(6):381.
Ramagopalan SV, Dyment DA, Ebers GC. Genetic epidemiology: the use of old and new tools for multiple sclerosis. Trends Neurosci. 2008;31(12):645.
Noffsinger D, Olson WO, Carhart R, et al. Auditory and vestibular aberrations in multiple sclerosis. Acta Otolaryngol Suppl (Stockh). 1972;303:7.
Ozunlu A, Mus N, Gulhan M. Multiple sclerosis: a cause of sudden hearing loss. Audiology. 1998;37:52.
Anagnostou E, Varaki K, Anastasopoulos D. A minute demyelinating lesion causing acute positional vertigo. J Neurol Sci. 2008;266(1–2):187.
Frohman EM, Kramer PD, Dewey RB, Kramer L, Frohman TC. Benign paroxysmal positioning ver- tigo in multiple sclerosis: diagnosis, pathophysiology and therapeutic techniques. Mult Scler. 2003;9(3): 250.
Cogan DG. Internuclear ophthalmoplegia typical and atypical. Arch Ophthalmol. 1970;84:583.
Averbuch-Heller L, Zivotofsky AZ, Das VE, DiScenna AO, Leigh RJ. Investigations of the pathogenesis of acquired pendular nystagmus. Brain. 1995;118:369.
Dam M, Thomsen J, Johnsen NJ, Zilstorff K. Vestibular aberrations in multiple sclerosis. Acta Neurol Scand. 1975;52:407.
Daugherty WT, Lederman RJ, Nodar RH, Conomy JP. Hearing loss in multiple sclerosis. Arch Neurol. 1983;40:33.
Papathanasiou ES, Piperidou C, Pantzaris M, et al. Vestibular symptoms and signs are correlated with abnormal neurogenic vestibular evoked potentials in patients with multiple sclerosis. Electromyogr Clin Neurophysiol. 2005;45(4):195.
Patkó T, Simó M, Arányi Z. Vestibular click-evoked myogenic potentials: sensitivity and factors determin- ing abnormality in patients with multiple sclerosis. Mult Scler. 2007;13(2):193.
Gass A, Steinke W, Schwartz A, Hennerici MG. High resolution magnetic resonance imaging in peripheral vestibular dysfunction in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1998;65:945.
96. Rovira A, Swanton J, Tintoré M, et al. A single, early magnetic resonance imaging study in the diagnosis of multiple sclerosis. Arch Neurol. 2009;66(5):587. | 107. | and neuropathological characteristics of gluten ataxia. Lancet. 1998;352:1582. Abele M, Schols L, Schwartz S, Klockgether T. |
97. Compston A. Methylprednisolone and multiple scle- | Prevalence of antigliadin antibodies in ataxia patients. | |
rosis. Arch Neurol. 1988;45:669. | Neurology. 2003;60:1674. | |
98. Filippini G, Brusaferri F, Sibley WA, et al. | 108. | Hadjivassiliou M, Maki M, Sanders DS, et al. |
Corticosteriods or ACTH for acute exacerbations | Autoantibody targeting of brain and intestinal trans- | |
in multiple sclerosis. Cochrane Database Sys Rev. | glutaminase in gluten ataxia. Neurology. 2006;66: | |
2002;4:CD001331. | 373. | |
99. Tselis AC, Lisak RP. Multiple sclerosis. Therapeutic | 109. | Selim M, Drachman DA. Ataxia associated with |
update. Arch Neurol. 1999;56:277. | Hashimoto’s disease: progressive non-familial adult | |
100. Wiendl H, Hohlfeld R. Multiple sclerosis therapeu- | onset cerebellar degeneration with autoimmune | |
tics: unexpected outcomes clouding undisputed suc- | thyroiditis. J Neurol Neurosurg Psychiatry. 2001;71: | |
cesses. Neurology. 2009;72(11):1008. | 81. | |
101. Vial T, Descotes J. Clinical toxicity of the interferons. | 110. | Shiihara T, Kato M, Konno A, Takahashi Y, Hayasaka |
Drug Safety. 1994;10:115. | K. Acute cerebellar ataxia and consective cerebelli- | |
102. Iwasaki H, Sato R, Shichiri M, Hirata Y. A patient | tis produced by glutamate receptor 2 autoantibody. | |
with type 1 diabetes mellitus and cerebellar ataxia | Brain Dev. 2007;29:254. | |
associated with high titer of circulating anti-glutamic | 111. | Shimokaze T, Kato M, Yoshimura Y, Takahashi Y, |
acid decarboxylase antibodies. Endocr J. 2001;48: | Hayasaka K. A case of acute cerebellitis accompa- | |
261. | nied by autoantibodies against glutamate receptor 2. | |
103. Rakocevic G, Raju R, Semino-Mora C, Dalakas | Brain Dev. 2007;29:224. | |
MC. Stiff person syndrome with cerebellar disease | 112. | Bataller L, Wade DF, Fuller GN, Rosenfeld MR, |
and high titer anti-GAD antibodies. Neurology. | Dalmau J. Cerebellar degeneration and autoim- | |
2006;67:1068. | munity to zinc-finger proteins of the cerebellum. | |
104. Pittock SJ, Yoshikawa H, Ahlskog JE, et al. Glutamic | Neurology. 2002;59:1985. | |
acid decarboxylase autoimmunity with brainstem, | 113. | Zuliani L, Sabater L, Saiz A, Baiges JJ, Giometto |
extrapyramidal, and spinal cord dysfunction. May | B, Graus F. Homer 3 autoimmunity in subacute | |
Clin Proc. 2006;81:1207. | idiopathic cerebellar ataxia. Neurology. 2007;68: | |
105. Pellecchia MT, Scala R, Filla A, De Michele G, Ciacci | 239. | |
C, Barone P. Idiopathic cerebellar ataxia associated | 114. | Mori M, Kuwabara S, Fukutake T, Hattori T. |
with celiac disease: lack of distinctive neurological | Intravenous immunoglobulin therapy for Miller | |
features. J Neurol Neurosurg Psychiatry. 1999;66: | Fisher syndrome. Neurology. 2007;68:1144. | |
32. | 115. | Hadjivassiliou M, Davies-Jones GA, Sanders DS, |
106. Hadjivassiliou M, Grunewald RA, Chattopadhyay | Grunewald RA. Dietary treatment of gluten ataxia. | |
AK, et al. Clinical, radiological, neurophysiological, | J Neurol Neurosurg Psychiatry. 2003;74:1221. |
![]()
VERTEBROBASILAR ISCHEMIA
Pathophysiology
Transient Ischemic Attacks (TIAs) Stroke Syndromes
Diagnosis Treatment
INTRALABYRINTHINE HEMORRHAGE
Diagnosis and Management
HEMORRHAGE INTO THE BRAIN STEM AND CEREBELLUM
Diagnosis and Management
VASCULAR COMPRESSION SYNDROMES
Vertebrobasilar Dolichoectasia
Vascular Compression by Normal Vessels (Vestibular Paroxysmia)
Rotational Vertebral Artery Syndrome
Pathophysiology
As discussed in Chapter 2, the vascular supply to the labyrinth, eighth nerve, brain stem, and cer- ebellum arises from a common source: the verte- brobasilar circulation. The vestibular system is subject to two general categories of vascular isch- emia: (1) hypoperfusion in the vertebrobasilar system, in which case multiple areas (both periph- eral and central) become simultaneously isch- emic, or (2) hypoperfusion in the distribution of a single smaller feeding vessel, in which case there is a circumscribed area of ischemia. In the former case the site of origin of hypoperfusion can be anywhere from the heart and major vessels in the chest and neck to the basilar artery, whereas in the latter case an occlusion is usually situated near the origin or, less frequently, in the smaller feed- ing vessel itself. These two categories of ischemia are not mutually exclusive, however; a patient with small-vessel disease may be as asymptomatic because of collateral circulation, but an added hypoperfusion in the vertebrobasilar system can lead to focal ischemia and or infarction.
The most common causes of vertebrobasilar ischemia (VBI) are large-artery atherosclerosis,
emboli, and penetrating small-vessel disease.1 Other less common causes include dissection, fibromuscular displasia, arteritis, polycythemia, thromboangiitis obliterans, and hypercoagula- tion syndromes. In rare cases, occlusion or stenosis of the subclavian or innominate arter- ies just proximal to the origin of the vertebral artery results in the so-called subclavian steal syndrome. In this syndrome, VBI results from siphoning of blood down the vertebral artery from the basilar system to supply the upper extremity. Occasionally, episodes of VBI are precipitated by postural hypotension, Stokes- Adams attacks, or mechanical compression from cervical spondylosis. Cervical spondylosis is extremely common in the elderly, but docu- mented cases of mechanical compression of the vertebral arteries by neck turning or exten- sion are rare (see later discussion).
Vertigo is the most common symptom of VBI, but it is not always clear which structure or combination of structures is ischemic.2 When vertigo is accompanied by other symp- toms of brainstem ischemia (e.g., slurred speech or a brainstem pattern of motor/sensory symptoms), one can reasonably assume that the vertigo is the result of ischemia of the vestibular nuclei in the lateral medulla or the
319
Isolated episodes of vertigo can also result from transient ischemia to the vestibular laby- rinth.4 Because the labyrinthine circulation is an end circulation with minimal collaterals from the otic capsule, the labyrinth is especially vulnerable to ischemia. Possibly, because of the small caliber of the anterior vestibular artery (see Fig. 2–9 in Chapter 2) and the gen- eral lack of collateralization, the superior part of the vestibular labyrinth is selectively vulner- able to ischemia.5 One would expect ischemia to the labyrinth to be accompanied by hearing loss, but patients may simply be unaware of a transient, mild, unilateral hearing loss when it is accompanied by a severe attack of vertigo.7
Infarction in the distribution of the posterior inferior cerebellar artery (PICA) or the ante- rior inferior cerebellar artery (AICA) is invari- ably associated with severe vertigo, nausea, and vomiting. The full-blown syndromes are easily recognized on the basis of the highly localized neurologic signs,3,6 but there might be more difficulty localizing the lesion to these distribu- tions in the case of a partial syndrome. Infarction in the lateral medullary region in the distribu- tion of PICA is usually associated with throm- bosis of the ipsilateral vertebral artery proximal to the take-off of PICA.1,8 Traumatic dissection of the distal vertebral artery is a common cause of lateral medullary infarction in younger patients.9–11 Infarction in the lateral pontomed- ullary distribution of the AICA is usually associated with occlusive disease of the distal vertebral artery and proximal basilar artery.7,12
Often these brainstem syndromes occur with- out associated cerebellar infarction because of the anastomotic channels between the cerebel- lar arteries. Isolated cerebellar infarction in the distribution of the superior, anterior inferior, or posterior inferior cerebellar arteries usually indicates an embolic source.13,14 Emboli typi- cally migrate to the most distal arterial branch— in this case, the cerebellar branches. Patchy areas of infarction in the cerebellum occurring at the junctions of the three major arteries may indicate hypoperfusion within the entire poste- rior circulation, resulting in “watershed infarc- tions.”15,16 These types of infarctions can also be seen with diffuse vasospasm associated with basilar migraine.
Transient Ischemic Attacks (TIAs)
Transient ischemia within the vertebrobasilar system is a common cause of episodic vertigo in older patients. It is typically abrupt in onset, usually lasting several minutes, and is fre- quently associated with nausea and vomiting. In their classical description of TIAs within the vertebrobasilar circulation, Williams and Wilson17 reported that vertigo was the initial symptom in 48% of patients. Invariably, the vertigo is associated with symptoms resulting from ischemia in the remaining territory sup- plied by the posterior circulation (those listed in Table 14–1). Next to vertigo, visual symp- toms are most common. Associated symptoms occur in episodes, either in combination with the vertigo or in isolation.
Fisher’s classic paper on this topic18 is remembered for emphasizing that episodes of vertigo without other neurologic symptoms is strongly suggestive of a disorder other than VBI. However, among his cases of VBI, he described several patients who manifested isolated episodes of vertigo at some time during their course. Fields and Weibel19 also described several patients with isolated episodes of vertigo and VBI. We reviewed the clinical findings of 42 patients who pre- sented to our neurotology clinic with vertigo due to VBI in 1989.4 The diagnosis was based on their having at least some episodes with a characteristic combination of symptoms (those listed in Table 14–1). There was a surprisingly
Table 14–1 Frequency of Symptoms Associated with Vertigo in 42 Patients with Vertebrobasilar Insufficiency
![]()
Symptom Patients
Visual (diplopia, visual illusions and 29
hallucinations, field defects, etc.)
Stroke Syndromes
LABYRINTHINE INFARCTION
Occlusion of the internal auditory artery leads to a sudden profound loss of both auditory and vestibular function. Hearing loss is usually permanent, and a vestibular imbalance
Drop attacks 14
Unsteadiness, incoordination 9
Extremity weakness 9
Confusion 7
Headache 6
Hearing loss 6
Loss of consciousness 4
Extremity numbness 4
Dysarthria 4
Tinnitus 4
Perioral numbness 2
![]()
Source: Data from Grad and Baloh.4
high incidence of isolated episodes of vertigo in these patients; 62% had at least one isolated episode of vertigo and, in 19%, their TIAs began with an isolated episode of vertigo. These patients with isolated episodes of vertigo also reported the same vertiginous sensation in combination with other symptoms at other times, which suggests that the vertigo was indeed due to transient ischemia with the posterior circulation. Subsequently, there have been many other reports emphasizing that isolated attacks of vertigo can be the only manifestation of a TIA within the verte- brobasilar system.20–22 As suggested earlier, it is still unclear whether these isolated episodes of vertigo originate from the brain or inner ear. As a general rule, vertigo may be an isolated initial symptom of VBI or may occur in isolation intermixed with more typical episodes, but VBI becomes extremely unlikely whenever recurrent episodes date back more than 3 months and are unaccompa- nied by other focal neurologic symptoms.1 However exceptions even to this more gener- ous rule do occur. In fact, we reported a patient having hundreds of attacks of vertigo and uni- lateral tinnitus over a 2-year period who was found to have a severe stenosis of the basilar artery just proximal to AICA.23 A stent was placed to open the stenosis and resulted in resolution of the attacks.
remains, although symptoms of dizziness and imbalance gradually improve with central compensation. Pathological studies in such patients show widespread necrosis of the inner ear tissues with subsequent proliferation of fibrous tissue and new bone formation.24 Most documented cases have been associated with ischemia in the distribution of the AICA, accompanied by infarction of the brain stem and/or cerebellum.25 Not infrequently, infarction of the labyrinth is preceded by epi- sodes of transient ischemia within the verte- brobasilar system, in some cases, manifested by isolated episodes of vertigo.7 The diagnosis should be considered in patients with impor- tant risk factors for stroke who present with sudden onset of unilateral deafness and vertigo, particularly if there is a history of TIA, stroke, or known atherosclerotic vascular disease.
The role of vascular occlusion in producing sudden one-sided deafness without associated vertigo is unclear. There is little reason to sus- pect that unilateral deafness in a young, healthy patient is caused by vascular disease. As noted in Chapter 9, most of these cases are probably due to isolated viral infections. However, the sudden onset of deafness without associated vertigo or brainstem signs in a patient with known vascular disease or a hypercoagulation syndrome should raise the suspicion for ischemia within the distribution of the com- mon cochlear artery or one of its branches (see Fig. 2–9 in Chapter 2).25 Sudden deafness (reversible or permanent) has been reported in patients with fat emboli,26 thromboangiitis obliterans,27 and macroglobulinemia.29 Athero- sclerotic disease is also associated with sudden deafness, but pathological confirmation of the site of vascular occlusion is often lacking.28 Examination of the cochlea at necropsy in such patients reveals a loss of the organ of Corti and degeneration of the stria vascularis, spiral ligament, and distal cochlear nerve fibers (findings similar to those seen in
animals that have had their labyrinthine artery occluded).24
Ischemia that is confined to the anterior ves- tibular artery distribution (see Fig. 2–9 in Chapter 2) can result in transient episodes of vertigo (lasting minutes) or a prolonged attack (lasting days) due to infarction of the vestibular labyrinth.5,22 The former may be associated with hyperviscosity syndromes such as hyperlipidemia, polycythemia, macroglobu- linemia, and sickle cell anemia.30,31 The clinical picture of infarction in the distribution of the anterior vestibular artery is that of the acute vestibular syndrome (i.e., sudden, severe vertigo without hearing loss or brainstem symptoms),2 and as such cannot be distin- guished from vestibular neuritis. After recover- ing from the acute manifestations, patients may develop episodes of typical benign positional vertigo months or years later. The positional vertigo presumably results from ischemic necrosis of the utricular macule, causing a release of otoconia that make their way into the long arm of the posterior semicir- cular canal. Since the posterior semicircular canal is supplied by a branch of the common
cochlear artery, it may be spared even though the superior part of the vestibular labyrinth is completely infarcted.5,32
LATERAL MEDULLARY INFARCTION
The zone of infarction producing the lateral medullary syndrome (Wallenberg’s syndrome) consists of a wedge of the dorsal lateral medulla just lateral to the olive (Fig. 14–1). Although the syndrome is commonly known as that of the PICA, as noted earlier, it usually results from occlusion of the ipsilateral vertebral artery, and only rarely from occlusion of the PICA. In young patients, dissection of the ver- tebral artery is a common cause, particularly if there is a history of trauma or neck manipula- tion.11,33–36 Symptoms may come on right at the time of the trauma or neck manipulation or they may be delayed for several days. Major symptoms include vertigo, nausea, vomiting, intractable hiccups, severe imbalance, ipsilat- eral facial numbness and weakness, diplopia, dysphagia, and dysphonia. A dissection is typi- cally accompanied by occipital headache or neck pain.36 Lesions involving the mid-level of
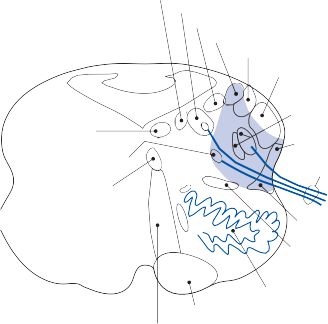
Dorsal efferent tract of nerve X Nucleus solitarius
Med. vestibular nucleus Inf. vestibular nucleus
Lat. cuneate nucleus
Restiform body
Nucleus of nerve XII
Nucleus ambiguus
Medial longitudinal fasciculus
Pyramid Medial lemniscus
Nucleus and root of nerve V
Vent. spinocerebellar tract
Root of nerve X
Lat. spinothalamic tract
Second ascending tract of nerve V
Inferior olive
Figure 14–1. Cross-section of the medulla illustrating the zone of infarction with Wallenberg’s syndrome (blue area).
![]()
Table 14–2 Mechanism of Symptoms and Signs Commonly Seen with Infarction in the Distribution of Posterior Inferior and Anterior Inferior Cerebellar Arteries
![]()
Symptoms and Signs Structures Involved with PICA Infarct
Vertigo, nystagmus Vestibular nuclei, posterior inferior
cerebellum
Structures Involved with AICA Infarct
Labyrinth, vestibular nerve, flocculus
Tinnitus, hearing loss None Cochlea, auditory nerve, cochlear nucleus
Gait and limb ataxia Ventral spinocerebellar tract, posterior
inferior cerebellum
Middle cerebellar peduncle, anterior inferior cerebellum
Dysphagia, decreased gag Vagal nuclei and nerve None
Facial hemianesthesia Fifth nerve and nucleus Fifth nerve and nucleus Facial paralysis Seventh nerve Seventh nerve
Crossed hemisensory loss Spinothalamic tract Spinothalamic tract
Horner’s syndrome Descending sympathetic fibers Descending sympathetic fibers
![]()
AICA, anterior inferior cerebellar artery; PICA, posterior inferior cerebellar artery.
the lateral medulla produce a characteristic combination of symptoms, including vertigo, nausea, vomiting, hiccups, and dysphagia.37
Neurological examination identifies multiple signs localizing to the lateral medulla (Table 14–2). The spontaneous nystagmus is typically horizontal-torsional with an increase in the horizontal component on gaze toward the side of the lesion and often a change in the direc- tion of the horizontal component with gaze away from the side of the lesion. Patients also may exhibit a transient ocular tilt reaction (ipsi- lateral head tilt, skew deviation, and ocular tor- sion, upper pole of the eye rotated toward the side of the lesion) that is associated with a devi- ation of the subjective visual-vertical in the direction of the head tilt.38 Even with their heads fixed in the true vertical, they perceive it as tilted opposite the direction of the tilt reac- tion. This phenomenon results from involve- ment of the otolith-ocular pathways at the level of the vestibular nucleus.39 Two distinct pat- terns of otolith dysfunction are seen depending on whether the cerebellar nodulus is involved. If the nodulus is infarcted, there is ocular tor- sion and skew deviation with falling and tilt of the subjective visual vertical to the contrale- sional side. If the nodulus is spared, falling and tilt of the subjective visual vertical is ipsilateral without ocular torsion and skew deviation.40
Patients with Wallenberg’s syndrome often suffer a prominent motor disturbance that causes them to deviate toward the side of the lesion as if being pulled by a strong external force.41,42 This so-called lateropulsion also
affects the oculomotor system, causing exces- sively large voluntary and involuntary saccades directed toward the side of the lesion, whereas saccades away from the lesion side are abnor- mally small.43,44
LATERAL PONTOMEDULLARY INFARCTION
Ischemia in the distribution of the AICA usually results in infarction of the dorsolateral pontomedullary region and the middle cerebellar peduncle (Fig. 14–2).6,7 As noted earlier, because the labyrinthine artery arises from the AICA in most cases, infarction of the membranous labyrinth is a common accompa- niment. Severe vertigo, nausea, and vomiting may be the initial and most prominent symp- toms. The key difference between the lateral medullary syndrome and the lateral pontomed- ullary syndrome is that the latter is typically associated with a profound unilateral or bilat- eral hearing loss that can be the result of infarc- tion of the labyrinth, eighth nerve, or even the eighth nerve root entry zone into the brain stem. Sudden unilateral or bilateral hearing loss and vertigo can be the initial or sole mani- festation of AICA infarction.45,46 Neurological examination typically reveals a profound unilateral hearing loss, unilateral facial paraly- sis, cerebellar ataxia, and ipsilateral loss of pain and temperature sensation on the face due to involvement of the trigeminal nucleus and tract, and a contralateral decrease in pain and
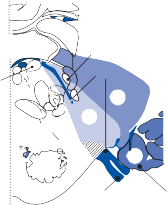
![]()
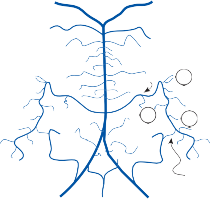
A PCA B
IAA
AICA
SCA
2
4th ventricle
VI
V
VIII
1B
MCP
1 3 1A
PICA
VA
PICA-AICA
anastamosis
VIII
3
Flocculus
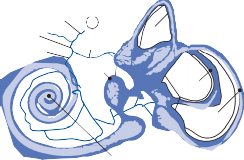
C
IAA CCA
AICA
2
AVA
ASC
D
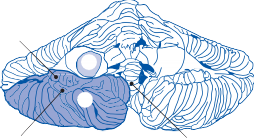
Flocculus
Saccule
1
HSC
3
PSC
Cochlea
Paraflocculus Nodulus
Figure 14–2. A Classical anterior inferior cerebellar artery (AICA) anatomy with AICA and posterior inferior cerebellar artery (PICA) of equal dominance. Numbers refer to the three zones of AICA supply shown in B, C, and D. Zone 1 is sup- plied by the recurrent penetrating arteries (RPA) off AICA, zone 2 by the internal auditory artery (IAA), and zone 3 by the terminal cerebellar branches of AICA. B Zones 1A and 1B represent the arterial supply to the rostral pons supplied by a premeatal and postmeatal RPA. The cross-hatched area represents the root entry zone of the facial and vestibuloco- chlear nerves. C Zone 2 represents the arterial supply to the inner ear. D Zone 3 is the part of the cerebellum supplied by AICA. ASC, anterior semicircular canal; AVA, anterior vestibular artery; CCA, common cochlear artery; HSC, horizontal semicircular canal; MCP, middle cerebellar peduncle; PCA, posterior cerebral artery; PSC, posterior semicircular canal; SCA, superior cerebellar artery; VA, vertebral artery; V, spinal trigeminal tract and nucleus; VI, abducens nucleus; VII, facial nerve; VIII, vestibulocochlear nerve.
temperature sensation on the body due to involvement of the crossed spinothalamic tract (Table 14–2).
Branches of the AICA supplying the ventro- lateral pons have few anastomoses with adja- cent vessels. By contrast, the root entry zones of the seventh and eighth cranial nerves have a rich network of anastomotic vessels arising from the lateral medullary artery, AICA, and the inferior lateral pontine artery. Thus, occlu- sion of the AICA reliably causes infarction of the lateral pons and middle cerebellar pedun- cle, but because of collateral vessels, the root entry zone of the seventh and eighth nerves is often spared.7
CEREBELLAR INFARCTION
Acute cerebellar infarction may present with prominent vertigo, vomiting, and ataxia; because typical lateral brainstem signs are not present, a mistaken diagnosis of an acute peripheral labyrinthine disorder might be made.3,47,48 We do not know the true proportion of patients with isolated vertigo who harbor an ischemic stroke because there is no large series of such patients who all receive magnetic reso- nance imaging (MRI) with diffusion weight sequences. One of the first studies to attempt to address this issue found that 25% (6 out of 24) of patients with risk factors for stroke
who present to an emergency medical setting with isolated severe vertigo, nystagmus, and postural instability had an infarction of the inferior cerebellum, but diffusion-weighted images were not available at the time so the acuity of the infarction could not be deter- mined.49 A more recent study using MRI with diffusion-weighted imaging found that 75% (25/33) of patients with the acute vestibular syndrome had a causative stroke, most involv- ing the cerebellum.50 However, this study only included patients with at least one stroke risk factor and also excluded patients with a recent viral illness, so that the population was at higher risk of stroke etiology than the general popula- tion of patients with acute vertigo.50 The key features suggesting a cerebellar infarction include prominent cerebellar signs, particu- larly severe truncal ataxia (i.e., inability to sit or stand without assistance), and direction-chang- ing gaze-evoked nystagmus (See Video 6–9). Patients with peripheral vestibular disorders will have imbalance, even to the point of falling when trying to walk during the acute phase, but they can sit unassisted and can typically stand unassisted.38,50 Nystagmus, in patients with an acute peripheral vestibular disorder, does not change direction with changes in gaze (See Video 6–4, Video 6–5, and Video 6–6). Spontaneous down-beating nystagmus localizes the lesion to the caudal midline cerebellum (See Video 6–7).
There are three major cerebellar arteries: posterior inferior, anterior inferior, and supe- rior (Figs. 14–2A and 14–3A). After supplying branches to the brain stem, each of these arter- ies supplies the part of the cerebellum indicated by its name. Typically, there are prominent anastomoses between these three cerebellar arteries on the cerebellar surface, so that occlusion near the take-off results only in brainstem infarction with sparing of the cere- bellum. There are also common anatomical variants, the most common being anterior infe- rior cerebellar dominance on one side and pos- terior inferior cerebellar dominance on the opposite side. The PICA typically has two major cerebellar branches—a medial branch and a lateral branch—with the former supply- ing midline structures, including the vermis. Infarction in the territory of the medial branch of PICA can mimic an acute peripheral vestibulopathy probably by interrupting nodulouvular inhibitory projections to the
vestibular nuclei.51 In addition to supplying the anterior inferior surface of the cerebellum, the AICA also supplies the flocculus and para- flocculus, structures known to be critical for visual–vestibular interaction (see Fig. 14–2D). Patients with infarction in the distribution of the superior cerebellar artery typically do not present with vertigo or nystagmus but rather severe ataxia and dysarthria.50,52 There are reports though of rather mild imbalance (able to stand and walk unassisted, but not able to walk in tandem) from a superior cerebellar stroke.50
Diagnosis
CLINICAL EXAMINATION
Vertebrobasilar ischemia can usually be diag- nosed with a careful history and examination. As discussed earlier, TIAs within the posterior circulation typically present with the character- istic symptoms, even though occasionally, diz- ziness or vertigo occurs in isolation. Transient ischemic attacks typically come on abruptly, without an apparent precipitating factor, last for a few minutes, and then end abruptly, usu- ally with minimal or no residual symptoms. The abrupt spontaneous (nonpositional) onset and minutes’ duration are not typical of any of the common inner ear disorders. The acute stroke syndromes within the posterior circulation are usually easily identified on the basis of their characteristic combination of neurological symptoms and signs (Table 14–3). Although cerebellar infarcts can masquerade as a more benign inner ear disorder, through a careful examination one should be able to identify pro- found truncal ataxia and a direction-changing gaze-evoked nystagmus, which do not occur with inner ear lesions. The suspicion for a cerebellar infarction also increases if uni- directional nystagmus is found but the head- thrust sign is negative.38,50,58 The history can provide risk factors for atherosclerosis, includ- ing a history of coronary artery disease, hyper- tension, diabetes mellitus or hyperlipidemia, and a family history of early-onset atheroscle- rosis. In young patients without obvious vascu- lar risk factors, the history should focus on the possibility of trauma (including neck manipula- tion) and other systemic illnesses that might predispose to hypercoagulation.
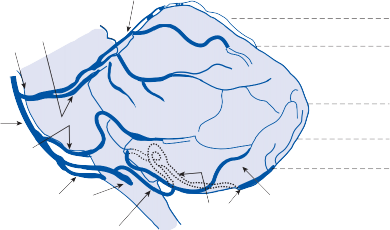
Superior cerebellar (SCA)
Lateral branch of SCA
Medial branch of SCA
1
2
3
4
Basilar (BA) 5
Anterior inferior
cerebellar (AICA) 6
Vertical (VA)
Posterior inferior cerebellar (PICA)
Medial branch of PICA
Lateral branch of PICA
B


1 2 3
AICA
MSCA
LSCA
MSCA
LSCA MSCA
MPICA
LSCA

4 5 6
AICA AICA
LSCA
LPICA
MSCA
MPICA
LSCA
MPICA MPICA
Figure 14–3. A Branches of the three main cerebellar arteries. B Magnetic resonance imaging horizontal axial sections from rostral to caudal (1 to 6) showing territory supplied by each branch. L, lateral; M, medial.
BRAIN IMAGING
Magnetic resonance imaging is the procedure of choice for viewing brain structures supplied by the vertebrobasilar system.1,59,61 An MRI is usually normal in patients with TIAs within the posterior circulation, although occasionally such patients show evidence of old, silent infarcts, particularly in the occipital poles or
the cerebellar hemispheres. Specific stroke syndromes, such as the lateral medullary and lateral pontomedullary syndromes, are all eas- ily identified with MRI (Figs. 14–4 and 14–5).
The typical appearance is a T2 and FLAIR intense lesion within the brain stem in the
distribution of either the posterior inferior or the anterior inferior cerebellar arteries.
![]()
Table 14–3 Differentiating among Common Acute Vertigo Syndromes
![]()
Syndrome Spontaneous Nystagmus
Gait Other Possible Signs
Acute peripheral vestibulopathy
Unidirectional, beats toward intact side
Falls toward lesion side but can walk
Unilateral hearing loss
Cerebellar infarction Usually changes
direction with gaze
Severe ataxia, may not be able to sit or stand
Scanning speech, extremity incoordination
Lateral medullary infarction
![]()
ipsi, ipsilateral.
Usually changes direction with gaze
Falls toward lesion side, may not be able to walk
Decreased gag reflex, ipsifacial numbness, ipsi-incoordination, ipsi-Horner’s syndrome, hypophonia
Diffusion-weighted images (DWIs) may show abnormalities in the first few hours after the acute stroke syndrome that will not show up on T2-weighted MRI scans for several hours.60
However, even DWIs can give false-negative
images in the first 24–48 hours after infarc- tion.38,50,61–63 Typically, areas of infarction do
not enhance with contrast. The territories sup- plied by the different cerebellar artery branches are shown in Figure 14–3B. The sensitivity of computed tomography (CT) scan for acute infarction is extremely low, meaning that a neg- ative result does not alter the probability of infarction in a meaningful way.61 CT scans can
Figure 14–4. Magnetic resonance images showing infarction in the lateral medulla (PICA territory) (A) and in the pontine cerebellar peduncle (AICA territory) (B). T2-weighted axial sections.
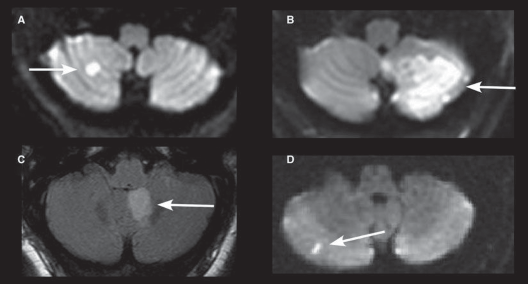
Figure 14–5. Posterior inferior cerebellar artery (PICA) acute strokes. A variety of PICA infarctions are seen from four different patients. Arrows point to the region of infarction. A, shows a small discrete acute stroke on a diffusion weighted image (DWI) sequence; B, shows a large PICA stroke on a DWI sequence; C, show a midline PICA infarction on a fluid- attenuated inversion-recovery sequence; and D, shows a very small acute stroke on DWI.
be useful in monitoring for swelling in the posterior fossa once the diagnosis of infarction has been made.
Deciding which patients with acute pro- longed vertigo should be considered for an MRI can be difficult. However the probability of an infarction is extremely low when there is unidirectional horizontal nystagmus, a corre- sponding positive head-thrust test, and no other neurologic signs or symptoms. Red flags for a stroke include any pattern of nystagmus other than unidirectional horizontal nystagmus, normal head-thrust test (indicating the periph- eral vestibular system is intact), inability to sit unassisted, and substantial risk factors for stroke. Red flags for transient ischemic attacks include new onset spontaneous episodes last- ing minutes and a crescendo pattern.
ULTRASOUND
Ultrasound studies of the heart and great ves- sels are used to search for an embolic source in patients with TIAs or stroke of presumed embolic origin.1 Echocardiography is used to search for mural thrombi and can also detect cardiac septal defects that allow direct commu- nication between the left and right heart chambers. Transesophageal echocardiography (TEE) is useful for identifying embolic sources
in the aortic arch. Amarenco and colleagues64 identified echogenic masses in the aortic arch on TEE in 6 of 12 patients with posterior circu- lation infarcts of unknown cause. Ultrasound studies of the neck (duplex scanning and Doppler) can identify an occlusion of the ver- tebral arteries within their bony canal that may be a source of artery-to-artery emboli within the posterior circulation.65 Through transcra- nial doppler (TCD) imaging, one can assess the intracranial vertebral arteries, but TCD is less accurate for identifying lesions in the basilar artery. TCD is not useful for assessing small branches of the vertebrobasilar system. More than any of the other ultrasound studies, TCD reliability is highly dependent on the skill of the performing technician.
ANGIOGRAPHY
Magnetic resonance angiography (MRA) is the procedure of choice for assessing the verte- brobasilar circulation.1,59 Overall, there is a good correlation between MRA and conven- tional angiography, although MRA is relatively limited for evaluating smaller branches within the vertebrobasilar system. Depending on ana- tomical variations and the positioning of the subject, the distal vertebral artery–basilar artery junction may be difficult to visualize
adequately. The neuroradiologist should be alerted in advance when it is important to visu- alize these structures so that the technician can properly position the patient. High-quality CT angiography (CTA) is also a method for imag- ing the posterior circulation and can be used in place of MRA, particularly if MR is contraindi- cated (e.g., patient with pacemaker).1 CTA is particularly useful for evaluating patients with suspected basilar artery occlusion because CTA takes substantially less time than MRA. Conventional contrast angiography is reserved for patients in whom the pathophysiology remains unclear after MRA or CT angiography (Figs. 14–6 and 14–7).
The main risk of conventional angiography is a TIA or infarction within the distribution of the injected vessel. The mechanism is usually vasospasm, although pieces of atherosclerotic plaque may be dislodged, resulting in a shower of emboli. Less common complications include an allergic reaction, a localized hemorrhage, or, rarely, infection at the site where the cath- eter enters the artery. Patients with a history of
migraine headaches are at greater risk for developing vasospasm.
Treatment
TRANSIENT ISCHEMIC ATTACKS
The urgency regarding any TIA is that 10%–15% of patients diagnosed with this transient event will suffer a completed stroke within 3 months, and half of these occur within the first 48 hours.66,67 It is not clear whether the risk of stroke is higher or lower for posterior circula- tion TIAs compared with anterior circulation TIAs.69 As a way to estimate the risk of future stroke in individual TIA patients, the ABCD2 score was developed and validated.67 To deter- mine the ABCD2 score, patients presenting with TIA are assigned points for the following five factors: age 60 years; blood pressure
140/90 mmHg; clinical features of unilateral weakness or speech impairment; duration 60 minutes or 10–59 minutes; and diabetes.
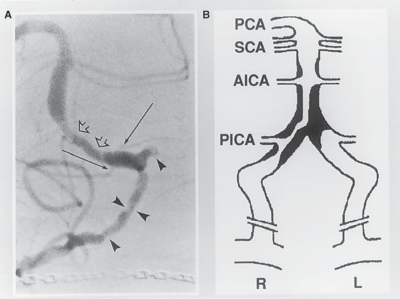
Figure 14–6. (A) Conventional cerebral angiogram in a patient with recurrent episodes of vertigo due to vertebrobasi- lar insufficiency. Magnetic resonance angiography showed an absent left vertebral artery but it was unclear whether this represented a normal variant or occlusion. Right vertebral artery injection (anterior posterior view). Arrowheads show nar- rowing of the right vertebral artery and the left vertebral stump (the left vertebral artery is blocked). Hollow arrows show basilar artery narrowing. The long thin arrows point to the anterior inferior cerebellar arteries. (B) Schematic diagram of the anterior posterior view of the vertebrobasilar system shown in the angiogram. AICA, anterior inferior cerebellar artery; PCA, posterior cerebral artery; PICA, posterior inferior cerebellar artery; SCA, superior cerebellar artery. (From Fife TD, Baloh RW, Duckwiler GR. Isolated dizziness in vertebrobasilar insufficiency: clinical features, angiography, and follow up.
Stroke Cerebrovasc Dis 4:4, 1994, with permission.)
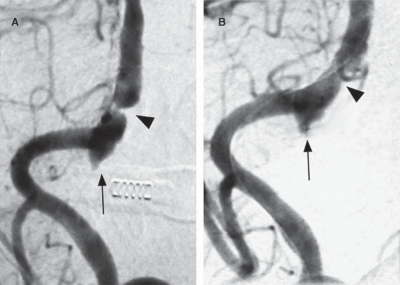
Figure 14–7. Conventional angiogram of a patient with recurrent vertigo attacks and basilar stenosis. (A) Pre-stent angio- gram (lateral view), demonstrating basilar artery stenosis proximal to the anterior inferior cerebellar arteries (arrow head) and right vertebral artery occlusion (arrow). (B) Corresponding post-stent angiogram showing the treated vessel after angioplasty and stenting. (Kerber KA, et al. Neurology. 2005;65:962).
Based on the number of points, the risk of stroke at 90 days can be determined. However, the validity of this risk assessment tool in poste- rior circulation TIA is not clear. As with all diz- ziness, vertigo, and imbalance presentations, there are likely to be problems with the reli- ability of the diagnostic classification particu- larly in TIA populations from claims databases or large cohort studies such as those used to derive and validate the ABCD2 score.67
Consensus guidelines for the evaluation and management of TIA have been published.66,68 The first step includes initiating an antiplatelet agent and other steps to control risk factors for stroke.68 Noninvasive imaging of the head and neck vessels is recommended to search for a critical stenosis. However, this is less relevant in posterior circulation stroke because there are no good trials to support use of surgical approaches to vessel stenosis in the posterior circulation, whereas there is high-level evi- dence to support the use of surgical procedures for patients with symptomatic carotid stenosis. Echocardiography is used to search for a car- diac embolic source, which is particularly com- mon in posterior circulation events.
Anticoagulation is recommended over anti- platelet agents in patients with atrial fibrillation or a prosthetic heart valve.68 Anticoagulation can also be considered when a cardioembolic
source is suspected based on finding an intracardiac clot or severe dilated cardiomyo- pathy.68 There is uncertainty about the use of anticoagulants versus antiplatelet agents in the management of patients having recurrent ver- tebrobasilar artery ischemic events. Retrospective studies have suggested warfarin may have efficacy in patients with vertebrobasi- lar disease.70–72 However, in the Warfarin ver- sus Aspirin in Symptomatic Intracranial Disease (WASID) trial, warfarin therapy did not reduce the risk of stroke even in the subgroup with stenosis of the posterior circulation.73 Furthermore, the WASID trial was stopped prematurely because warfarin was associated with significantly higher rates of adverse events (i.e., increased death, major hemorrhage, and myocardial infarction or sudden death). A sub- group analysis of the Ticlopidine Aspirin Stroke Study (TASS) found that patients classified as having vertebrobasilar symptom TIAs had a lower risk of stroke on ticlopidine compared to aspirin,74 but this finding has not been repli- cated and ticlopidine is rarely used because of the potential for adverse effects.
Transient vertigo episodes can be a warning sign of impending basilar artery occlusion— particularly when new in onset, increasing frequency and in patients with significant stroke risk factors, or when other neurologic
features (particularly motor weakness or speech disturbance) are reported.78–80 If a severe pos- terior circulation stenosis (50%) is identified, endovascular therapies become a management option (Fig. 14–7). But randomized trials are lacking for the use of endovascular therapies in treating patients with vertebrobasilar stenosis. Nonetheless, these procedures hold promise in the management of patients with recurrent posterior circulation TIAs or stroke. The Phase I trials of stenting suggest a high technical suc- cess rate (>95%) with a low periprocedural rate of complications (about 6%).75–77 Re-stenosis rates have varied from 7.5% to 35% at 6 months. Hopefully some of the important questions about the use of endovascular treatments will be answered by a large 60-site randomized controlled trial of stenting in intracranial steno- sis, the Stenting and Aggressive Medical Management for Preventing Recurrent stroke in Intracranial Stenosis (SAMMPRIS) trial (ClinicalTrials.gov Identifier: NCT00576693).
INFARCTION
Randomized controlled studies and consensus guideline statements support the use of intra- venous thrombolytic therapy for acute ischemic stroke patients when used less than 3 hours from stroke onset.82,83 More recent findings from a large randomized trial indicate the ben- efits of IV thrombolytics outweigh the risks for up to 4.5 hours from symptom onset.84 Patients with posterior circulation stroke, however, were less likely to be enrolled in these acute stroke trials, with about 5% of the patients in the original thrombolytic trial having a poste- rior circulation stroke.85 In subsequent trials, patients with posterior circulation stroke have either been excluded or were likely to be underrepresented.85 The reasons for this are largely because posterior circulation stroke is less common than anterior circulation stroke and also because it can be more difficult to dis- criminate between patients with posterior cir- culation stroke and patients with stroke-mimics such as peripheral vestibular disorders or even nonspecific symptom presentations.
The time window for the thrombolytic treat- ment of stroke is generally extended when basilar artery occlusion is identified, though this remains a controversial topic because of the lack of large randomized controlled data. The main reason that the window is extended
to even up to 24 hours is because the prognosis of basilar occlusion is so poor. One recent small randomized trial found that patients receiving intra-arterial thrombolysis had a good outcome in 4 of 8 patients receiving thrombolysis com- pared with only 1 of 8 patients in the control group.86 A systematic review of published case series of basilar artery occlusion treated with thrombolysis (either intravenous or intra- arterial) found that an outcome of death or dependency was more than 75% among 420 patients, and it did not differ on the adminis- tration route.87 Recanalization was achieved more frequently with intra-arterial thromboly- sis (225 of 344; 65%) than with intravenous
thrombolysis (40 of 76; 53%), but death and dependency rates did not differ. Only 24% of patients with intra-artertial thrombolysis and 22% with intravenous thrombolysis had a good outcome (e.g., modified Rankin score 2, meaning slight disability but able to walk unas- sisted and able to care for self without assis- tance). Without recanalization the likelihood of a good outcome was only 2%. A similar rate (68%) of a poor outcome (death or depen- dency) was also found in a large international registry study of basilar artery occlusion, and no statistically significant superiority was found for any treatment strategy.88 Factors associated with a better outcome include recanalization, treatment within 8 hours of symptom onset, and a Glasgow Coma Scale score 9 at presen- tation (generally meaning not comatose).85,87,88 Importantly, patients with radiographic evi- dence of brainstem infarction have been excluded from many of the studies reporting on the effects of thrombolytic treatment in basilar occlusion.87
After a latent interval of several days, some patients with cerebellar infarction may develop progressive brainstem dysfunction due to com- pression by the swollen cerebellum or due to the production of hydrocephalus.53,54 A large infarction in the cerebellar distribution of the PICA poses the greatest risk for brainstem compression. Surgical management should be a consideration in these cases because relent- less progression to quadriplegia, coma, and death can ensue.55–57 However, uncertainty exists about the timing and selection of patients for surgical management.56,57
Management of labyrinthine infarction is pri- marily symptomatic. Antivertiginous medica- tions can help relieve the acute vertigo and
nausea. Vestibular rehabilitation exercises should be started as soon as the patient is able to cooperate. The clinical course for patients sur- viving a brainstem or cerebellar infarction is typically that of gradual but incomplete recov- ery. Vertigo may persist for months because of damage to central structures important for com- pensation. Many patients complain of disabling oscillopsia due to spontaneous nystagmus and damaged central vestibular and cerebellar path- ways. Antivertiginous medications are less effec- tive for controlling vertigo than with peripheral vestibular lesions and vestibular rehabilitation exercises are often of minimal benefit. One exception to this overall generally poor progno- sis is the recovery seen in young patients with lateral medullary infarction due to vertebral artery dissection. Many of these patients have a complete return to normal and the great major- ity have only minimal residual dysfunction.
Intralabyrinthine hemorrhage can be associ- ated with multiple systemic disorders, includ- ing leukemia, vasculitis, and cocaine abuse.24,91–93 Sudden deafness due to intrlabyrinthine hemorrhage is common in patients with endo- lymphatic sac tumors associated with von Hippel-Lindau disease.94 A small percentage of patients with idiopathic sudden deafness show MRI features of intralabyrinthine hemor- rhage.95 Pathologic examination of the inner ear reveals hemorrhage into the perilymphatic space with smaller focal hemorrhages in the endolymphatic space.24 The vestibular and cochlear end organs, although morphologically intact, are rendered nonfunctional, apparently from altered fluid chemistry. A similar condi- tion may follow from a blow to the head with- out the occurrence of a bony fracture (so-called labyrinthine concussion).
Diagnosis and Management
Diagnosis of intralabyrinthine hemorrhage is based on finding a sudden auditory and vestibular loss in a patient with an underlying predisposing condition (see earlier discussion). Hearing loss and vestibular loss (documented
with audiometric and electronystagmography tests) are profound and usually permanent. MRI may reveal findings suggestive of intral- abyrinthine hemorrhage, including increased signal on precontrast T1 sequences and also increased signal on T2 or fluid-attenuated inversion recovery (FLAIR) sequences. However, these imaging findings could also indicate increased protein concentration rather than hemorrhage. A more advanced FLAIR sequence, three-dimensional (3D) FLAIR, might be a more sensitive test for identifying hemorrhage or increased protein content in the inner ear but requires more detailed study to determine the reliability and validity of the test.95,96 Management consists of symptomatic treatment of the vertigo (see Chapter 19) and correcting an underlying bleeding diathesis when possible.
HEMORRHAGE INTO THE BRAIN STEM AND CEREBELLUM
Spontaneous intraparenchymal hemorrhage into the brain stem or cerebellum produces a dramatic clinical syndrome frequently pro- gressing to loss of consciousness and death.97,98 The cause of hemorrhage is hypertensive vas- cular disease in approximately two-thirds of patients. Anticoagulation therapy, cryptic arte- riovenous malformations, and bleeding diathe- sis are also important etiologic factors, whether alone or in combination with hypertension. Cerebellar hemorrhage can be a remote effect of spinal or supratentorial surgery associated with drainage of large volumes of CSF.99
Vertigo may be an initial symptom with brain stem hemorrhage, but it is never an isolated symptom and is usually only fleeting as the patient rapidly plunges into coma. Hemorrhage into the pons typically results in a rapid onset of coma, flaccid quadriplegia, loss of horizontal eye movements, pinpoint reactive pupils, and ocular bobbing.48 Hemorrhage into the medulla is associated with rapid cardiorespiratory failure and death.
Because of its potential reversibility, cere- bellar hemorrhage deserves particular empha- sis.98 The initial symptoms of acute cerebellar hemorrhage are vertigo, nausea, vomiting, headache, and inability to stand or walk. As with cerebellar infarction, these symptoms
might be confused with an acute peripheral vestibular lesion. Unlike the latter, however, examination in the initial period usually reveals nuchal rigidity, prominent cerebellar signs, ipsilateral facial paralysis, and ipsilateral gaze paralysis. Pupils are often small bilaterally but reactive. Approximately 50% of patients lose consciousness within 24 hr of the initial symp- toms, and 75% become comatose within 1 week of onset.101 The condition is often fatal unless surgical decompression is performed.102 Midline cerebellar hemorrhage is particularly difficult to diagnose because it produces bilat- eral signs and generally runs a more fulminant course than a lateralized hemorrhage. Such patients have profound ataxia, usually being unable to stand—a finding not associated with benign peripheral vestibular lesions.
Diagnosis and Management
The diagnosis of hemorrhage into the brain stem and cerebellum has been revolutionized with the introduction of CT and MR scanning. Computed tomography is usually superior to MR for identifying intraparenchymal blood (Fig. 14–8). An imaging study is recommended in any patient presenting with the acute onset of vertigo not meeting criteria for a peripheral vestibular disorder and who also exhibits prominent ataxia, either of the trunk and or extremities.
As indicated earlier, hemorrhage into the cere- bellum is often fatal unless surgical decompression is performed.103 The earlier the syndrome is recog- nized, the more likely the surgery will be success- ful. Once the patient is comatose, almost none
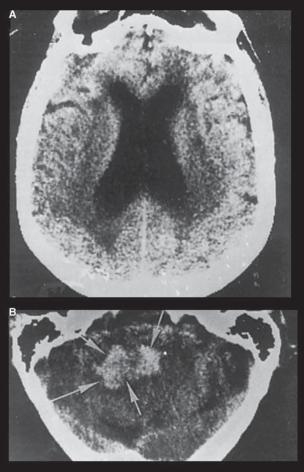
Figure 14–8. Computed tomography scan showing hydrocephalus (A) secondary to a cerebellar hemorrhage (arrows) (B).
survive. Small hemorrhages into the brain stem and cerebellum may spontaneously resolve.
VASCULAR COMPRESSION SYNDROMES
Vertebrobasilar Dolichoectasia
Dolichoectasia refers to an enlargement and elongation of the basilar artery (Fig. 14–9). It is a common finding on MR images of the brain and on cerebral angiography, typically unasso- ciated with any clinical symptoms. However, there are reports suggesting that vertebrobasi- lar dolichoectasia can be implicated in neuro- logic symptoms, both by compression of the brain stem and cranial nerves or by producing TIAs.104–106 In some cases, both compression and ischemia coexist in the same patient. Patients with vertebrobasilar dolichoectasia can have evidence of both peripheral and cen- tral vestibular dysfunction, most commonly due to compression of the vestibular nerves.107 The pathophysiology for cerebral ischemia is uncertain, but obstruction by atheromata, intraluminal thrombi, or artery-to-artery emboli have been proposed.108 Distortion of small branches of the basilar artery due to elongation and tortuosity and hemodynamic factors related to reduction in flow velocity in the enlarged basilar artery may also contribute to the isch- emia. There is still debate as to whether verte- brobasilar dolichoectasia represents a congeni- tal anomaly or is the result of atherosclerotic
degeneration of the vascular wall. Probably both factors can lead to the production of ver- tebrobasilar dolichoectasia, but often there is a more generalized vascular ectasia of cerebral vessels, suggesting a more diffuse arterial defect. Transient ischemic attacks associated with vertebrobasilar dolichoectasia are treated like other causes of TIAs. The role of percuta- neous transluminal angioplasty and stenting in symptomatic dolichoectasia is not clear.81
Vascular Compression by Normal Vessels (Vestibular Paroxysmia)
There is controversy as to whether audioves- tibular symptoms can result from compression of the eighth cranial nerves by normal arteries. Jannetta et al.109 reported improvement in patients with “disabling positional vertigo” after surgically removing vascular loops that were compressing the eighth cranial nerve near the root entry zone. However, the clinical syn- drome was ill defined and there were no specific diagnostic tests prior to surgical explo- ration of the posterior fossa. Furthermore, vascular loops, particularly loops of the AICA, are common in the cerebellopontine angle in normal subjects without symptoms. Brandt and Dieterich111 described a syndrome characterized by brief episodes of vertigo— vestibular paroxysmia—which they attributed to neurovascular compression of the eighth cranial nerve. Some patients had associated auditory symptoms and signs, whereas others did not. Episodes could occur spontaneously
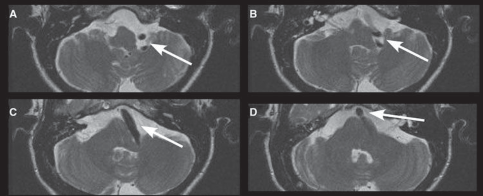
Figure 14–9. Vertebro-basilar dolichoectasia. Magnetic resonance images show an ectatic vertebrobasilar artery. Arrows point to the artery. The large and tortuous artery can be seen lateral to the medulla (A), and then compressing the cerebellar peduncle and pons (B and C), before coursing back to the midline (D).
or after head turns or position change. A more recent follow-up study of 32 patients with vestibular paroxysmia found that most responded to either carbamazepine (mean dose 568 mg/day) or oxcarbazepine (mean dose 870 mg/day).112 Patients with vestibular paroxysmia may also respond to low-dose gabapentin.110
Rotational Vertebral Artery Syndrome
Although rare, vertebrobasilar ischemia can be caused by vertebral artery occlusion during neck rotation.113 Nearly always one vertebral artery is hypoplastic or occluded and the other becomes compressed by an osteophyte, usually at the C level, when the head is turned to the
Lee H, Sohn SI, Cho YW, Lee SR, Ahn BH, Park BR, Baloh RW. Cerebellar infarction presenting iso- lated vertigo: frequency and vascular topographical patterns. Neurology. 2006;67(7):1178.
Grad A, Baloh RW. Vertigo of vascular origin: clinical and electronystagmographic features in 84 cases. Arch Neurol. 1989;46:281.
Lee H, Kim HJ, Koo JW, Kim JS. Superior divisional vestibular paresis in anterior inferior cerebellar artery infarction. J Neurol Sci. 2009;285:250.
Lee H, Kim JS, Chung E, et al. Infarction of the terri- tory of anterior inferior cerebellar artery: spectrum of audiovestibular loss. Stroke. 2009;40:3745.
Oas JG, Baloh RW. Vertigo and the anterior infe- rior cerebellar artery syndrome. Neurology. 1992;42: 2274.
Fisher CM, Karnes WE, Kubik CS. Lateral med- ullary infarction–the pattern of vascular occlusion. J Neuropath Exp Neurol. 1961;20:323.
Hosoya T, Nagahata M, Yamaguchi K. Prevalence of artery dissection in Wallenberg’s syndrome: neurora- dilogical analysis of 93 patients in the Tohoku District, Japan. Radiat Med. 1996;14:241.
Chen WL, Chern CH, Wu YL, Lee CH. Vertebral
side
5-6
opposite
the intact vertebral artery.
artery dissection and cerebellar infarction following chi-
ropractic manipulation. Emerg Med J. 2006;23(1):e1.
However, there is a report of a patient who compressed both vertebral arteries with head turn to one side.114
Choi et al.115 described a stereotypical nys- tagmus that was triggered by head turn to the side opposite the intact vertebral artery. This nystagmus was mostly downbeat with horizontal and torsional components beating toward the compressed vertebral artery side. A dynamic conventional angiogram can be performed to identify the vertebral artery compression on head turning, but a dynamic CT scan may be an option as well.116,117
Bartels E. Dissection of the extracranial vertebral artery: clinical findings and early noninvasive diagnosis in 24 patients. J Neuroimaging. 2006;16(1):24.
Amarenco P, Hauw J-J. Cerebellar infarction in the ter- ritory of the anterior inferior cerebellar artery: a clini- copathological study of 20 cases. Brain. 1990;113:139.
Caplan LR. Brain embolism, revisited. Neurology. 1993;42:1281.
Terao S, Miura N, Osano Y, et al. Multiple cerebel- lar infarcts: clinical and pathophysiologic features. J Stroke Cerebrovasc Dis. 2005;14(5):193.
Amarenco P, Kase CS, Rosengart A, et al. Very small (border zone) cerebellar infarcts: distribution, causes, mechanisims and clinical features. Brain. 1993;116:161.
Kikuchi S, Yamasoba T. Neuro-otological findings
When rotational vertebral artery syndrome is identified there is still uncertainty about the best management. No long-term studies in a large enough sample of patients has been per- formed so that we can understand the natural history of the disorder. In theory, patients could learn to prevent these attacks by avoiding the movements that trigger them. Surgical decompression of the vertebral artery may be an option for alleviating refractory symptoms and could be important for reducing the risk of stroke.
REFERENCES
Savitz SI, Caplan LR. Vertebrobasilar disease. N Engl J Med. 2005;352(25):2618.
Hotson JR, Baloh RW. Acute vestibular syndrome.
N Engl J Med. 1998;339:680.
in patients with very small (border zone) cerebellar infarcts. Acta Otolaryngol Suppl. 2007;(559):56.
Williams D, Wilson TG. The diagnosis of the major and minor syndromes of basilar insufficiency. Brain. 1962;85:741.
Fisher CM. Vertigo in cerebrovascular disease. Arch Otolaryngol. 1967;85:855.
Fields WS, Weibel J. Effects of vascular disorders on the vestibular system. In: Fields WS, Alford BR, eds. Neurological Aspects of Auditory and Vestibular Disorders. Spring-field, IL: Charles C Thomas; 1964.
Fife TD, Baloh RW, Duckwiler GR. Isolated dizzi- ness in vertebrobasilar insufficiency: clinical feature, angiography and follow-up. J Stroke Cerbrovasc Dis. 1994;4:4.
Gomez CR, Cruz-Flores S, Malkoff MD, Sauer CM, Burch CM. Isolated vertigo as a manifestation of ver- tebrobasilar ischemia. Neurology. 1996;47:94.
Kim HA, Lee SR, Lee H. Acute peripheral vestibular syndrome of a vascular cause. J Neurol Sci. 2007;254 (1-2):99.
Kerber KA, Rasmussen PA, Masaryk TJ, Baloh RW. Recurrent vertigo attacks cured by stenting a basilar artery stenosis. Neurology. 2005;65:962.
Schuknecht HE. Pathology of the Ear. 2nd ed. Philadelphia: Lea & Febiger; 1993.
Lee H, Baloh RW. Sudden deafness in vertebrobasi- lar ischemia: clinical features, vascular topographi- cal patterns and long-term outcome. J Neurol Sci. 2005;228(1):99.
Jaffe B. Sudden deafness—a local manifestation of systemic disorders: fat emboli, hypercoagulation and infections. Laryngoscope. 1970;80:788.
Kirikae I, Nomura Y, ShitaraT, Kobayashi T. Sudden deafness due to Buerger’s disease. Arch Otolaryngol. 1962;75:502.
Lee H, Yi HA, Cho YW, et al. Nodulus infarction mim- icking acute peripheral vestibulopathy. Neurology. 2003;60(10):1700.
Ruben R, Distenfeld A, Berg P, Carr R. Sudden sequential deafness as the presenting symptom of macroglobulinema. JAMA. 1969;209:1364.
Andrews JC, Hoover LA, Lee RS, Honrubia V. Vertigo in hyper-viscosity syndrome. Otolaryngol Head Neck Surg. 1988;98:144.
Saadah HA. Vestibular vertigo associated with hyper- lipidemia: response to antilipidemic therapy. Arch Intern Med. 1993;153:1846.
Kim JS, Lopez I, DiPatre PL, Liu F, Ishiyama A, Baloh RW. Internal auditory artery infarction: clinicopatho- logic correlation. Neurology. 1999;52:40.
Frumkin LR, Baloh RW. Wallenberg’s syndrome fol- lowing neck manipulation. Neurology. 1990;40:611.
de Bray M, Penisson-Benier I, Dubas F, Emile J. Extracranial and intracranial vertebrobasilar dissec- tions: diagnosis and prognosis. J Neurol Neurosurg Psychiatry. 1997;63:46.
Auer A, Felber S, Schmidauer C, Waldenberger P, Aichner F. Magnetic resonance angiographic and clin- ical features of extracranial vertebral artery dissection. J Neurol Neurosurg Psychiatry. 1998;64:474.
Arnold M, Bousser MG, Fahrni G, et al. Vertebral artery dissection: presenting findings and predictors of outcome. Stroke. 2006; 37:2499.
Park MH, Kim BJ, Koh SB, Park MK, Park KW, Lee DH. Lesional location of lateral medullary infarction presenting hiccups (singultus). J Neurol Neurosurg Psychiatry. 2005;76(1):95.
Kattah JC, Talkad AV, Wang DZ, Hsieh Y, Newman- Toker DE. HINTS to diagnose stroke in the acute vestibular syndrome. Three step bedside oculomotor examination more sensitive than early MRI diffusion weighted imaging. Stroke. 2009;40:3504.
Brandt T, Dieterich M. Pathological eye-head coordi- nation in roll: tonic ocular tilt reaction in mesenceph- alic and medullary lesions. Brain. 1987;110:649.
Kim HA, Lee H, Yi HA, Lee SR, Lee SY, Baloh RW. Pattern of otolith dysfunction in posterior inferior cer- ebellar artery territory cerebellar infarction. J Neurol Sci. 2009;280(1–2):65.
Bjerner K, Silfverskold BJ. Lateropulsion and imbal- ance in Wallenberg’s syndrome. Acta Neurol Scand. 1968;44:91.
Nowak DA, Topka HR. The clinical variability of Wallenberg’s syndrome. The anatomical cor- relate of ipsilateral axial lateropulsion. J Neurol. 2006;253(4):507.
Kommerell G, Hoyt WF. Lateropulsion of saccadic eye movements: electro-oculographic studies in a
patient with Wallenberg’s syndrome. Arch Neurol. 1973;28:313.
Choi KD, Kim HJ, Cho BM, Kim JS. Saccadic adapta- tion in lateral medullary and cerebellar infarction. Exp Brain Res. 2008;188(3):475.
Lee H, Ahn BH, Baloh RW. Sudden deafness with vertigo as a sole manifestation of anterior inferior cer- ebellar artery infarction. J Neurol Sci. 2004;222(1–2): 105.
Lee H, Yi HA, Baloh RW. Sudden bilateral simulta- neous deafness with vertigo as a sole manifestation of vertebrobasilar insufficiency. J Neurol Neurosurg Psychiatry. 2003;74(4):539.
Huang CY, Yu YL. Small cerebellar strokes may mimic labyrinthine lesions. J Neurol Neurosurg Psychiatry. 1985;48:263.
Edlow JA, Newman-Toker DE, Savitz SI. Diagnosis and initial management of cerebellar infarction. Lancet Neurol. 2008;7(10):951.
Norrving B, Magnusson M, Holtas S. Isolated acute vertigo in the elderly: vestibular or vascular disease? Acta Neurol Scand. 1995;91:43.
Newman-Toker DE, Kattah JC, Alvernia JE, Wang DO. Normal head impulse test differentiates acute cerebellar strokes from vestibular neuritis. Neurology. 2008; 70:2378.
Lee H, Yi HA, Cho YW, et al. Nodulus infarction mim- icking acute peripheral vestibulopathy. Neurology. 2003;60(10):1700.
Sohn SI, Lee H, Lee SR, Baloh RW. Cerebellar infarc- tion in the territory of the medial branch of the supe- rior cerebellar artery. Neurology. 2006;66(1):115.
Duncan GW, Parker SW, Fisher CM. Acute cer- ebellar infarction in the PICA territory. Arch Neurol. 1975;32:364.
Sypert GW, Alvord EC, Jr. Cerebellar infarction: a clinicopathological study. Arch Neurol. 1975; 32:357.
Kudo H, Kawaguchi T, Minami H, Kuwamura K, Miyata M, Kohmura E. Controversy of surgical treatment for severe cerebellar infarction. J Stroke Cerebrovasc Dis. 2007;16(6):259.
Pfefferkorn T, Eppinger U, Linn J, et al. Long-term outcome after suboccipital decompressive craniec- tomy for malignant cerebellar infarction. Stroke. 2009;40:3045.
Juttler E, Schweickert S, Ringleb PA, Huttner HB, Kohrmann M, Aschoff A. Long term outcome after surgical treatment for space-occupying cerebellar infarction: Experience in 56 patients. Stroke. 2009;9: 3060.
Lee H. Neuro-otological aspects of cerebellar stroke syndrome. J Clin Neurol. 2009;5(2):65.
Seynaeve P, Hasso AN, Thompson JR, Hinshaw DB Jr. Basilar and distal vertebral artery occlusive dis- ease: correlation of MR imaging and MR angiography. J Beige Radiol. 1996;79:61.
Kitis O, Calli C, Yunten N, Kocaman A, Sirin H. Wallenberg’s lateral medullary syndrome: diffusion- weighted imaging findings. Acta Radiol. 2004;45(1): 78.
Chalela JA, Kidwell CS, Nentwich, et al. Magnetic res- onance imaging and computed tomography in emer- gency assessment of patients with suspected acute stroke: a prospective comparison. Lancet. 2007;369: 293.
Frey LC, Sung GY, Tanabe J. Early false-negative diffusion-weighted imaging in brainstem infarction. J Stroke Cerebrovasc Dis. 2002;11(1):51.
Oppenheim C, Stanescu R, Dormont D, et al. False-negative diffusion weighted MR findings in acute ischemic stroke. Am J Neuroradiol. 2000;21: 1434.
Amarenco P, Cohen A, Baudrimont M, Bousser M-
Transesophageal echocardiographic detection of aortic arch disease in patients with cerebral infarction. Stroke. 1992;23:1005.
von Büdingen HC, Staudacher T, von Büdingen HJ. Ultrasound diagnostics of the vertebrobasilar system. Front Neurol Neurosci. 2006;21:57.
Easton JD, Saver JL, Albers GW. Definition and evaluation of transient ischemic attack. Stroke. 2009;40:2276.
Johnston SC, Rothwell PM, Nguyen-Huynh MA. Validation and refinement of scores to predict very early stroke risk after transient ischemic attack. Lancet. 2007;369:283.
Sacco RL, Adams R, Albers G, et al. Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attacks. Circulation. 2006;113:e409.
Flossman E, Rothwell PM. Prognosis of vertebrobasi- lar transient ischaemic attack and minor stroke. Brain. 2003;126:1940.
Browne TR, Poskanzer DC. Treatment of strokes. I.
N Engl J Med. 1969;281:594.
Qureshi AI, Ziai WC, Yahia AM, et al. Stroke-free survival and its determinants in patients with symp- tomatic vertebrobasilar stenosis: a multicenter study. Neurosurgery. 2003;52(5):1033.
Chimowitz MI, Kokkinos J, Strong J, et al. The Warfarin-Aspirin Symptomatic Intracranial Disease Study. Neurology. 1995;45:1488.
Kasner SE, Lynn MJ, Chimowitz MI, et al. Warfarin vs aspirin for symptomatic intracranial stenosis. Subgroup analyses from WASID. Neurology. 2006;67:1275.
Grotta JC, Norris JW, Kamm B. TASS Baseline and Angiographic Data Subgroup. Prevention of stroke with ticlopidine: who benefits more? Neurology. 1992;42:111.
Stenting of symptomatic atherosclerotic lesions in the vertebral or intracranial arteries (SSYLVIA): study results. Stroke. 2004;35:1388.
Bose A, Hartmann M, Henkes H, et al. A novel, self- expanding, nitinol stent in medically refractory intrac- ranial atherosclerosis stenosis: the wingspan study. Stroke. 2007;38:1531.
Turan TN, Derdeyn CP, Fioralla D, Chimowitz MI. Treatment of atherosclerotic intracranial arterial dis- ease. Stroke. 2009;40:2257.
Ferbert A. Bruckmann H, Drummen R. Clinical fea- tures of proven basilar artery occlusion. Stroke. 1990; 21:1135.
Von Campe G, Regli F, Bogousslavsky J. Heralding manifestations of basilar artery occlusion with lethal or severe stroke. J Neurol Neurosurg Psychiatry. 2003;74:1621.
Voetsch B, DeWitt D, Pessin MS, Caplan LR. Basilar artery occlusive disease in the New England Medical Center Posterior Circulation registry. Arch Neurol. 2004;61:496.
Fiorella D, Chow MM, Anderson M, Woo H, Rasmussen PA, Masaryk TJ. A 7-year experience with balloon-mounted coronary stents for the treatment of symptomatic vertebrobasilar intracranial atheroma- tous disease. Neurosurgery. 2007;61(2):236.
The National Institute of Neurological Disorders and Stroke rt-PA Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333:1581.
Adams HP, del Zoppo G, Alberts MJ, et al. Guidelines for the early management of adults with ischemic stroke. Stroke. 2007;38:1655.
Hacke W, Kaste M, Bluhmki E, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317.
Tsao JW, Hemphill JC, Johnston JC, Smith WS, Bonovich DC. Initial Glasgow coma scale score predicts outcome following thombolysis for posterior circulation stroke. Arch Neurol. 2005;62:1126.
Macleod MR, Davis SM, Mitchell PJ, et al. Results of a multicentre, randomized controlled trial of intra- arterial urokinase in the treatment of acute poste- rior circulation ischemic stroke. Cerebrovasc Dis. 2005;20:12.
Lindsberg P, Mattle HP. Therapy of basilar artery occlusion: a systematic analysis comparing intra- arterical and intravenous thrombolysis. Stroke. 2006;37:922.
Schonewille WJ, Wijman CC, Michel P, et al. Treatment and outcomes of acute basilar artery occlu- sion in the Basilar Artery International Cooperation Study (BASICS): a prospective registry study. Lancet Neurol. 2009;8:724.
Grond M, Rudolf J, Schmulling S, Stenzel C, Neveling M, Heiss WD. Early intravenous thrombolysis with recombinant tissue-type plasminogen activa- tor in vertebrobasilar ischemic stroke. Arch Neurol. 1998;55:466.
Montavont A, Nighoghossian N, Derex L, et al. Intravenous r-tPA in vertebrobasilar acute infarcts. Neurology. 2004;62:1854.
Cherchi M, Huo E, Nelson N, Frankfurt O, Russell E, Raizer J. Gradual hearing loss with bilateral labyrin- thine hemorrhage in chronic myelogenous leukemia. Neurology. 2006;67(1):177.
Sugiura M, Naganawa S, Teranishi M, Sato E, Kojima S, Nakashima T. Inner ear hemorrhage in systemic lupus erythematosus. Laryngoscope. 2006;116(5):826.
Nicoucar K, Sakbani K, Vukanovic S, Guyot JP. Intralabyrinthine haemorrhage following cocaine con- sumption. Acta Otolaryngol. 2005;125(8):899.
Butman JA, Kim HJ, Baggenstos M, et al. Mechanisms of morbid hearing loss associated with tumors of the endolymphatic sac in von Hippel-Lindau disease. JAMA. 2007;298(1):41.
Sugiura M, Naganawa S, Teranishi M, Nakashima T. Three-dimensional fluid-attenuated inversion recov- ery magnetic resonance imaging findings in patients with sudden sensorineural hearing loss. Laryngoscope. 2006;116(8):1451.
Yoshida T, Sugiura M, Naganawa S, Teranishi M, Nakata S, Nakashima T. Three-dimensional fluid- attenuated inversion recovery magnetic resonance imaging findings and prognosis in sudden sensorineu- ral hearing loss. Laryngoscope. 2008;118:1433.
Dinsdale HB. Spontaneous hemorrhage in the poste- rior fossa: a study of primary cerebellar and pontine hemorrhage with observations on the pathogenesis. Arch Neurol. 1964;10:200.
Elijovich L, Patel PV, Hemphill JC, III. Intracerebral hemorrhage. Semin Neurol. 2008;28(5):657.
Brockmann MA, Groden C. Remote cerebellar hem- orrhage: a review. Cerebellum. 2006;5(1):64.
Kushner MJ, Bressman SB. The clinical manifes- tations of pontine hemorrhage. Neurology. 1985; 35:637.
Brennen RW, Bergland RM. Acute cerebellar hem- orrhage. Analysis of clinical findings and outcome in 12 cases. Neurology. 1977;27:527.
Pollak L, Rabey JM, Gur R, Schiffer J. Indication to surgical management of cerebellar hemorrhage. Clin Neurol Neurosurg. 1998;100:99.
Mayer SA, Rincon F. Treatment of intracerebral hae- morrhage. Lancet Neurol. 2005;4(10):662.
Passero S, Nuti D. Auditory and vestibular findings in patients with vertebrobasilar dolichoectasia. Acta Neurol Scand. 1996;93:50.
Passero S, Filosomi G. Posterior circulation infarcts in patients with vertebrobasilar dolichoectasia. Stroke. 1998;29:653.
Ubogu EE, Zaidat OO. Vertebrobasilar dolichoecta- sia diagnosed by magnetic resonance angiography and risk of stroke and death: a cohort study. J Neurol Neurosurg Psychiatry. 2004;75(1):22.
Nuti D, Passero S, Di Girolaamo S. Bilateral vestibu- lar loss in vertebrobasilar dolichoectasia. J Vestib Res. 1996;6:85.
Besson G, Bogousslavsky J, Moulin T, Hommel M. Vertebrobasilar infarcts in patients with dolichoec- tatic basilar artery. Acta Neurol Scand. 1995;91:37.
Jannetta PJ, Moller MB, Moller ARC. Disabling posi- tional vertigo. N Engl J Med. 1984;310:1700.
Russell D, Baloh RW. Gabapentin responsive audiovestibular paroxysmia. J Neurol Sci. 2009;281 (1-2):99.
Brandt Th, Dieterich M, Danek A. Vestibular parox- ysmia. Bailliere’s Clin Neurol. 1994;3:565.
Hüfner K, Barresi D, Glaser M, et al. Vestibular par- oxysmia: diagnostic features and medical treatment. Neurology. 2008;71(13):1006.
Brandt T, Baloh RW. Rotational vertebral artery occlusion: a clinical entity or various syndromes? Neurology. 2005;65(8):1156.
Kuether TA, Nesbit GM, Clark WM, Barnwell SL. Rotational vertebral artery occlusion: a mechanism of vertebrobasilar insufficiency. Neurosurgery. 1997;41:427.
Choi KD, Shin HY, Kim JS, et al. Rotational vertebral artery syndrome: oculographic analysis of nystagmus. Neurology. 2005;65(8):1287.
Bulsara KR, Velez DA, Villavicencio A. Rotational vertebral artery insufficiency resulting from cervical spondylosis: case report and review of the literature. Surg Neurol. 2006;65(6):625.
Petridis AK, Barth H, Buhl R, Mehdorn HM. Vertebral artery decompression in a patient with rotational occlusion. Acta Neurochir (Wien). 2008;150(4):391.
![]()
TUMORS OF THE MIDDLE EAR AND TEMPORAL BONE
Glomus Body Tumors (Paragangliomas) Diagnosis
Management
TUMORS OF THE INTERNAL AUDITORY CANAL AND CEREBELLOPONTINE ANGLE
Schwannomas Meningiomas
Epidermoid Cysts (Primary Cholesteatomas) Cholesterol Granulomas
Metastatic Tumors Diagnosis Management BRAIN TUMORS
Brain Stem Fourth Ventricle Cerebellum
Diagnosis and Management
TUMORS OF THE MIDDLE EAR AND TEMPORAL BONE
A wide variety of benign and malignant tumors involve the middle ear and temporal bone.1 Tumors involving the middle ear produce symptoms of fullness or conductive hearing loss early, whereas tumors in the temporal bone outside the middle ear can become quite large without producing symptoms. The tumor may not become apparent until it erodes into the external auditory canal (producing a conductive hearing loss) or through the mastoid cortex into the skin. Anterior extension into the cavernous sinus produces ophthalmoplegia from involve- ment of the third, fourth, and sixth nerves. Malignant tumors in this region tend to spread locally to the regional lymph nodes; distant metastasis is unusual. Ultimately, the tumor can be seen in the nasopharynx, middle ear, or neck.
Malignant Tumors
Squamous cell carcinoma is the most frequent histologic type of malignant tumor involving the middle ear and mastoid.2,3 It typically arises from epidermal cells of the auricle, external auditory canal, or the middle ear and mastoid. The prognosis is good for tumors confined to the auricle and external canal but not for those invading the middle ear and mastoid. The latter are frequently associated with prominent ear symptoms that include vertigo, hearing loss, pain, otorrhea, mastoid swelling, and facial paralysis. Squamous cell carcinomas often begin in an ear with previous otologic disease, partic- ularly chronic suppurative otitis media with a mastoid cavity. Other, less common, tumors originating in the external auditory canal and middle ear include adenoid cystic carcinoma, basal cell carcinoma, mucoepidermoid carci- noma, and ceruminoma. In general, these tumors are less malignant but occasionally will
339
Generally, carcinomas occur in an elderly age group, whereas sarcomas occur in the young. Both osteogenic sarcoma and chondrosarcoma occur as primary tumors of the temporal bone, running fulminant courses in older children and young adults. Rhabdomyosarcoma is the most common middle ear malignant tumor in the young, typically occurring in children under the age of 5.4 The initial symptom is often facial paralysis, which may be misdiagnosed as idio- pathic Bell’s palsy. In later stages, the tumor extends beyond the middle ear to involve the petrous apex and may invade the posterior or middle cranial fossi. Rhabdomyosarcoma should be considered in any infant presenting with idiopathic facial paralysis.
Metastatic involvement of the temporal bone is common with several different tumor types but, because of the enchondral layers’ resis- tance, neoplasms rarely invade the bony laby- rinth.5 The most common sites of origin for metastatic tumors in order of frequency are breast, kidney, lung, stomach, larynx, prostate, and thyroid gland.6 Metastatic tumors from the breast and prostate commonly incite new bone formation.
Glomus Body Tumors (Paragangliomas)
Glomus tumors are the most common tumor of the middle ear, and next to schwannomas they are the most common tumor of the temporal bone.7 Glomus tumors arise in the glomera of the chemoreceptor system, which may be found along the vagus nerve, glossopharyngeal nerve, Jacobson’s nerve (tympanic branch of the ninth nerve), and the nerve of Arnold (postauricular branch of the tenth nerve). Most are hamartomas with tissue components found in the normal glomus body.8 Genetic factors are likely important in pathogenesis, and in some families glomus tumors are inherited in an autosomal dominant fashion with genomic imprinting (the gene only results in tumors if inherited from the father).9 The most common tumor sites are the glomus jugulare (jugular bulb), glomus tympanicum (middle ear), and
glomus vagale (along the course of the vagus nerve). Glomus vagale and jugulare tumors often involve the labyrinth and cranial nerves, whereas glomus tympanicum tumors usually produce only local symptoms such as conductive hearing loss, pulsatile tinnitus, and rhinorrhea because of the tumor bulk in the middle ear. Invasion of the labyrinth is an uncommon but serious prognostic sign and is often associated with extension to the petrous apex and into the middle and posterior cranial fossae. The jugu- lar foramen syndrome consisting of ninth, tenth, and eleventh nerve involvement occurs with glomus jugulare and vagale tumors. Involvement of the twelfth nerve is an ominous sign, indicating destruction of the jugular fora- men with tumor extension into the hypoglossal canal and usually into the posterior fossa.
Diagnosis
Tumors of the middle ear space and temporal bone can often be identified on careful physical examination. A malignant tumor may be visible after it has eroded into the external auditory canal or through the mastoid cortex into the skin. Nearby lymph nodes are often enlarged. A biopsy of these lesions should lead to the cor- rect histologic diagnosis. Glomus body tumors are often visible through the tympanic mem- brane (see Fig. 6–1C). If not all the borders are visible, the tumor may be either a large glomus tympanicum tumor or a much larger glomus jugulare tumor that has extended from the jug- ular bulb into the middle ear. If cranial nerve deficits are present, the tumor is most likely a glomus jugulare type.
Although the presence of a tumor involving the middle ear or temporal bone can often be identified on physical examination, computer- ized tomography (CT) and magnetic resonance (MR) scanning are used to assess the extent of the tumor. Computed tomography is the diag- nostic procedure of choice for determining bony involvement, and MR is most useful for determining the soft tissue extent. Contrast should be used with either procedure to visual- ize vascular elements.10 Magnetic resonance angiography (MRA) is particularly useful for defining the features of a glomus body tumor.11,12 Individuals in families with glomus tumors should be screened after age 16 to detect early asymptomatic tumors.9
Malignant tumors confined to the ear and external auditory canal can often be surgically resected with minimal cosmetic and functional disability.13 Much more extensive surgical procedures with greater cosmetic and func- tional disabilities are required for tumors invading the middle ear and mastoid, and often only subtotal resection is possible. Most patients are treated with postsurgical radiation, but the long-term prognosis in these patients is poor.
Although some small glomus tympanicum tumors, when the borders are clearly demar- cated, can be removed via the external auditory canal, more extensive procedures are usually required for these tumors. Glomus jugulare and vagale tumors are much more difficult to remove because they are highly vascular and closely interrelated with key neural and vascu- lar structures.7,14 A “wait-and-see” policy has been recommended for most of these tumors.12,15 Radiation therapy may be useful for management of tumor recurrences and for unresectable lesions.16
TUMORS OF THE INTERNAL AUDITORY CANAL AND CEREBELLOPONTINE ANGLE
Tumors arising in the narrow confines of the internal auditory canal typically produce a gradual compression of the seventh and eighth cranial nerves. Sensorineural hearing loss, tin- nitus, and facial paresis insidiously evolve usually over months to years. Vertigo is uncom- mon with such lesions because the nervous system is able to adapt to the gradual loss of vestibular function. Lesions within the cerebel- lopontine (CP) angle produce a similar, slowly progressive compression of the seventh and eighth cranial nerves, although if the tumor arises in the angle, it can grow to a much larger size before critical compression occurs. Most often, tumors begin in the internal auditory canal and grow outward into the CP angle, inasmuch as it is the path of least resistance. Next to the seventh and eighth nerves, the fifth nerve is most commonly involved with CP-angle tumors, causing ipsilateral facial numbness.
In later stages of progression, involvement of the sixth, ninth, and tenth nerves may give rise to diplopia, dysphonia, and dysphagia. Compression of the brain and cerebellum results in ipsilateral gaze dysfunction and dys- metria of the extremities.
In a series of over 2000 tumors of the CP angle, 92% were vestibular schwannomas (acoustic neuromas); 3%, meningiomas; 2.5%, epidermoid cysts; and 1%, facial nerve schwannomas.17
Schwannomas
Tumors arising from the sheaths of the cranial and peripheral nerves have been called neuro- mas, neurilemmomas, and neurofibromas, but convincing evidence that most represent pro- liferation of the sheath-producing schwann cells makes schwannoma a more appropriate term.6 These tumors comprise about 5% of intracranial neoplasms and are by far the most common tumor found in the temporal bone. They arise from the vestibular nerve in more than 90% of cases, and much less frequently from the facial, acoustic, or trigeminal nerves. The general term acoustic neuroma, therefore, is inappropriate on two accounts. Mostly, schwannomas are circumscribed and encapsu- lated, encroaching on and displacing neural structures as they grow, without direct invasion of tissue. Vascularity is variable, but they are usually less vascular than meningiomas. Vestibular schwannomas typically arise at the myelin–glial junction near the porous acousti- cus, producing symptoms by exerting pressure on surrounding neurovascular structures. Infrequently, the tumor arises from the vestib- ular nerve terminals near or in the end organ, in which case end-organ destruction occurs, or it may arise from the nerve after it leaves the canal in the angle, in which case it can be rela- tively large before producing symptoms and signs. Intralabyrinthine schwannomas typically begin in the cochlea and later spread to the vestibular labyrinth.18 Schwannomas usually grow very slowly but occasionally hemorrhage into the tumor; cyst formation or associated edema produces clinical evidence of more rapid growth. Malignant schwannomas are rare; approximately half of them occur with neurofibromatosis type II.
VESTIBULAR SCHWANNOMAS (ACOUSTIC NEUROMA)
The incidence of vestibular schwannomas has been estimated at about 1 per 100,000 person/ years.19,20 Although several studies identified an increasing incidence over the past 25 years, this increase can probably be attributed to easier access to magnetic resonance imaging (MRI) particularly in elderly patients, in whom small intracanalicular tumors predominate.21 To date, there is no conclusive evidence that cel- lular phone use leads to an increased incidence of vestibular schwannomas.22 By far the most common symptom associated with a vestibular schwannoma is a slowly progressive unilateral hearing loss.23 Occasionally, patients will expe- rience fluctuating or sudden hearing loss, apparently from compression of the labyrin- thine vasculature. Often patients will complain of an inability to understand speech when using the telephone even before they are aware of a loss of hearing. Unilateral tinnitus is the next most common symptom. True vertigo occurs in
<20% of patients, although about half will com- plain of some mild impairment of balance.24 When vertigo is reported in acoustic neuroma patients, it can sometimes be difficult to deter- mine whether the tumor is the cause of the symptom because bothersome dizziness symp- toms, including vertigo, are common symptoms in the general population.25 At the bedside a positive head-thrust test can be seen if the tumor is causing sufficient dysfunction. Next to the auditory nerve, the most commonly involved cranial nerves (by compression) are the seventh and fifth, producing facial weakness and numb- ness, respectively. Involvement of the sixth, ninth, tenth, eleventh, and twelfth nerves occurs only in the late stages of disease with massive tumors. Large vestibular schwannomas
may also produce increased intracranial pres- sure from obstruction of cerebrospinal fluid (CSF) outflow, resulting in severe headaches and vomiting.
FACIAL NERVE SCHWANNOMAS
Facial nerve schwannomas typically present with a slowly progressive facial paralysis devel- oping over months to years—although rare cases with sudden onset of paralysis, fluctuat- ing paresis, and facial tic have been reported. Hearing loss from compression of the cochlear nerve in the internal auditory canal is the sec- ond most common presenting symptom. When the tumor is confined to the internal auditory canal, differentiating vestibular from facial nerve schwannomas is impossible prior to surgery.26 Conductive hearing loss can occur when the tumor arises in the middle ear, where it can disrupt the ossicular chain. In these cases, the tumor mass may be visible behind the pos- terior superior tympanic membrane.
BILATERAL VESTIBULAR SCHWANNOMAS AND NEUROFIBROMATOSIS TYPE 2
Neurofibromatosis type 2 (NF2) is an auto- somal dominantly inherited syndrome charac- terized by the development of bilateral vestibular schwannomas; schwannomas of other cranial, spinal, and cutaneous nerves; and men- ingiomas involving cranial and spinal nerves.27 It occurs in about one in 25,000 live births and is nearly 100% penetrant by age 60. About half are due to de novo mutations. Since about 10% to 20% of patients with NF2 present with a unilateral vestibular schwannoma, treatment of these patients should be based on the knowl- edge that they will eventually develop a ves- tibular schwannoma on the opposite side (Table 15–1). The key features that suggest the diag- nosis of NF2 in a patient with a unilateral ves- tibular schwannoma are a family history of NF2 or at least two other neural tumors, particularly meningiomas.28
The NF2 gene is a tumor-suppressor gene located on chromosome 22q.29 It codes for the protein schwannomin/merlin (S/M) that inter- acts with the extracellular matrix to regulate cell cycle processes.30,31 A wide range of muta- tions in the NF2 gene have been described, from small and large deletions to missense and
Table 15–1 Average Risk per Decade of Patients Presenting with Vestibular Schwannoma Having Neurofibromatosis Type 2
Age (years) | NF2 (%) | NF2 UVS at Diagnosis (%) | NF2 with no FH and no Other NF2 Features (%) |
10–19 | 33 (22–50) | 6 (2.6–9) | 1 (0.3–1.4) |
20–29 | 15 (8–22) | 2.7 (0.9–4) | 0.45 (0.1–0.6) |
30–39 | 5 (3–8) | 0.9 (0.3–1.4) | 0.15 (0.03–2.2) |
40–49 | 2 (1–3) | 0.36 (0.1–0.54) | 0.06 (0.01–0.08) |
50–59 | 1 (0.5–1.5) | 0.18 (0.06–0.27) | 0.03 (0.007–0.05) |
FH, family history; NF2, neurofibromatosis type 2; UVS, unilateral vestibular schwannoma.
(From Evans DGR, et al. Probability of bilateral disease in people presenting with unilateral vestibular schwannoma.
J Neurol Neurosurg Psychiatry. 1999;66:764–767, with permission.)
nonsense mutations. Only about half of the patients who meet the diagnostic criteria for NF2 have identifiable mutations in the NF2 gene in peripheral blood, probably because of somatic mosaicism with the NF2 mutation being present in only a proportion of somatic cells. A mutation that is present in an insuffi- cient proportion of cells to detect in lympho- cyte DNA may still be found as an identical mutation in all tumors from that patient. Sporadic schwannomas lack normal S/M protein either due to a spontaneous mutation in the NF2 gene or activation of a protease cascade that leads to ineffective S/M.31
Of patients with a vestibular schwannoma, about 5% have NF2 and of patients with NF2, about 15% are likely to present initially with a unilateral tumor.28 Therefore, <1% of patients who present with a unilateral vestibular schwan- noma will go on to develop a tumor on the other side. The likelihood of developing the second bilateral tumor in a patient presenting with a unilateral vestibular schwannoma decreases with age so that a patient presenting with a uni- lateral vestibular schwannoma between the ages of 10 and 20 without a family history or other features of NF2 has about a 1% chance of developing bilateral tumors. In contrast, a patient presenting with a unilateral vestibular schwannoma after the age of 30 without a fam- ily history or other features of NF2 has less than a 0.1% chance of developing bilateral tumors.28
Meningiomas
Meningiomas comprise about 14% of intracra- nial tumors and after schwannomas are the most common primary tumor of the CP
angle.32,33 Meningiomas arise from arachnoid fibroblasts, usually in the posterior aspect of the petrous pyramid near the sigmoid and pet- rosal sinuses. They displace cranial nerves and compress the brain stem and cerebellum but do not invade brain tissue. In the posterior fossa, the lobulated variety is more common than the flat (enplaque) type. Meningiomas in the CP angle are frequently calcified and induce osteoblastic reaction in adjacent bone. Because these tumors usually arise outside of the internal auditory canal, they often become very large before producing symptoms and signs. As with schwannomas, the most common symptoms are auditory—hearing loss and tin- nitus. Large tumors compress the brain stem and cerebellum and stretch the fifth and sev- enth cranial nerves, producing facial numbness and weakness. In the rare cases that originate in the internal auditory canal, symptoms and signs are identical to schwannomas.34
Epidermoid Cysts (Primary Cholesteatomas)
Epidermoid cysts arise from congenital epithe- lial inclusion rests in the area of the petrous apex.35,36 They slowly enlarge to fill the CP angle, stretching nearby cranial nerves and eventually compressing the brain stem and cerebellum. Because these cysts are slow grow- ing, the symptoms do not become manifest until the second to fourth decade of life. As with other CP-angle tumors, involvement of the eighth nerve is a common early feature, but unlike other tumors in this area, hemifacial spasm is a frequent early distinguishing feature.
Cholesterol granulomas arise in the pneuma- tized spaces of the temporal bone when a small hemorrhage into the air cells causes a foreign body reaction and progressive granuloma for- mation.36,37 The lesion within the temporal bone expands and can produce compression of the structures in the CP angle. As with other CP-angle lesions, eighth nerve involvement is most common, with hearing loss being the most frequent presenting symptom. This is an impor- tant lesion to recognize because the surgical management is quite different from that of other CP-angle tumors (see later discussion).
Metastatic Tumors
The internal auditory canal is a frequent site of metastatic tumor growth.5 From this site, tumor cells destroy the seventh and eighth nerves and extend into the inner ear or into the CP angle. The rapid onset of hearing loss and vertigo, fol- lowed by other signs of cranial nerve compres- sion and brainstem dysfunction, suggests the likelihood of a malignant tumor rather than the more common benign CP-angle tumors. The lung and breast are the most common sites for the primary neoplasm.6
Diagnosis
By far the most common presentation of a CP-angle tumor is a slowly progressive unilat- eral sensorineural hearing loss (Fig. 15–1). Unilateral tinnitus is the next most common symptom, with vertigo being an infrequent symptom. The neurologic examination should focus on the involved and nearby cranial nerves—including the vestibular component of cranial nerve eight (nystagmus and the head thrust test) and the fifth and seventh nerves. If there are findings localizing to the internal auditory canal or CP angle, one would proceed directly to neuroimaging, beginning with an MRI. The sudden onset of vertigo and unilat- eral hearing loss can occasionally be seen with benign tumors, but a malignant tumor should also be considered whenever a mass is identi- fied and the symptoms are abrupt in onset or rapidly progressive.5,38
In those cases in which the neurologic exam- ination is nonlocalizing (other than audioves- tibular deficits), the first diagnostic test should be an audiogram. If a progressing unilateral sensorineural hearing loss is documented, then one would proceed directly to an MRI with contrast because there is an important likeli- hood of an expanding lesion. If the audiogram is normal or if there is a unilateral sensorineu- ral hearing loss of unknown duration, then one can consider a brainstem auditory-evoked response (BAER). For some time, BAER was a widely used test in the workup for acoustic neuroma. Early small studies in patients with known vestibular schwannomas reported BAER to be abnormal in 85% to 95% of the patients (see Fig. 8–8 in Chapter 8).39,40 However, currently the role and the value of BAER is uncertain because more recent stud- ies (including systematic reviews of studies) show that when BAER results are compared with MRI, the sensitivity and specificity of BAER ranges widely with values, indicating it can be very accurate to values indicating a high risk for false-positive and false-negative results.42 BAER is most sensitive when tumors are 1 cm, though false-positive rates remain a concern.42 If the BAER is used and the results are normal, then the patient should be followed with at least one repeat audiogram in 6 months. Whether further repeat studies are necessary, should the second audiogram be normal, depends on whether the patient notes a subjec- tive progression of either the hearing loss or tinnitus. If the BAER is abnormal, then one would proceed directly to an MRI with con- trast with emphasis on the internal auditory canal and CP angles.
Magnetic resonance imaging with contrast is the imaging procedure of choice for identifying a vestibular schwannoma (Fig. 15–2).42 It can identify small tumors a few millimeters in diameter that would be missed without con- trast.41,42 MRI can also result in false-positive and false-negative results as well.42 The dilemma is often deciding which patients with audio-vestibular symptoms or signs should have an MRI to evaluate for a tumor. Studies on symptoms with acoustic neuroma demon- strate the wide variability in symptoms and that these tumors are even incidental findings on an imaging study done for other purposes in a patient with no audiovestibular symptoms.42 One study used the prevalence of symptoms in
Unilateral hearing loss or tinnitus
Neurologic examination
Focal findings
Normal
Repeat in 6 months
Normal
Audiogram
Unilateral sensorineural, unknown duration | |
Unilateral sensorineural, progressing
Unilateral sensorineural, unknown duration | |
Unilateral sensorineural, progressing
MRI
Normal
Abnormal
Normal
Abnormal
Typical schwannoma
Typical schwannoma
Atypical tumor
Meningioma, epidermoid cyst, cholesterol granuloma
Meningioma, epidermoid cyst, cholesterol granuloma
CT
Figure 15–1. Algorithm for diagnosis of tumors of the cerebellopontine angle. CT, computerized tomography; MRI, magnetic resonance imaging.
the general population and the prevalence of vestibular schwannomas (or any CP angle mass) to estimate that approximately 2500 MRI stud- ies would need to be performed to detect one vestibular schwannoma in patients with non- specific dizziness.43 Among patients with dizzi- ness and subjectively normal hearing, 9307 MRI scans would be need to be performed to identify one vestibular schwannoma. Using more stringent criteria of patients with dizzi- ness and asymmetric hearing loss (defined as asymmetry of 15dB at two or more pure-tone frequencies), it would take 638 MRI scans to diagnose one vestibular schwannoma.
Computed tomography scanning is most useful for identifying bony erosion and/or
calcification within tumors. Meningiomas are usually more dense than vestibular schwanno- mas on CT. With contrast infusion they appear very dense and homogeneous, a feature that differentiates them from schwannomas.32 There may be calcification within the tumor, and the nearby temporal bone may be thick- ened. Epidermoid cysts also have a characteris- tic profile on CT; they are less dense than brain and do not enhance after intravenous contrast material. They have an irregular scalloped surface contour and are usually eccentric to the opening of the internal auditory canal. Cholesterol granulomas appear as a punched- out lesion in the temporal bone with a central density the same as that of brain and with a rim
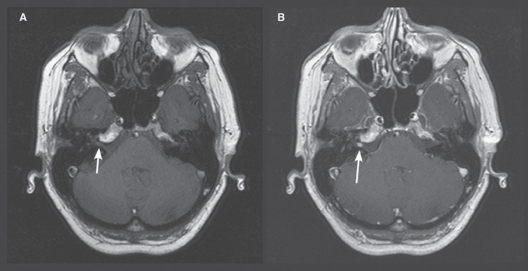
Figure 15–2. Vestibular schwannoma (acoustic neuroma). Pre-contrast (A) and postcontrast (B) magnetic resonance images show that no lesion is seen on the precontrast image (A), but the small schwannoma is clearly seen on the postcon- trast image.
of enhancement after contrast infusion.36 The lesions are smooth walled and the contralateral petrous bone is always well pneumatized. On MRI they give a high-intensity signal in both T1-and T2-weighted images.36,44
Management
Vestibular schwannomas are generally very slow-growing tumors. Over time, there is the chance that if left untreated the hearing loss would become severe, which is important because substantial unilateral hearing loss is associated with disability, including difficulty hearing on the affected side, in noisy environ- ments, sounds at a distance, and difficulty judg- ing the direction of sound.45,46 But there is also a very reasonable chance that the tumor will not grow or may even regress over time. In fact, systematic reviews have found that about 50% of tumors do not demonstrate growth and about 6% show regression.42 There is the pos- sibility that surgical treatment could protect one against future hearing loss and also reduce the chance for other deficits from the tumor, but the surgery itself could lead to hearing loss, facial paralysis, or the many other complica- tions that can arise from surgery in general. No one would want to subject a patient whose tumor will not grow to the surgical risk. But we
also should not wait too long to intervene in a growing tumor since the chance of surgical complications increases with increasing size of the tumor. There are no large randomized con- trolled trials of surgical versus conservative management of vestibular schwannoma, and there are also no validated predictive models to assess the likelihood of future problems from an untreated vestibular schwannoma in an individual patient.
There is a recent trend toward more conser- vative management of patients with vestibular schwannomas since clinical outcomes appear no different in patients who fail conservative management versus those who receive primary treatment without conservative manage- ment.47–49 Patients with an intracanalicular or small (2 cm) CP angle vestibular schwannoma can be followed with serial MRIs without treat- ment. This approach is particularly recom- mended for older patients, asymptomatic patients with “incidental” tumors,50 and patients with underlying medical problems at increased risk for surgery. Large tumors (generally con- sidered >2 cm) are more likely to grow than small tumors, but there are no reliable clinical indicators to predict growth. Surgical or radiation treatment may lead to total unilateral hearing loss, whereas patients treated conservatively often show no change in hearing even after years of follow-up.47 If the tumor
demonstrates initial growth (>2 mm/year), then there is increased risk that it will continue to grow; however, about 40% of tumors with ini- tial growth will not demonstrate future growth. In patients managed conservatively, an initial follow-up MRI is recommended at 6 months, then annually for 2 years, then 2 years later and then every 5 years.49
There are three general surgical approaches to the CP angle: (1) translabyrinthine, (2) sub- occipital, and (3) middle fossa.23,51 The trans- labyrinthine approach destroys the labyrinth but often allows complete removal of the tumor without endangering other nearby neural struc- tures, particularly the facial nerve. This would be the procedure of choice for a patient with severe hearing loss and a tumor under 3 cm in size. With the suboccipital and middle fossa approaches, residual hearing can be saved, as the labyrinth is not destroyed during the surgi- cal procedure. With the introduction of the modern operating microscope, preservation of hearing is now a distinct possibility with either of these procedures.52 Traditionally, the suboc- cipital approach has been performed by neuro- surgeons, whereas the middle fossa approach was developed by otologic surgeons. The mid- dle fossa approach is more likely to save hear- ing and avoid damage to the facial nerve when the tumor is <3 cm in size. For tumors >3 cm in diameter, a combined translabyrinthine sub- occipital approach is commonly used. This procedure allows the surgeon to reduce the size of the tumor from behind by working between the tumor capsule and the brain stem. Furthermore, large tumors are often adjacent to or attached to the basilar artery; with the combined approach the surgeon can dissect the artery from the tumor capsule under direct vision. As many as 10% of patients with vestib- ular schwannomas will have tumor recurrence after surgery mainly due to regrowth of microf- ragments left in the operative field along the course of the facial nerve or at the surface of the pons.53
Stereotaxic radiosurgery using ionizing radi- ation provides another alternative for manag- ing vestibular schwannomas, particularly smaller tumors confined to the internal audi- tory canal.54,55 Radiosurgery provides a better chance of preserving hearing, is more cost effective, and has less impact on patient’s activ- ities of daily living than traditional surgery.56 Although radiosurgery reliably stops the growth
of most tumors, long-term follow-up is still lacking. Stereotaxic radiosurgery is ideal for managing vestibular schwannomas associated with NF2, since eventually the tumors will be bilateral. Subach et al.57 were able to preserve hearing in about half of the patients with vestibular schwannomas and NF2 treated with radiosurgery. In addition, normal facial nerve function was preserved in most patients.
Cholesterol granulomas involving the petrous apex can often be managed without surgery, particularly when the symptoms are stable or improving.58 When symptoms are progressive, however, a transmastoid extra- dural approach with simple drainage into the mastoid sinus or middle ear can be accom- plished with low morbidity. Cholesterol granu- lomas do not need to be resected. Solid tumors involving the petrous apex, however, do require surgery for removal, usually via a middle fossa or infratemporal fossa approach.59
Brain Stem
Gliomas of the brain stem usually grow slowly and infiltrate the brainstem nuclei and fiber tracts, producing multiple symptoms and signs. Although brainstem gliomas are five to ten times more common in children than in adults, they still make up approximately 1% of adult intracranial tumors.60,61 The neurologic symp- toms and signs of childhood brainstem gliomas do not differ in essence from those of adults. The typical history is that of relentless progres- sive involvement of one brainstem center after another, often ending with destruction of the vital cardiorespiratory centers of the medulla. Vestibular and cochlear symptoms and signs are common (occurring in approximately 50% of cases), the brainstem origin of which is usu- ally obvious because of the multiple associated findings. Tumors originating in the pons or midbrain usually cause long tract signs, cranial nerve deficits, and ataxia. Spontaneous, gaze-evoked, and central paroxysmal positional nystagmus all occur, and impairment of sac- cade and pursuit eye movements further sug- gests an intrinsic brainstem disorder. Although less common, gliomas originating in the
medulla may present with recurrent vertigo and vomiting.
Cerebral cavernous malformations (CCM; cavernomas) are common vascular malforma- tions of the brain that occur as a sporadic or as a familial autosomal dominant disorder. So far, mutations in three genes have been identified in families with cavernomas: CCA1/KRIT1, CCA2/MGC4607, and CCA3/PDCD10.62 A
small percentage will bleed and can produce dramatic symptoms and signs, particularly when the cavernoma is located in the brain stem. Bleeding into a cavernoma near the ves- tibular nuclei or outflow tracts to the oculomo- tor nuclei can produce episodes of vertigo and nystagmus. Patients are often thought to have either an infarct or primary hemorrhage prior to identifying the characteristic vascular mal- formation on MRI.63 Patients can have primary position upbeat nystagmus, downbeat nystag- mus, and central types of positional nystagmus associated with cavernomas of the brain stem. Brainstem metastasis also commonly leads to a dramatic clinical syndrome that may mimic a stroke or a primary hemorrhage. Hemorrhage into the metastatic tumor may account for the sudden symptoms and signs. As with other metastatic brain tumors, the most common pri- mary sites are lung and breast.
Fourth Ventricle
Many tumors arising in the fourth ventricular region compress the vestibular nuclei and pro- duce vestibular symptoms. Medulloblastomas, occurring primarily in children and adoles- cents, are rapidly growing, highly cellular tumors that arise in the posterior midline or vermis of the cerebellum and invade the fourth ventricle and adjacent cerebellar hemi- spheres.64,65 Vertigo and disequilibrium are common initial complaints. Headaches and vomiting also occur early from an obstructive hydrocephalus and associated increased intrac- ranial pressure. An attack of headache, vertigo, vomiting, and visual loss may result from a change in head position, producing transient CSF obstruction (Bruns’ symptom). In Nylen’s classic study66 of patients with subtentorial tumors, 17 of 27 patients with medulloblastoma demonstrated positional nystagmus, and in two cases it was the only focal neurologic sign.
Grand67 reported two cases of medulloblastoma in which paroxysmal positional nystagmus was the initial abnormal neurologic sign. Other fourth ventricular tumors that produce similar clinical pictures include ependymomas, papil- lomas, teratomas, epidermoid cysts, and, in endemic areas, cysticercosis.
Cerebellum
The most common cerebellar tumor in adults is metastasis, whereas in children the most common tumor is an astrocytoma. A metastatic cerebellar tumor can be the initial sign of a distant tumor, or it can occur many years after treatment of a primary cancer.68 The location of the tumor in the cerebellum is the key to determining the nature and severity of symptoms and signs. A tumor in the cerebel- lar hemisphere can grow large with relatively few symptoms, whereas a tumor near the midline will usually lead to early symptoms and signs due to either brainstem compression or CSF obstruction. In children, gliomas can infiltrate a large part of the cerebellum, being relatively silent until they become large enough to obstruct CSF circulation or compress the brain stem.69 The characteristic symptoms include new-onset severe headache, position-provoked vertigo and vomiting, and gait imbalance. Most patients will develop papilledema from increased intracranial pres- sure. As with medulloblastoma, occasionally positional vertigo can be the initial symptom of a cerebellar tumor.70 Other tumors of the cer- ebellum that can produce identical symptoms and signs include teratomas, hemangiomas, and hemangioblastomas.
Diagnosis and Management
Magnetic resonance imaging is the diagnostic procedure of choice for identifying brainstem and cerebellar tumors71 (Fig. 15–3). Gliomas of the posterior fossa are particularly difficult to identify with non-contrast CT because they are often isodense; the only evidence for a lesion is enlargement of the brain stem or compression of the fourth ventricle (the tumor shown in Fig. 15–3 was not seen on CT scanning). Magnetic resonance imaging, however, can reliably identify
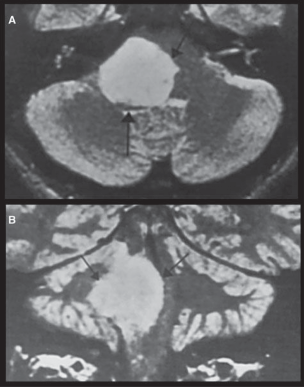
Figure 15–3. Magnetic resonance scans (T2 weighted) showing a brain stem glioma (arrows) involving the root entry zone of the right eighth nerve. (A) Transverse section. (B) Coronal section.
both brainstem and cerebellar gliomas as well as the other tumor types mentioned earlier. In some cases, CT scanning can complement MRI by identifying calcification and helping differentiate between tumor and associated edema. If an MRI scan is not possible, then a CT scan with contrast is also an accurate test for identifying a tumor in the posterior fossa.
When possible, biopsy and surgical resection of the tumor is the treatment of choice. Microsurgical resection of superficial caverno- mas may be possible.72 For metastatic tumors, the primary tumor can be biopsied if it can be found. Occasionally, a solitary metastatic lesion can be resected. For nonresectable tumors, radiation therapy may be an option. Prolonged survival (>5 years) is not uncommon with low- grade astrocytomas. Medulloblastomas are also very sensitive to radiation therapy.73
major concern for radiation therapy of cerebellar and brainstem tumors is delayed neurological damage from the radiation. This can be a particularly difficult problem in a
young patient who presents with minimal defi- cits at the time of diagnosis. Radiation to the brain stem or cerebellum can produce late radiation damage that is slowly progressive and unresponsive to treatment.71,73
REFERENCES
Devaney KO, Boschman CR, Willard SC, Ferlito A, Rinaldo A. Tumours of the external ear and temporal bone. Lancet Oncol. 2005;6(6):411.
Lobo D, Llorente JL, Suárez C. Squamous cell car- cinoma of the external auditory canal. Skull Base. 2008;18(3):167.
Madsen AR, Gundgaard MG, Hoff CM, et al. Cancer of the external auditory canal and middle ear in Denmark from 1992 to 2001. Head Neck. 2008;30(10): 1332.
Sbeity S, Abella A, Arcand P, Quintal MC, Saliba I. Temporal bone rhabdomyosarcoma in children. Int J Pediatr Otorhinolaryngol. 2007;71(5):807.
Cureoglu S, Tulunay O, Ferlito A, Schachern PA, Paparella MM, Rinaldo A. Otologic manifestations of metastatic tumors to the temporal bone. Acta Otolaryngol. 2004;124(10):1117.
Schuknecht HF. Pathology of the Ear. 2nd ed. Philadelphia: Lea & Febiger; 1993.
Heys SD, Brittenden J, Atkinson P, Eremin O. Glomus tumour. An analysis of 43 patients and review of the literature. Br J Surg. 1992;79:345.
Gombos Z, Zhang PJ. Glomus tumor. Arch Pathol Lab Med. 2008;132(9):1448.
McCaffrey TV, Meyer FB, Michels VV, Piepgras DG, Marion MS. Familial paragangliomas of the head and neck. Arch Otolaryngol Head Neck Surg. 1994;120(11):1211.
Vogl T, Bruning R, Scedel H, et al. Paragangliomas of the jugular bulb and carotid body. MR imaging with short sequences and Gd-DTPA enhancement. AJR Am J Roentgenol. 1989;153:583.
Stein AM, Lewin JS, Maniglia AJ. Value of magnetic resonance angiography in the evaluation of head and neck neoplasms. Otolaryngol Head Neck Surg. 1996;114:125.
van den Berg R. Imaging and management of head and neck paragangliomas. Eur Radiol. 2005;15(7): 1310.
Chang CH, Shu MT, Lee JC, Leu YS, Chen YC, Lee KS. Treatments and outcomes of malignant tumors of external auditory canal. Am J Otolaryngol. 2009;30(1):44.
Anand VK, Leonetti JP, al-Mefty O. Neurovascular considerations in surgery of glomus tumors with intracranial extensions. Laryngoscope. 1993;103:722.
van der May AG, Frijns JH, Cornelisse CJ, et al. Does intervention improve the natural course of glomus tumors? A series of 108 patients seen in a 32-year period. Ann Otol Laryngol. 1992;101:635.
Krych AJ, Foote RL, Brown PD, Garces YI, Link MJ. Long-term results of irradiation for paraganglioma. Int J Radiat Oncol Biol Phys. 2006;65(4):1063.
Brackman DE, Bartels LJ. Rare tumors of the cer- ebellopontine angle. Otolaryngol Head Neck Surg. 1980;88:555.
Tieleman A, Casselman JW, Somers T, et al. Imaging of intralabyrinthine schwannomas: a retrospective study of 52 cases with emphasis on lesion growth. AJNR Am J Neuroradiol. 2008;29(5):898.
Urben SL, Benninger MS, Gibbens ND. Asymmetric sensorineural hearing loss in a community-based pop- ulation. Otolaryngol Head Neck Surg. 1999;120:809.
Propp JM, McCarthy BJ, Davis FG, Preston-Martin S. Descriptive epidemiology of vestibular schwannomas. Neuro Oncol. 2006;8(1):1.
Stangerup SE, Tos M, Caye-Thomasen P, Tos T, Klokker M, Thomsen J. Increasing annual incidence of vestibular schwannoma and age at diagnosis. J Laryngol Otol. 2004;118(8):622.
Ahlbom A, Feychting M, Green A, et al. Epidemiologic evidence on mobile phones and tumor risk: a review. Epidemiology. ePub ahead of print, July 10, 2009.
Mattox DE. Vestibular schwannomas. Otolaryngol Clin North Am. 1987;20:149.
Selesnick SH, Jackler RK, Pitts LW. The changing clinical presentation of acoustic tumors in the MRI era. Laryngoscope. 1993;103:431.
Neuhauser HK, von Brevern M, Radtke, et al. Epidemiology of vestibular vertigo: a neurotologic survey of the general population. Neurology. 2005;65:898.
Park HY, Kim SH, Son EJ, Lee HK, Lee WS. Intracanalicular facial nerve schwannoma. Otol Neurotol. 2007;28(3):376.
Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet. 2009;373(9679): 1974.
Evans DGR, Lye R, Neary W, et al. Probability of bilat- eral disease in people presenting with a unilateral ves- tibular schwannoma. J Neurol Neurosurg Psychiatry. 1999;66:764.
Bourn D, Carter SA, Mason S, et al. Germline muta- tion in the neurofibromatosis type 2 tumour suppres- sor gene. Hum Mol Genet. 1994;5:813.
Welling DB, Packer MD, Chang LS. Molecular studies of vestibular schwannomas: a review. Curr Opin Otolaryngol Head Neck Surg. 2007;15(5): 341.
Roche PH, Bouvier C, Chinot O, Figarella-Branger D. Genesis and biology of vestibular schwannomas. Prog Neurol Surg. 2008;21:24.
Granick MS, Martuza RL, Parker SW, et al. Cerebellopontine angle meningiomas: clinical mani- festations and diagnosis. Ann Otol Rhinol Laryngol. 1985;94:34.
Roser F, Nakamura M, Dormiani M, Matthies C, Vorkapic P, Samii M. Meningiomas of the cerebello- pontine angle with extension into the internal auditory canal. J Neurosurg. 2005;102(1):17.
Nakamura M, Roser F, Mirzai S, Matthies C, Vorkapic P, Samii M. Meningiomas of the internal auditory canal. Neurosurgery. 2004;55(1):119.
Sabin HI, Bordi LT, Symon L. Epidermoid cysts and cholesterol granulomas centered on the posterior fossa: twenty years of diagnosis and management. Neurosurgery. 1987;21(6):798.
Mafee MF, Kumar A, Heffner DK. Epidermoid cyst (cholesteatoma) and cholesterol granuloma of the temporal bone and epidermoid cysts affecting the brain. Neuroimaging Clin North Am. 1994;4: 561.
Eisenberg MB, Haddad G, Al-Mefty O. Petrous apex cholesterol granulomas: evolution and management. J Neurosurg. 1997;86:822.
Chaimoff M, Nageris BI, Sulkes J, Spitzer T, Kalmanowitz M. Sudden hearing loss as a present- ing symptom of acoustic neuroma. Am J Otolaryngol. 1999;20:157.
Hirsch A, Anderson H. Audiologic test results in 96 patients with tumors affecting the eighth nerve: a clinical study with emphasis on the early audiologi- cal diagnosis. Acta Otolaryngol Suppl (Stockh). 1980; 369:1.
Musiek FE, Josey AF, Glasscock ME. Auditory brain stem response in patients with acoustic neuromas. Arch Otolaryngol. 1986;112:186.
Schmalbrock P, Chakeres DW, Monroe JW, et al. Assessment of internal auditory canal tumors: a com- parison of contrast-enhanced T1-weighted and steady- state T2-weighted gradient-echo MR imaging. AJNR Am J Neuroradiol. 1999;20:1207.
Fortnum H, O’Neill C, Taylor R, et al. The role of magnetic resonance imaging in the identification of suspected acoustic neuroma: a systematic review of clinical and cost effectiveness and natural history. Health Technol Assess. 2009;13(18):iii, ix, 1.
Gizzi M, Riley E, Molinari S. The diagnostic value of imaging in patients with dizziness: a Bayesian approach. Arch Neurol. 1996;53:1299.
Bonneville F, Savatovsky J, Chiras J. Imaging of cerebellopontine angle lesions: an update. Part 2: intra-axial lesions, skull base lesions that may invade the CPA region, and non-enhancing extra-axial lesions. Eur Radiol. 2007;17(11):2908.
McLeod B, Upfold L, Taylor A. Self reported hearing difficulties following excision of vestibular schwanno- mas. Int J Audiol. 2008;47:420.
Brooker J, Burney S, Fletcher J, Dally M. A qualitative exploration of quality of life among individuals diag- nosed with an acoustic neuroma. Br J Health Psychol. 2009;14:563.
Smouha EE, Yoo M, Mohr K, Davis RP. Conservative management of acoustic neuroma: a meta-analysis and proposed treatment algorithm. Laryngoscope. 2005;115(3):450.
Hajioff D, Raut VV, Walsh RM, et al. Conservative management of vestibular schwannomas: third review of a 10-year prospective study. Clin Otolaryngol. 2008;33(3):255.
Martin TP, Senthil L, Chavda SV, Walsh R, Irving RM. A protocol for the conservative management of vestibular schwannomas. Otol Neurotol. 2009;30(3): 381.
Jeyakumar A, Seth R, Brickman TM, Dutcher P. The prevalence and clinical course of patients with ‘incidental’ acoustic neuromas. Acta Otolaryngol. 2007;127(10):1051.
Kim HN, Jenkins HA. Vestibular schwannomas and other cerebellopontine tumors. In: Baloh RW, Halmagyi CM, eds. Disorders of the Vestibular System. New York: Oxford University Press; 1996.
Snyder WE, Pritz MB, Smith RR. Suboccipital resection of a medial acoustic neuroma with hearing preservation. Surg Neurol. 1999;51:548.
Roche PH, Ribeiro T, Khalil M, Soumare O, Thomassin JM, Pellet W. Recurrence of vestibular schwannomas after surgery. Prog Neurol Surg. 2008; 21:89.
Lin VY, Stewart C, Grebenyuk J, et al. Unilateral acoustic neuromas: long-term hearing results in patients managed with fractionated stereotactic radio- therapy, hearing preservation surgery, and expectantly. Laryngoscope. 2005;115(2):292.
Iwai Y, Yamanaka K, Kubo T, Aiba T. Gamma knife radiosurgery for intracanalicular acoustic neuromas. J Clin Neurosci. 2008;15(9):993.
Pollock BE. Vestibular schwannoma management: an evidence-based comparison of stereotactic radiosur- gery and microsurgical resection. Prog Neurol Surg. 2008;21:222.
Subach BR, Knodziolka D, Lunsford LD, et al. Stereotactic radiosurgery in the management of acous- tic neuromas associated with neurofibromatosis type 2. J Neurosurg. 1999;90:815.
Brodkey JA, Robertson JH, Shea JJ, III, Gardner G. Cholesterol granulomas of the petrous apex: com- bined neurosurgical and otological management. J Neurosurg. 1996;85:625.
Muckle RP, De la Cruz A, Lo WM. Petrous apex lesions. Am J Otol. 1998;19:219.
Laigle-Donadey F, Doz F, Delattre JY. Brainstem gliomas in children and adults. Curr Opin Oncol. 2008;20(6):662.
Pfister S, Witt O. Pediatric gliomas. Recent Results Cancer Res. 2009;171:67.
Labauge P, Denier C, Bergametti F, Tournier- Lasserve E. Genetics of cavernous angiomas. Lancet Neurol. 2007;6(3):237.
Tomlinson FH, Houser OW, Sheithauer BW, et al. Angiographically occult vascular malformations: a correlative study of features on magnetic resonance imaging and histological examination. Neurosurgery. 1994;34:792.
Pobereskin I, Treip C. Adult medulloblastoma.
J Neurol Neurosurg Psychiatry. 1986;49:39.
Johnston DL, Keene D, Bartels U, et al. Medulloblastoma in children under the age of three years: a retrospective Canadian review. Neurooncol. 2009;94(1):51.
Nylen CO. The oto-neurological diagnoses of tumors of the brain. Acta Otolaryngol Suppl (Stockh). 1939;33:81.
Grand W. Positional nystagmus: an early sign of medulloblastoma. Neurology. 1971;21:1157.
Ammirati M, Samii M, Skaf G, Sephernia A. Solitary brain metastasis 13 years after removal of renal adeno- carcinoma. J Neurooncol. 1993;15:87.
Geissinger JD, Bucy PC. Astrocytomas of the cer- ebellum in children. Long-term study. Arch Neurol. 1971;24:125.
Gregorius FK, Crandall PH, Baloh RW. Positional vertigo in cerebellar astrocytoma: report of two cases. Surg Neurol. 1976;6:283.
Frazier JL, Lee J, Thomale UW, Noggle JC, Cohen KJ, Jallo GI. Treatment of diffuse intrinsic brain- stem gliomas: failed approaches and future strategies. J Neurosurg Pediatr. 2009;3(4):259.
Ferroli P, Sinisi M, Franzini A, Giombini S, Solero CL, Broggi G. Brainstem cavernomas: long-term results of microsurgical resection in 52 patients. Neurosurgery. 2005;56(6):1203.
Packer RJ, Vezina G. Management of and prognosis with medulloblastoma: therapy at a crossroads. Arch Neurol. 2008;65(11):1419.
This page intentionally left blank
![]()
TRAUMA TO THE TEMPORAL BONE
Fracture
Labyrinthine Concussion Posttraumatic Positional Vertigo Diagnosis
Management
PERILYMPH FISTULA
Pathophysiology Diagnosis Management
SEMICIRCULAR CANAL DEHISCENCE SYNDROME
Pathophysiology Diagnosis Management BRAIN TRAUMA
Intracranial Complications Associated with Temporal Bone Fractures
Dizziness Due to Brainstem Trauma Postconcussion Syndrome Whiplash Injuries
Diagnosis Management
Fracture
Fractures of the temporal bone most com- monly result from direct lateral blunt trauma to the skull in the parietal region of the head.1,2 Traffic accidents are by far the most common cause.3 Because the otic capsule surrounding the inner ear is very dense bone, the fracture usually courses around it to involve the major foramina in the skull base, the most common being that of the carotid artery and the jugular bulb. Fractures commonly occur near the root of the external auditory canal and run parallel along the petrous apex, extending anteriorly to the foramen lacerum and the carotid artery. They may also extend into the temporal man- dibular joint region. Ecchymosis in the mastoid region (Battle’s sign) occurs when the fracture line extends into the mastoid.
Although temporal bone fractures have been classified as longitudinal and transverse, mod- ern high-resolution computed tomography (CT) scanning shows that most fractures are actually oblique.4,5 The fracture line typically extends anteriomedially to the skull base, mak- ing its way through the weakest places in the skull base but avoiding the compact bone of the otic capsule surrounding the labyrinth. Transient or persistent facial paralysis occurs in about two-thirds of patients.3 Conductive hear- ing loss due to ossicular chain dislocation occurs in more than half, but sensorineural hearing loss occurs in less than 20%. Overall, the audi- tory system is more vulnerable to temporal bone fracture than the vestibular system.
Labyrinthine Concussion
Auditory and vestibular symptoms (either isolated or in combination) can follow blows to the head that do not result in temporal
353
bone fracture. The absence of associated brain- stem symptoms and signs and the usual rapid improvement in symptoms following injury support a peripheral localization for the lesion. The pathology is presumably microscopic hem- orrhages in the membranous labyrinth.6 Although protected by a bony capsule, the del- icate labyrinthine membranes are susceptible to blunt trauma. Of 57 cases of labyrinthine damage due to blunt head trauma reported by Davey,7 51% resulted from blows to the occipi- tal region; 26%, to the frontal region; and 23%, to other areas.
Sudden deafness following a blow to the head without associated vestibular symptoms is often partially or completely reversible.8 It is probably caused by intense acoustic stimula- tion from pressure waves created by the blow, which are transmitted through the bone to the cochlea just as pressure waves are transmitted from air through the conduction mechanism.9 Supporting this suggestion is the observation that the pathologic changes in the cochlea pro- duced by experimental head blows in animals are similar to those produced by intense air- borne sound stimuli.10 These changes consist of degeneration of hair cells and cochlear neurons in the middle turns of the cochlea. Pure-tone hearing loss is usually most pronounced at 4000 and 8000 Hz.
Posttraumatic Positional Vertigo
Benign positional vertigo is a common sequela after any kind of head injury. The most com- mon cause of head trauma is motor vehicle accidents followed by common falls.11 Otoconia are dislodged from the otolithic membrane and typically enter the posterior semicircular canal, where they move under the influence of gravity and trigger episodes of vertigo. Animal studies have shown that otoconia are easily dislodged from the otolithic membrane with high accel- erations. Karl Wittmaack performed a series of experiments on guinea pigs in Germany in the 1930s, showing that the otolithic membrane could be denuded of otoconia with centrifugal accelerations.12 He also noted that these ani- mals with free-floating otoconia in their laby- rinth developed positional nystagmus, which he attributed to loading of the posterior semi- circular canal cupula. As described in Chapter 10, we now know that the otoconial debris
usually enters the long arm of the posterior semicircular canal, where it moves under the influence of gravity to trigger bouts of vertigo and nystagmus. Benign positional vertigo after head trauma can be delayed since not only does the otoconial debris have to be free floating, but patients must get their head in a critical position so that the debris can enter the long arm of the posterior semicircular canal (back and to the side). Patients may be unconscious or be heavily medicated for multiple injuries, so they may only recognize the positional ver- tigo once they begin to recover and are able to make the critical positional changes that will trigger vertigo. Benign positional vertigo occurs after nearly half of head injuries associated with temporal bone fractures.13 Bilateral involve- ment and multiple recurrences are more com- mon in posttraumatic positional vertigo than from other causes of positional vertigo.11,14
Diagnosis
Signs of a temporal bone fracture can often be identified on physical examination.2 If the fracture line transverses the tympanic annulus, the tympanic membrane is lacerated, produc- ing a step-like deformity in the external auditory canal (see Fig. 6–1D in Chapter 6). Cerebrospinal and hemorrhagic otorrhea are common, and the combination of laceration of the tympanic membrane, ossicular damage, and hemotympanum produces a conductive hearing loss. If the fracture line passes through the vestibule of the inner ear, the membranous labyrinth may be torn and the vestibular and cochlear nerves lacerated, producing complete loss of vestibular and cochlear function. Vertigo, nausea, and vomiting are prominent for several days after the fracture, typical of any acute peripheral vestibular lesion. The facial nerve may also be lacerated and loss of function may be permanent unless surgical repair is insti- tuted. Examination of the ear can reveal hemo- tympanum, but bleeding from the ear occurs infrequently because the tympanic membrane remains intact. Cerebrospinal fluid (CSF) may fill the middle ear and drain through the eusta- chian tube into the nasopharynx. Meningitis is a late complication of all types of temporal bone fractures.
The diagnosis of labyrinthine concussion rests on the finding of acute vertigo and/or
High-resolution CT scanning with bone windows is the radiologic procedure of choice for evaluating trauma to the temporal bone and skull base.2 With CT scanning, one is able to identify fracture lines throughout the base of the skull (Fig. 16–1). Magnetic resonance (MR) scanning can be of some use for identifying soft tissue lesions but is of little use for identifying fractures. Once the patient stabilizes, a systematic evaluation of the audi- tory, vestibular, and facial nerves should be undertaken.
Management
There is no treatment for sensorineural hear- ing loss secondary to temporary bone trauma unless there is evidence for a perilymphatic fis- tula (discussed later). If on the basis of audio- metric testing and tympanometric studies a conductive hearing loss is identified, surgical intervention may lead to restoration of normal hearing. Separation of the incudostapedial joint with or without dislocation of the body of the incus from the articulation with the malleus head is the most common type of ossicular dis- location seen with temporal bone injury.15 The surgeon can usually deal with these problems through a transcanal route under local anesthe- sia using a tympanomeatal flap.
Damage to the vestibular apparatus results in acute symptoms, with gradual improvement as central compensation occurs. Symptomatic treatment of vertigo is helpful initially, and the
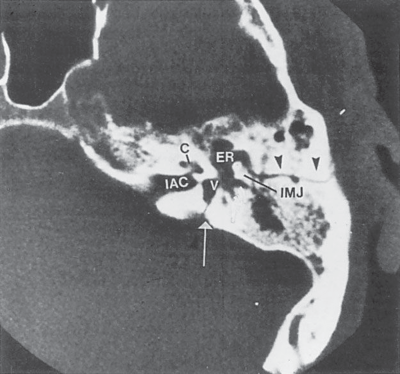
Figure 16–1. Computed tomography scan of the temporal bone showing longitudinal and transverse fractures in the same patient. The longitudinal fracture (arrowheads) crosses the middle ear, disrupting the ossicular chain, and the transverse fracture (white arrow) enters the vestibule, damaging the membranous labyrinth. C, cochlea; ER, epitympanic recess; IAC, internal auditory canal; IMJ, incudomalleal joint; V, vestibule.
As suggested earlier, most fractures do not interrupt the continuity of the facial nerve. In these cases, it is appropriate to observe the facial weakness closely with regular follow-up from the time of the accident. If function does not return within 4 to 6 months, surgical inter- vention is probably indicated.17 As a general rule, regeneration of a damaged but uninter- rupted nerve occurs at a rate of about 1 mm a day. There is marked variability in this rate, however; some patients show evidence of con- tinued improvement for a year or longer after injury. In cases of transverse fracture of the temporal bone with associated severe auditory and vestibular loss, the likelihood of mechani- cal disruption of the facial nerve is great, so the decision regarding surgical intervention is eas- ier. In these cases, translabyrinthine decom- pression and repair of the facial nerve may be achieved because there is already a total loss of auditory and vestibular function. Some cases of severe blunt head injury may result in a tear of the facial nerve at the root entry zone into the brain stem. These patients invariably have a prolonged period of unconsciousness at the time of the accident, and there are nearly always associated symptoms and signs of brain- stem injury. Obviously, there is little prospect for recovery of facial nerve function with this type of lesion.
A perilymph fistula results when there is a break in the bony labyrinth or the oval or round win- dows, allowing perilymph to leak out.18 The most common symptoms associated with a perilymph fistula are vertigo, tinnitus, and hearing loss,
typically associated with a change in pressure such as occurs with coughing, sneezing, or straining.19 The symptoms and signs are remark- ably variable; a perilymph fistula should be con- sidered in the differential diagnosis of sudden hearing loss, recurrent vestibulopathy (particu- larly with associated hearing loss), Meniere’s syndrome, congenital sensorineural hearing loss, posttraumatic hearing loss and vertigo, and sta- pedectomy failure (Fig. 16–2).20
History
Sudden onset hearing loss, tinnitus, vertigo associate with:
Head trauma
Barotrauma
Cough, sneeze, straining, exercise
Cholesteatoma
Post-stapedectomy
Congenital malformation
Examination
Hearing loss – conductive or sensorineural
Positive fistula test
ENG – Spontaneous nystagmus, caloric hypoexcitability
Management
Bed rest, elevate head, antivertiginous medication, avoid straining
Recurrent symptoms
Explore middle ear
Figure 16–2. Algorithm for diagnosis and management of a perilymph fistula. ENG, electronystagmography.
The cause of a fistula is obvious when there is a disruption of the otic capsule or a tear in the bony labyrinth associated with trauma, sur- gery, or infection; spontaneous fistulae are more difficult to explain.19 A sudden negative or positive pressure change in the middle ear from violent nose blowing, sneezing, or barotraumas could lead to rupture of the round window. Flying with a common cold and poor middle ear equalization should be avoided.21 A sudden increase in cerebrospinal fluid (CSF) pressure associated with lifting, straining, coughing, or vigorous activity leads to a change in CSF pressure that is transmitted to the inner ear via the cochlear aqueduct and/or internal auditory canal (see Chapter 2). A direct force through the auditory canal that damages the ossicular chain may rupture the oval window, producing a large perilymph fistula.22,23 Perilymph fistulae may also be associated with developmental abnormalities of the middle or inner ear (e.g., Mondini malformation, defects in the stapes footplate, malformations of the stapedial arch). Recurrent meningitis can result from a congenital perilymph fistula.24
Pathophysiology
How leakage of perilymph leads to fluctuating vestibular and auditory symptoms is unclear. Removal of the round window in animals has little effect on cochlear electrical potentials,25 and the perilymphatic space is routinely entered during stapedectomy surgery, usually without sequelae. The cochleosacculotomy operation for Meniere’s syndrome produces a round-window membrane puncture, yet the incidence of postoperative sensorineural hearing loss is less than 25%.26 Although the sensorineural hearing loss associated with peri- lymph fistulae is usually not reversible, patients have occasionally been reported with dramatic recovery of hearing as long as 10 years after the onset of hearing loss.27 Obviously, such patients could not have had irreversible damage of their sensory epithelium. Presumably, the perilymph fistula leads to aberrant or inefficient transmis- sion of mechanical energy within the cochlea.
Vestibular symptoms are even more difficult to explain on the basis of perilymph fistulae. How would such a leakage lead to stimulation of the vestibular receptors? Pressure changes presumably displace fluid toward the site of the
leak, which may either deflect the cupula of one of the semicircular canals or create a shear force on one of the maculae.18 More persistent dizziness may be the result of permanent damage to the vestibular sensory receptors.
Diagnosis
The classical presentation of an acute peri- lymph fistula is a sudden audible pop in the ear immediately followed by hearing loss, vertigo, and tinnitus, which occur in the setting of trauma or substantial pressure change (e.g., decent on airplane, sneeze, etc.). The key to the diagnosis is to identify the characteristic precipitating factors listed in Figure 16–2.
Nonspecific imbalance and disequilibrium aggravated by quick head movements or sud- den turning may result from chronic perilymph fistulae. Patients report aggravation of these symptoms in certain head positions, preferring to sleep on one side rather than on the other to avoid an ill-defined uncomfortable “dizzy” sen- sation. The latter feature might suggest benign positional vertigo, but such patients do not show the characteristic torsional vertical parox- ysmal positional nystagmus. Occasionally, peri- lymph fistulae are discovered during middle ear exploration for other reasons in a patient with auditory or vestibular symptoms.
Unfortunately, there is no pathognomonic test for a perilymphatic fistula. A positive fistula test (see Chapter 6) is suggestive but not spe- cific. False negatives are common, and false positives occur with Meniere’s syndrome and after stapedectomy. Furthermore, dizziness and imbalance are occasionally reported by normal subjects when the air pressure is changed in the external auditory canal during routine pneuma- toscopy. Auditory examination may identify either a conductive or sensorineural hearing loss, and a unilateral caloric hypoexcitability may appear on electronystagmography (ENG); these findings are not unique to a perilymph fis- tula. Even when the middle ear is explored at the time of surgery, serum or local anesthetic introduced into the middle ear may be mistak- enly identified as leaking perilymph. Mini- endoscopes allow the surgeon to explore the middle ear under more natural conditions, but this technique has not proved to be sensitive or specific for perilymph fistula.28 Fluid identified in the middle ear can be analyzed for substances
found in CSF and perilymph but not in serum (e.g., beta 2 transferrin, prostaglandin D syn- thase).29,30 It is hoped that these newer diagnos- tic studies will enable the surgeon to identify perilymph fistula before surgery. Computed tomography scanning of the temporal bone can identify erosion of the otic capsule by infection or tumor (see Fig. 9–2 in Chapter 9).
Management
The great majority of perilymph fistulae heal spontaneously without intervention. For this reason, most authors advocate conservative management with an initial period of bed rest, sedation, head elevation, and measures to decrease straining. The one exception to this conservative approach might be acute barotrau- mas in which immediate exploration has been advocated,31 but randomized controlled trials are lacking. Persistent auditory and vestibular symptoms after the classical presentation of a perilymph fistula are indications for explora- tion of the middle ear after an initial trial of conservative management. The rate of occur- rence of spontaneous fistulae continues to be a controversial issue. Endoscopic studies indi- cate a much lower incidence than has been generally reported in the literature.32 The peri- lymph leak may become apparent only when the patient is placed in the Trendelenburg position, after performing a Valsalva maneuver, after jugular vein compression, or with manip- ulation of the ossicular chain.
The goal of surgery is to stabilize the hearing loss and relieve vestibular symptoms. The middle ear is typically entered through a posterior tym- panotomy.20 Most often the fistula is in the area of the oval window. Most surgeons will patch the oval window region with perichondrium or fascia and cover the graft with gelatin sponge (Gelfoam). Recurrence of symptoms after repair occurs in at least 10% of cases; rarely, intractable symptoms will necessitate destructive surgery with laby- rinthectomy or nerve section.
SEMICIRCULAR CANAL DEHISCENCE SYNDROME
Normally there is a bony capsule completely surrounding the semicircular canals. Dehiscence of this bony capsule can cause a wide range of vestibular and auditory symptoms. Unlike a
perilymph fistula, where perilymph leaks from the inner ear, dehiscence of the bony capsule sur- rounding the semicircular canals results in inap- propriate activation of the affected canal when pressure waves enter the inner ear. Episodes of vertigo can be triggered by sound (Tullio phe- nomenen) or positive or negative pressure changes in the ear or CSF.33 Eye movement recordings during the episodes of vertigo show bursts of nystagmus in the plane of the affected semicircular canal. The anterior (superior) canal is most frequently involved since normally there is only a thin plate of bone separating the apex of the canal from the middle fossa. In some patients the bone is so thin it cannot be seen on CT (on either side). Presumably this thin bony plate can be fractured by minor trauma. Typically infec- tiom or cholesteotoma are associated with poste- rior or horizontal canal involvement.34,35
Vertigo attacks may occur after loud noise, exertion, straining, coughing, or sneezing. Exposure to continuous loud noise can cause prolonged nonspecific dizziness.33,36 Sudden drop attacks can occur even without vertigo.37 The most common auditory symptoms are auto- phony of voice and sensation of blocking in the ear.36 Bone conduction is increased and air con- duction decreased, leading to the characteristic air/bone gap on audiometry. Vibration any- where in the body can often be heard in the affected ear. Patients may hear their pulse or even their eye movements. This clinical presen- tation can mimic middle ear disease and patients may undergo inappropriate middle ear surgery.
Pathophysiology
The symptoms and signs of semicircular canal dehiscence can be explained by the presence of a pathological “third window.”33,34 The inner ear fluid compartments are normally completely sur- rounded by the bone of the otic capsule. Since the inner ear fluids are incompressible, an inward dis- placement of the stapes at the oval window is accompanied by an outward displacement of the round window. This fluid flow produces a pres- sure gradient along the length of the basilar mem- brane. The presence of a third window allows a portion of the sound energy to be shunted away from the cochlea, activating the affected semicir- cular canal and resulting in loss of hearing sensitiv- ity to air-conducted sound. Any positive or negative pressure change in the external ear canal or CSF can activate the affected canal (Fig. 16–3).38

Endolymphatic sac
Dura mater
Superior canal
Posterior
canal
CSF
Cochlear aqueduct
Endolymphatic duct
Scala vestibuli Perilymph
Horizontal
canal
Endolymph
Utricle
Oval window
Cochlear duct Scala tympani
Saccule
Rounded window
![]()
![]()

C D


Figure 16–3. Mechanism of symptoms and signs with superior semicircular canal dehiscence syndrome. (A) Schematic drawing of inner ear. There are normally two windows in the bony capsule—the oval window (filled by the stapes foot plate) and the round window. A third window in the bony wall of the superior semicircular canal leads to vertigo and nystagmus with loud sounds or pressure changes in the middle ear or CSF (B and C). If the window is large enough, the superior semi- circular canal can be blocked (D). See text for details. (From Baloh RW. Superior semicircular canal dehiscence syndrome: Leaks and squeaks can make you dizzy. Neurology. 2004;62:684 with permission).
Bone-conducted hearing normally results because of higher impedance at the round win- dow compared to the oval window.34 A patho- logical third window on the vestibular (oval window) side of the cochlear partition lowers the impedance on the vestibular side (round window), increasing the cochlear response to bone conduction. Thus, the air/bone gap typi- cally seen with semicircular canal dehiscence is due to a combination of decreased air conduc- tion and increased bone conduction.
Diagnosis
The diagnosis of semicircular canal dehiscence rests on finding the characteristic symptoms and signs (Table 16–1) in a patient with dehis- cence of a semicircular canal on CT of the tem- poral bone. Loud noise or pressure changes in the ear or CSF may trigger brief bursts of nystagmus in the plane of the affected canal. For the most common superior canal syn- drome, positive pressure in the external ear canal or a Valsalva maneuver against pinched nostrils (forcing air into the middle ear through the eustachian tube) causes inward displace- ment of the stapes and excitatory ampullofugal displacement of the cupula of the superior semicircular canal, producing an upbeating torsional vertical nystagmus in the plane of the canal (see Fig. 16–3A). Conversely, negative
Table 16–1 Symptoms and signs of dehiscence of the superior semicircular canal
![]()
![]()
Symptoms Signs
pressure in the external ear canal or a Valsalva maneuver against a closed glottis (taking a deep breath and bearing down) results in an inhibi- tory ampullopetal displacement of the cupula of the superior canal, producing torsional downbeat nystagmus (see Fig. 16–3B).38 Vibration applied to the suboccipital cranium may also trigger nystagmus.39 Nine out of ten patients have an air/bone gap (conductive hear- ing loss) on audiometric testing (Fig. 16–4A,B). Many of these show bone-conduction thresh- olds less than 0 dB at 250 and 500 Hz. Key findings that separate canal dehiscence from middle ear causes of conductive hearing loss are a normal acoustic reflex and a hyperactive VEMP (decreased threshold). These reflexes are usually absent with middle ear causes of conductive hearing loss.36,40,41 High-resolution CT with thin cuts reformatted in the planes of the semicircular canals identifies areas of dehis- cence (Fig. 16–4C,D).33,42,43 False positives are not uncommon, however, so the report of dehiscence alone without clear clinical symp- toms and signs does not make the diagnosis.44
Management
Conservative management is a valid option and one that many patients select, since patients can learn to avoid symptom triggers and the most common symptoms are brief and transient. Several different surgical procedures have been used to treat semicircular canal dehiscence, but randomized controlled trials and studies with long-term follow-up are lacking. For the supe- rior semicircular canal syndrome, initially a middle fossa approach was used to resurface
Sound-induced vertigo—“Tullio phenomenon”
Pressure-induced vertigo—coughing, blowing nose, straining
Hyperacusis to bone- conducted sound— hear pulse, eye movements
Sound-induced nystagmus, eye movement, head tilt
Pressure-induced nystagmus, eye movement, head tilt
Conductive hearing loss
the canal but delayed hearing loss occurred in several patients.33,45 Subsequently plugging or capping the canal produced overall better results.46,47 More recently a transmastoid approach was reported to be as effective as the middle fossa approach without the need for a prolonged hospital admission.48 Vestibular nerve section and occlusion of the round win- dow are other options for refractory cases.49
Chronic disequilibrium Increased vestibular-
evoked myogenic potentials
Oscillopsia Decreased VOR gain in plane of superior canal
The most common mechanism of brain injury with blunt head trauma is movement and
![]()
250
Right ear Left ear
500 1k 2k 4k 8k 250 500 1k 2k 4k 8k

![]()
0
20
40
60
Hearing level in dB
Hearing level in dB
80
100
B 250
500 1k 2k
4k 8k 250 500 1k 2k 4k 8k
0
20
40
60
80
100

Frequency in Hz
Figure 16–4. Audiograms from 2 patients with superior semicircular canal dehiscence syndrome (A) and (B) show a low-frequency air-bone gap on the affected side. High-resolution CT images of the temporal bones (C) and (D) show dehiscence of the superior semicircular canal on the affected side. Arrows point to where there is normally a thin bony roof to the superior canal. (From Brantberg K, Ishiyama A, Baloh RW. Drop attacks secondary to superior canal dehiscence syndrome. Neurology. 2005;64:2126 with permission).
deformation of the brain within the skull. When the rapidly moving head is suddenly stopped, the viscoelastic brain continues to move and may rotate in the skull around the axis of the brain stem. The internal shearing and stress forces traumatize neurons and disrupt axons and blood vessels. The latter may cause multi- focal petechial hemorrhages or even massive intracerebral hemorrhage. The term concus- sion refers to a brief loss of consciousness after head trauma unassociated with focal neuro- logic signs or radiologic evidence of structural
brain damage. Usually the loss of conscious- ness lasts for only a few minutes, although residual symptoms may last for months to years (see later discussion). The mechanism for such a brief loss of consciousness and rapid recovery is unknown. Often the terms concussion and mild traumatic brain injury are used inter- changeably.50
Most patients who suffer concussion proba- bly have some injury to brain cells, but nearly all rapidly regain function within minutes of the injury. They can usually remember the
Intracranial Complications Associated with Temporal Bone Fractures
In most cases of temporal bone fracture, brain injuries overshadow injuries to the labyrinth or eighth nerve. In a series of 43 patients treated for temporal bone fractures at Temple University in Philadelphia over 3 years, 19 patients (44%) required an open neurosurgical procedure.52 Subarachnoid hemorrhage, sub- dural hemorrhage, and cerebral edema were the most common brain findings. Four patients died (all due to neurological causes) and seven required institutional care after discharge. Most patients had a prior history of significant alcohol or drug use.
Dizziness Due to Brainstem Trauma
Brainstem injury from blunt head trauma is not a common cause of isolated auditory and ves- tibular symptoms. Severe head blows may pro- duce hemorrhage or infarction in the brain stem, but these pathologic changes are invari- ably associated with alteration in the level of consciousness and multiple neurologic signs. In their classic study, Mitchell and Adams53 sectioned the brain stem in 100 cases of fatal blunt head injury. Only 18 patients showed no evidence of increased intracranial pressure, and of these, only 7 had abnormalities in the brain stem attributable to the primary impact. In these seven patients, other areas of the brain were damaged, suggesting to the authors that so-called primary brainstem injury does not exist but, rather, is one aspect of diffuse brain damage. As a general rule, isolated episodes of vertigo occurring after brain trauma should not be attributed to brainstem injury.
Caloric examination can be particularly help- ful in evaluating the brainstem status in patients who are comatose from blunt head injuries.
These patients do not produce saccades, so a “normal” response is a conjugate tonic devia- tion of the eyes toward the side of a cold stimu- lus or away from the side of a warm stimulus. Absence of this tonic deviation affirms that the brainstem vestibulo-ocular reflex pathways have been damaged, assuming that the eighth nerve and end organs are intact. Unilateral loss of tonic deviation or nonconjugate deviation indicates focal involvement of the reflex path- ways. The absence of caloric responses after an acute head injury is a poor prognostic sign.54
Postconcussion Syndrome
The postconcussion syndrome has long been the center of medical–legal controversy.55–57 Symptoms include dizziness, headache (usually diffuse), increased irritability, insomnia, forget- fulness, mental obtuseness, and loss of initia- tive—all of which occur after a concussion. Because of the ill-defined nature of these symp- toms, it is difficult to localize the site of lesion and the patient is frequently diagnosed as being psychoneurotic (compensation neurosis). The dizziness associated with the postconcussion syndrome is nearly always nonspecific; patients use terms such as swimming, light-headed, float- ing, rocking, and disoriented to describe the sensations they feel. If vertigo is present, an addi- tional labyrinthine lesion should be suspected.
Rutherford and colleagues58 followed 145 patients with concussion from minor head inju- ries to assess the type and frequency of symp- toms and to evaluate whether the symptoms correlated with the severity of injury, associ- ated neurologic signs, or other circumstances related to the injury. Concussion was defined as a period of amnesia—no matter how brief— caused by a blow to the head. All of the patients were released from the hospital after a brief observation. Table 16–2 lists the symptoms and their frequency of occurrence reported by the
145 patients 6 weeks after the concussion. Approximately one-half were symptom-free, but the other half complained of one or more symptoms. In those patients with multiple symptoms, no consistent pattern was found to support the concept of a postconcussion syndrome. A significant correlation existed between the presence of multiple symptoms at 6 weeks and the occurrence of positive neurologic signs and symptoms within 24 hr of
Table 16–2 Symptoms Reported 6 Weeks after Concussion in 145 Patients58
![]()
Symptom n (%)
Headache | 36 | 24.8 |
Anxiety | 28 | 19.3 |
Insomnia | 22 | 15.2 |
Dizziness | 21 | 14.5 |
Irritability | 13 | 9.0 |
Fatigue | 13 | 9.0 |
Loss of concentration | 12 | 8.3 |
Loss of memory | 12 | 8.3 |
Hearing defect | 10 | 6.9 |
Sensitivity to alcohol | 9 | 6.2 |
Depression | 8 | 5.5 |
Visual defect | 7 | 4.8 |
Anosmia | 4 | 2.8 |
Epilepsy | 3 | 2.1 |
Diplopia | 2 | 1.4 |
Other | 16 | 11.0 |
No symptoms | 71 | 49.0 |
the concussion. Postconcussion symptoms were more frequent in women and in patients who blamed their employers or large impersonal organizations for their accidents. The authors concluded that both organic and psychosomatic factors were involved in the pathogenesis of postconcussion symptoms. Although modern neuropsychiatric and electrophysiologic stud- ies can identify subtle brain abnormalities in most patients with postconcussion syndrome, the type and degree of abnormality do not cor- relate with the severity of symptoms, and psy- chosocial factors continue to be important in long-term prognosis.57,59
Whiplash Injuries
A perplexing problem because of the frequency of occurrence and the medical–legal ramifica- tions is the role of soft-tissue injuries of the neck in producing dizziness and disequilib- rium.60,61 Patients often describe the dizziness in nonspecific terms such as lightheaded, swim- ming, off-balance, floating, and rocking; as with psychophysiologic dizziness, they may describe a sensation of spinning inside the head unas- sociated with an illusion of movement or with spontaneous nystagmus. The dizziness may last for months or years after the injury, although it
usually disappears as the swelling and pain sub- side. The occasional finding of unilateral caloric hypoexcitability is likely due to associated laby- rinthine trauma.
From the known anatomic substrate for neck–vestibular interaction (see Chapter 3), it is unlikely that lesions involving only soft tis- sues of the neck could produce vertigo and dis- equilibrium. The major neck afferent input to the vestibular nuclei arises from the paraverte- bral joints and capsules, with relatively minor input from the paravertebral muscles. The skin and superficial muscles do not appear to provide any input to the vestibular system. In addition, the relative contribution of neck affer- ent input to the vestibular nuclei is small com- pared with the direct labyrinthine and indirect visual signals transmitted via other brainstem nuclei and the cerebellum. Lesions involving the neck afferents in primates are rapidly com- pensated, and therefore prolonged dizziness after neck injuries of any type would be diffi- cult to explain on the basis of damage to the neck afferent input to the vestibular nuclei.
Diagnosis
Persistent dizziness after a blunt head injury often poses a difficult diagnostic dilemma (Table 16–3). The diagnosis of postconcussion syndrome rests on finding the characteristic symptoms in the absence of focal neurologic findings. A careful neurotologic examination should identify most specific syndromes that require individualized treatment. Examination of the ear may reveal evidence of a temporal bone fracture or perilymph fistula, positional testing may reveal benign paroxysmal posi- tional nystagmus, and neurologic examination may identify signs of brainstem damage. Standard audiometric, brainstem auditory- evoked response (BAER), and ENG testing methods are useful to assess the functional sta- tus of the auditory and vestibular systems and thus can be used to document intact physiolog- ical and quantifiable laboratory measures of the audio-vestibular system, though false- positive and false-negative results are not uncommon. Neuroimaging is usually helpful only when there are focal findings on the neu- rologic examination.
Computed tomography scanning is most useful for evaluating the base of the skull and
Table 16–3 Differential Diagnosis of Persistent Dizziness after Head Trauma
![]()
![]()
Diagnosis Dizziness Exam Laboratory
Benign positional vertigo
Labyrinthine concussion
Brief episodes, position induced
Severe initially, gradual improvement
Fatigable positional nystagmus
Peripheral spontaneous nystagmus
Normal
Caloric vestibular paresis, unilateral hearing loss
Perilymph fistula Fluctuating, induced by
coughing, sneezing, straining
Positive fistula test Caloric vestibular paresis,
unilateral hearing loss
Brain stem contusion
Postconcussion syndrome
Severe, associated brain stem symptoms
Continuous, associated headaches, irritability, etc.
Focal neurologic signs MR scan shows focal lesions Normal Normal
![]()
for identifying blood in the subarachnoid or subdural space and within the brain paren- chyma in the acute setting. Magnetic resonance scanning is best for identifying brain contusion and edema.
Management
Management of traumatic brain injuries has become increasingly sophisticated with the development of electronic and chemical moni- toring devices and advances in critical care medicine. So far, however, clinical trials of neuroprotective drugs have been largely unsuc- cessful.62 Acute surgical intervention is usually aimed at relieving intracranial pressure most commonly associated with hemorrhage. The degree of recovery after a closed head injury typically depends on the severity of the injury. As a general rule, the major part of the recov- ery occurs within the first 6 months. Physical rehabilitation should begin as soon as practical after the acute effects of the head injury have subsided. It is important that the patients and their families are counseled regarding the expected rate of recovery and whether perma- nent deficits are expected.
Treatment of the postconcussion syndrome begins by providing the patient with reassurance that there is no evidence of structural damage, that the symptoms are not unusual after head injury, and that they nearly always resolve over time. The patient should be encouraged to return to a normal exercise level gradually, even though initially the dizziness and other symp- toms may be aggravated.63 Tranquilizers such as diazepam and alprazolam can be useful for a
transient period, but prolonged use should be avoided as dependency is common. Endogenous depression is a common sequela after brain injury, and antidepressant medications (e.g., tri- cyclic amines, selective serotonin reuptake inhibitors [SSRIs]) may be helpful in severe cases. Although recovery is the rule, some patients will have persistent symptoms for years after a concussion. Although there is a positive correlation between the severity of head injury and the length of postconcussion symptoms, one cannot reliably judge the prognosis for recovery based on the nature of the head injury.
Soft-tissue injuries of the neck are usually associated with focal muscle tenderness and spasm. Initial management consists of rest and immobilization with a soft collar to allow the muscle contusions to heal. Once serious prob- lems have been ruled out, the patient should be reassured that there is no evidence of neu- rologic damage and that the symptoms nearly always spontaneously disappear. As in patients with postconcussion syndrome, it is important to begin a gradual exercise program as the acute soft-tissue injuries heal. Heat and mas- sage along with judicious use of pharmacologic muscle relaxation provide relief of the muscle spasm, which may come and go for weeks to months. Active range-of-motion exercises per- formed on a regular basis provide the best long-term relief of muscle spasm. Patients should be encouraged to sleep with a single flat pillow and avoid long periods of hyperflexion of the neck. In our experience, the nonspecific dizziness associated with whiplash injuries improves as the local symptoms of muscle spasm and stiffness subside (assuming the medicolegal aspects can be resolved).
REFERENCES
Cannon CR, Jahrsdoerfer RA. Temporal bone fractures. Review of 90 cases. Arch Otolaryngol. 1983;109:285.
Johnson F, Semaan MT, Megerian CA. Temporal bone fracture: evaluation and management in the modern era. Otolaryngol Clin North Am. 2008;41(3):597, x.
Yetiser S, Hidir Y, Gonul E. Facial nerve problems and hearing loss in patients with temporal bone fractures: demographic data. J Trauma. 2008;65(6): 1314.
Yeakley JW. Temporal bone fractures. Curr Prob Diagn Radiol. 1999;28:65.
Saraiya PV, Aygun N. Temporal bone fractures. Emerg Radiol. 2009;16(4):255.
Schuknecht HF. Mechanisms of inner ear injury from blows to the head. Ann Otol. 1969;78:253.
Davey LM. Labyrinthine trauma in head injury. Conn Med. 1965;29:250.
Ulug T, Ulubil SA. Contralateral labyrinthine con- cussion in temporal bone fractures. J Otolaryngol. 2006;35(6):380.
Igarashi M, Schuknecht H, Myers E. Cochlear pathol- ogy in humans with stimulation deafness. J Laryngol. 1964;78:115.
Schuknecht H, Neff W, Perlman H. An experimental study of auditory damage following blows to the head. Ann Otol Rhinol Laryngol. 1951;60:273.
Gordon CR, Levite R, Joffe V, Gadoth N. Is post- traumatic benign paroxysmal positional vertigo different from the idiopathic form? Arch Neurol. 2004;61(10):1590.
Wittmaack K. Kopfstellungsnystagmus (comment).
Acta Otolaryngol (Stockh). 1927;11:156.
Barber H. Positional nystagmus especially after head injury. Laryngoscope. 1964;74:891.
Baloh RW, Honrubia V, Jacobson K. Benign positional vertigo: clinical and oculographic features in 240 cases. Neurology. 1987;37:371.
Lancaster JL, Alderson DJ, Curley JW. Otological complications following basal skull fractures. J R Coll Surg Edinb. 1999;44:87.
Guerrissi JO. Facial nerve paralysis after intratempo- ral and extratemporal blunt trauma. J Craniofac Surg. 1997;8:431.
Gantz BJ, Rubenstein JT, Gidley P, Goodworth GG. Surgical management of Bell’s palsy. Laryngoscope. 1999;109:1177.
Minor LB. Labyrinthine fistulae: pathobiology and management. Curr Opin Otolaryngol Head Neck Surg. 2003;11(5):340.
Goto F, Ogawa K, Kunihiro T, Kurashima K, Kobayashi H, Kanzaki J. Perilymph fistula—45 case analysis. Auris Nasus Larynx. 2001;28(1):29.
Shott SR, Pensak ML. Perilymphatic fistula. Ear Nose Throat J. 1992;71:568.
Klokker M, Vesterhauge S. Perilymphatic fistula in cabin attendants: an incapacitating consequence of flying with common cold. Aviat Space Environ Med. 2005;76(1):66.
Suzuki M, Shigemi H, Mogi G. The leaking laby- rinthine lesion resulting from direct force through the auditory canal: report of five cases. Auris Nasus Larynx. 1999;26:29.
Hatano A, Rikitake M, Komori M, Irie T, Moriyama
Traumatic perilymphatic fistula with the luxation of the stapes into the vestibule. Auris Nasus Larynx. 2009;36(4):474.
Rupa V, Rajshekhar V, Weider DJ. Syndrome of recur- rent meninigitis due to congenital perilymph fistula with two different clinical presentations. Int J Pediatr Otorhinolaryngol. 2000;54(2-3):173.
Weisskopf A, Murphy JT, Merzenich MM. Genesis of the round window rupture syndrome: some experimental observations. Laryngoscope. 1978; 88:389.
Schuknecht HF. Cochleosacculotomy for Meniere’s disease: theory, technique and results. Laryngoscope. 1982;92:853.
Shannon DA, Blum SL. Surgical treatment of long term sensorineural hearing loss due to labyrinthine fistula. J Am Audiol Soc. 1979;5:1.
Selmani Z, Pyykkö I, Ishizaki H, Marttila TI. Role of transtympanic endoscopy of the middle ear in the diagnosis of perilymphatic fistula in patients with sensorineural hearing loss or vertigo. ORL J Otorhinolaryngol Relat Spec. 2002;64(5):301.
Bassiouny M, Hirsch BE, Kelly RH, Kamerer DB, Cass SP. Beta 2 transferrin application in otology. Am J Otol. 1992;13:552.
Michel O, Petereit H, Klemm E, Walther LE, Bachmann-Harildstad G. First clinical experience with beta-trace protein (prostaglandin D synthase) as a marker for perilymphatic fistula. J Laryngol Otol. 2005;119(10):765.
Pullen FW, Rosenberg GH, Cabeza CH. Sudden hearing loss in divers and fliers. Laryngoscope. 1979;84:1373.
Friedland DR, Wackym PA. A critical appraisal of spontaneous perilymphatic fistulas of the inner ear. Am J Otol. 1999;20:261.
Minor LB, Solomon D, Zinreich JS, Zee DS. Sound- and/or pressure-induced vertigo due to bone dehis- cence of the superior semicircular canal. Arch Otolaryngol Head Neck Surg. 1998;124:249.
Merchant SN, Rosowski JJ. Conductive hearing loss caused by third-window lesions of the inner ear. Otol Neurotol. 2008;29(3):282.
Brantberg K, Bagger-Sjöbäck D, Mathiesen T, Witt H, Pansell T. Posterior canal dehiscence syndrome caused by an apex cholesteatoma. Otol Neurotol. 2006;27(4):531.
Zhou G, Gopen Q, Poe DS. Clinical and diagnos- tic characterization of canal dehiscence syndrome: a great otologic mimicker. Am J Otol. 2007;28(7): 920.
Brantberg K, Ishiyama A, Baloh RW. Drop attacks secondary to superior canal dehiscence syndrome. Neurology. 2005;64(12):2126.
Baloh RW. Superior semicircular canal dehiscence syndrome: leaks and squeaks can make you dizzy. Neurology. 2004;62(5):684.
White JA, Hughes GB, Ruggieri PN. Vibration- induced nystagmus as an office procedure for the diag- nosis of superior semicircular canal dehiscence. Am J Otol. 2007;28(7):911.
Welgampola MS, Myrie OA, Minor LB, Carey JP. Vestibular-evoked myogenic potential thresholds normalize on plugging superior canal dehiscence. Neurology. 2008;70(6):464.
Roditi RE, Eppsteiner RW, Sauter TB, Lee DJ. Cervical vestibular evoked myogenic potentials (cVEMPs) in patients with superior canal dehiscence syndrome (SCDS). Otolaryngol Head Neck Surg. 2009;141(1):24.
Crane BT, Minor LB, Carey JP. Three-dimensional computed tomography of superior canal dehiscence syndrome. Am J Otol. 2008;29(5):699.
Krombach GA, DiMartino E, Schmitz-Rode T, et al. Posterior semicircular canal dehiscence: a morpho- logic cause of vertigo similar to superior semicircular canal dehiscence. Eur Radiol. 2003;13(6):1444.
Cloutier JF, Bélair M, Saliba I. Superior semicircular canal dehiscence: positive predictive value of high- resolution CT scanning. Eur Arch Otorhinolaryngol. 2008;265(12):1455.
Peterson EC, Lazar DA, Nemecek AN, Duckert L, Rostomily R. Superior semicircular canal dehiscence syndrome: successful treatment with repair of the mid- dle fossa floor: technical case report. Neurosurgery. 2008;63(6):E1207.
Crane BT, Minor LB, Carey JP. Superior canal dehiscence plugging reduces dizziness handicap. Laryngoscope. 2008;118(10):1809.
Vlastarakos PV, Proikas K, Tavoulari E, Kikidis D, Maragoudakis P, Nikolopoulos TP. Efficacy assess- ment and complications of surgical management for superior semicircular canal dehiscence: a meta- analysis of published interventional studies. Eur Arch Otorhinolaryngol. 2009;266(2):177.
Deschenes GR, Hsu DP, Megerian CA. Outpatient repair of superior semicircular canal dehiscence via the transmastoid approach. Laryngoscope. ePub ahead of print, June 24, 2009.
Silverstein H, Van Ess MJ. Complete round window niche occlusion for superior semicircular canal dehis- cence syndrome: a minimally invasive approach. Ear Nose Throat J. 2009;88(8):1042.
Bigler ED. Neuropsychology and clinical neurosci- ence of persistent post-concussive syndrome. J Int Neuropsychol Soc. 2008;14(1):1.
Yang CC, Hua MS, Tu YK, Huang SJ. Earlyclinicalchar- acteristics of patients with persistent post-concussion symptoms: a prospective study. Brain Inj. 2009;23(4): 299.
Alvi A, Bereliani A. Acute intracranial complications of temporal bone trauma. Otolaryngol Head Neck Surg. 1998;119:609.
Mitchell DE, Adams JH. Primary focal impact damage to the brain stem in blunt head injuries. Does it exist? Lancet. 1973;2:215.
Poulsen J, Zilstrorff K. Prognostic value of the caloric vestibular test in the unconscious patient with cranial trauma. Acta Neurol Scand. 1972;48:282.
Symonds C. Concussion and its sequelae. Lancet. 1962;1:1.
Binder LM. Persisting symptoms after mild head injury: a review of the post-concussive syndrome. J Clin Exp Neuropsychiatry. 1986;8:323.
Mickeviciene D, Schrader H, Obelieniene D, et al. A controlled prospective inception cohort study on the post-concussion syndrome outside the medicolegal context. Eur J Neurol. 2004;11(6):411.
Rutherford WH, Merrett JD, McDonald JR. Sequelae of concussion caused by minor head injuries. Lancet. 1977;1:1.
Fenton GW. The postconcussional syndrome reap- praised. Clin Electroencephalogr. 1996;27:174.
Partheni M, Constantoyannis C, Ferrari R, Nikiforidis G, Voulgaris S, Papadakis N. A prospective cohort study of the outcome of acute whiplash injury in Greece. Clin Exp Rheumatol. 2000;18(1):67.
Rowlands RG, Campbell IK, Kenyon GS. Otological and vestibular symptoms in patients with low grade (Quebec grades one and two) whiplash injury. J Laryngol Otol. 2009;123(2):182.
Xiong Y, Mahmood A, Chopp M. Emerging treat- ments for traumatic brain injury. Expert Opin Emerg Drugs. 2009;14(1):67.
Willer B, Leddy JJ. Management of concussion and post-concussion syndrome. Curr Treat Options Neurol. 2006;8(5):415.
![]()
DIZZINESS AND SYSTEMIC METABOLIC DISORDERS
Diabetes Mellitus Uremia Hypothyroidism
Alcohol and Thiamine Deficiency Management
METABOLIC DISORDERS OF THE TEMPORAL BONE
Otosclerosis Paget’s Disease Other Disorders Diagnosis
Management
OTOTOXINS
Aminoglycosides “Loop” Diuretics
Anti-inflammatory Drugs Platinum Compounds Diagnosis
Management
NEUROTOXINS
Heavy Metals Organic Solvents Diagnosis Management
DIZZINESS AND SYSTEMIC METABOLIC DISORDERS
Diabetes Mellitus
Auditory and vestibular symptoms and signs are common in patients with diabetes mellitus, but convincing evidence does not exist for a specific type of diabetic lesion of the inner ear or eighth nerve.1–5 In those diabetic patients with audiove- stibular dysfunction whose temporal bones and nervous systems have been studied at necropsy, pathologic changes can be explained on the basis of associated vascular disease.6–8 Three types of vascular changes occur with diabetes mellitus:
microangiopathy with thickening of the base- ment membrane of small vessels, (2) arterioscle- rotic narrowing of small arteries and arterioles, and (3) atherosclerotic narrowing of the large arteries. These vascular changes may damage the auditory and vestibular system from the peripheral end organs and eighth nerve to their
diffuse central nervous system (CNS) connec- tions. The most common finding in the labyrinth at necropsy in patients with diabetes mellitus is a thickening of the capillary walls, which is most prominent in the vascular stria of the cochlea, where it may account for the progressive, bilat- eral high-frequency hearing loss characteristic of the disease.7 Similar changes are found in the vestibular end organs; these, along with degen- eration of vestibular nerve and ganglion, could explain complaints of chronic disequilibrium and dizziness in diabetic patients.8
Sudden onset of hearing loss and/or vertigo in patients with diabetes mellitus can result from occlusion of the vessels to the labyrinth or the eighth nerve. The prevalence of diabetes mellitus was significantly higher in patients with idiopathic sudden sensorineural hearing loss compared with age- and sex-matched con- trols.9 Cranial nerve mononeuropathies are a well-known clinical phenomenon associated with diabetes mellitus; they are most likely due
367
Uremia
Multiple causes of auditory and vestibular symptoms can be identified in patients with chronic renal disease. The same pathologic process can affect both the kidneys and the labyrinths, as seen in Alport’s syndrome (hered- itary nephritis and deafness; see Chapter 18), diabetes mellitus, and Fabry’s disease. Immu- nosuppressive treatment either of the primary renal disorder or to avoid transplant rejection predisposes the patient to otologic infections, often with exotic or saprophytic organisms.11 Patients with renal disease are particularly vul- nerable to the ototoxic effects of aminoglyco- side antibiotics and loop diuretics because of their inability to clear these substances from the blood; ototoxicity is probably the most com- mon cause of auditory and vestibular symptoms in uremic patients (see “Ototoxins”).
Hyponatremia causes reversible hearing loss and tinnitus in patients undergoing chronic hemodialysis. There is a high degree of correla- tion between the hearing loss and serum sodium levels, irrespective of the blood urea level.12 The hearing loss can be corrected in most patients by returning the serum sodium level to normal. Patients undergoing chronic hemodi- alysis and those receiving kidney transplants often experience ill-defined fluctuating auditory and vestibular symptoms. Oda and colleagues13 performed necropsy studies on temporal bones of eight patients with chronic uremia who had undergone long-term hemodi- alysis therapy (24 to 546 treatments). At least one kidney transplant was performed in seven of the eight patients. Vestibular symptoms occurred in five patients and audi- tory symptoms in three—all symptoms began after the start of hemodialysis. Abnormal con- cretions were found in the vascular stria of the cochlea and in the subepithelial connective
tissue of the maculae and cristae in seven of the eight patients. The source of these abnor- mal deposits is unknown. Chronic dialysis is also associated with several neurological syn- dromes, including the disequilibrium syn- drome, subdural hematoma, and Wernicke’s encephalopathy.14
Hypothyroidism
A symmetrical, mild to moderate sensorineural hearing loss is commonly associated with spo- radic, nonendemic hypothyroidism.15,16 Vertigo may also occur in hypothyroid patients, although there is no vertiginous syndrome that is characteristic of this disorder. Some investi- gators have found a high incidence of hypothy- roidism in patients with idiopathic Meniere’s syndrome but others have not.17,18 Thyroid hor- mone is critical for the normal development of both the auditory and vestibular systems. Congenital hypothyroidism in rats leads to loss of auditory and vestibular function, and replace- ment hormone in the early stages of postnatal development prevents the loss of function.19 Similarly, there appears to be a critical thera- peutic window for preventing hearing loss in children with congenital hypothyroidism.20 Auditory and vestibular abnormalities have also been documented in animals who have been made hypothyroid.21 However, these abnor- malities typically do not recover after hormone replacement.
Alcohol and Thiamine Deficiency
ACUTE TOXIC EFFECTS OF ALCOHOL
Acute alcohol intoxication is regularly associ- ated with unsteadiness of gait, slurring of speech, and, occasionally, vertigo. The gait ataxia and slurring of speech suggest cerebellar dysfunction, but an additional vestibular com- ponent may be involved. In animal studies, alcohol selectively interferes with synaptic transmission within the vestibular nuclei.22 Vestibular function testing with rotational stimulation in patients with alcohol intoxication has shown normal vestibulo-ocular reflex gain in the dark, but impaired fixation suppres- sion of vestibular nystagmus consistent with cerebellar dysfunction.23,24 Slowing of saccades
Slow phase velocity (deg/sec)
Slow phase velocity (deg/sec)
10 Right lateral position
0
800
10
0.1
Left lateral position
Right-beating nystagmus Left-beating nystagmus
Blood alcohol (percent)
Blood alcohol (percent)
0.05
0
0 400 800
Time (minutes)
Figure 17–1. Direction-changing positional nystagmus after a normal subject ingested 150 ml of whiskey. Nystagmus initially beat toward the ground (undermost ear) in both lateral positions, but as the blood alcohol level decreased, the nystagmus beat away from the ground in both lateral positions.
and smooth pursuit is consistently found in subjects after only moderate alcohol ingestion.25–27 Gaze-evoked nystagmus is a reli- able sign of intoxication, the magnitude of which is highly correlated with the blood alcohol concentration.28
Positional vertigo is another well-documented effect of alcohol on the vestibular system (Fig. 17–1).29 Within 30 min after ingesting a moderate amount of alcohol (e.g., 150 ml of whiskey), the subject develops a direction- changing static positional nystagmus often associated with vertigo. The positional nystag- mus beats to the right in the right lateral posi- tion, to the left in the left lateral position, and is inhibited by fixation. The primary phase of the positional nystagmus reaches its peak in about 2 hr, at approximately the time of peak blood alcohol level (near 0.1% for the earlier exam- ple). Four to 5 hr after alcohol ingestion, when the blood alcohol level is below 0.01%, posi- tional nystagmus is still present, but now it is right-beating in the left lateral position and left-beating in the right lateral position (sec- ondary phase). The positional nystagmus can last up to 12 hr, at which time alcohol cannot be detected in the blood.
Direction-changing positional nystagmus in the reverse direction of primary alcohol posi- tional nystagmus can be produced by giving subjects heavy water—H3O.30 When subjects
with alcohol direction-changing positional
nystagmus (primary phase) are given H3O, the nystagmus disappears. Alcohol and heavy-
water direction-changing positional nystagmus are likely due to a different rate of diffusion of alcohol and heavy water into the cupula and the surrounding endolymph.30 In the primary phase of alcohol positional nystagmus, alcohol rapidly diffuses into the base of the cupula because of the latter’s proximity to blood capil- laries while it slowly diffuses into the endo- lymph. The cupula then has a lower specific gravity than that of the endolymph and acts as a gravity-sensing organ, maintaining a slight deflection as long as the position is held. After approximately 3 hr, the endolymph and cupula have approximately the same alcohol concen- tration, and the positional nystagmus disap- pears. As the blood alcohol level falls, the reverse situation occurs, with the cupula being heavier than the surrounding endolymph, and the secondary phase of positional nystagmus occurs.
Three-dimensional recordings of alcohol positional nystagmus showed that the buoyancy mechanism accounts for only part of the observed nystagmus.31 After consuming alco- hol, subjects were positioned so that the hori- zontal canals were earth-vertical and then rotated in the plane of the horizontal canals about an earth-horizontal axis to either 45 degrees or 90 degrees, right or left ear down. Spatial analysis of the recorded eye movements
WERNICKE’S ENCEPHALOPATHY
This is a common clinical syndrome caused by thiamine deficiency. In the past it was almost exclusively associated with alcoholism but now is commonly recognized with other causes of mal- nutrition, including cancer, and its treatments and bariatric surgery.32–34 It is characterized by the subacute onset of confusion, ophthalmople- gia, and ataxia of stance and gait. Dizziness and vertigo are not common complaints. Hearing loss is a rare presenting symptom.35 The truncal ataxia is often dramatic, with the patient being unable to take even a few steps without sup- port, and yet standard cerebellar function test- ing with finger-to-nose and heel-knee-shin is often normal or minimally impaired. The ataxia is increased with eye closure or darkness. These findings suggest a combination of midline cer- ebellar and either proprioceptive or vestibular impairment. Caloric responses can be dimin- ished or absent.36,37 Magnetic resonance imaging (MRI) of the brain typically shows symmetrical fluid-attenuated inversion recov- ery imaging (FLAIR) hyperintensities in the thalami, mamillary bodies, tectal plate, and periaquaductal area.38
Experimental studies in thiamine-deficient rats39 and monkeys40 showed that the earliest pathologic changes originate in the vestibular nuclei, particularly in the lateral nucleus. The nerve terminals and axons degenerate without evidence of damage to the neuronal parikaria. Neurologic signs appear even before these early pathologic changes. Loss of transketolase activity (a thiamine-dependent enzyme) in the lateral pontine tegmentum, including the lat- eral vestibular nuclei, correlates better with the onset of clinical signs. Injection of thiamine promptly restores transketolase activity and improves clinical signs.
Pathologic changes are seen in the vestibular nuclei of patients with thiamine deficiency studied at necropsy. Vascular changes include endothelial swelling in small arteries and veins and fibrinoid degeneration and hemorrhage
involving arterioles and capillaries on the arte- rial side.41 The medial vestibular nucleus is most commonly involved, but the other nuclei are also involved in between 30% and 50% of cases.32 The changes in the vestibular nuclei are relatively mild, however, compared with the frank necrosis and demyelination occurring in other areas. In the cerebellum, Purkinje cell loss is most prominent.42 Apparently, most of the clinical findings (including impaired ves- tibular function) are secondary to thiamine- dependent enzyme loss in the brain stem and cerebellum, and only after prolonged and/or repeated episodes of deficiency do irreversible structural changes occur.
CEREBELLAR DEGENERATION
In 1959, Victor and associates43 reported a dra- matic clinical syndrome in 50 alcoholics that was manifested by severe truncal ataxia with relative sparing of the upper extremities. On clinical examination, all patients exhibited severe ataxia of stance and gait, with instability of the trunk while standing and severe incoor- dination on the heel-knee-shin test. On neuro- pathologic examination, atrophy was remark- ably localized to the superior cerebellar vermis, paramedian superior cerebellar hemispheres, and the flocculi. Subsequent pathological stud- ies have confirmed these findings,44 and neu- roimaging studies have documented shrinkage of the cerebellum, particularly the superior vermis in chronic alcoholics (Fig. 17–2).45,46 The cerebellar damage is most likely due to malnutrition rather than direct alcohol toxicity, as similar cellular changes have been seen in malnourished nonalcoholics47 and some of the symptoms and signs seen with acute cerebellar degeneration can be reversed with massive doses of thiamine.48 Still, ethanol may have direct toxic effects on the cerebellum or pro- mote the toxic effects of thiamine deficiency.49
Management
There is no specific treatment for the neuroto- logic manifestations of diabetes mellitus. Presumably the likelihood of vascular occlu- sion (small and large vessel) decreases with good control of blood glucose levels.49 Although it has been suggested that auditory and vestib- ular dysfunctions occur in the prediabetic state,
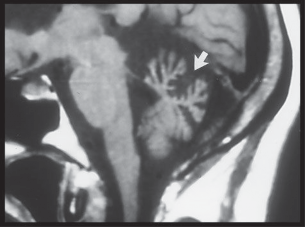
Figure 17–2. Magnetic resonance image of the brain in a patient with alcoholic cerebellar degeneration showing atrophy of the superior vermis (arrow). T1-weighted sagittal section.
as with the retinal and renal changes, there have been no controlled studies to support this supposition. The single most important aspect in preventing auditory and vestibular dysfunc- tions in patients with uremia is to avoid the use of potential ototoxic drugs. Careful manage- ment of electrolytes in patients undergoing chronic renal dialysis will prevent fluctuating auditory and vestibular symptoms. The bilat- eral sensorineural hearing loss associated with acquired hypothyroidism improves in a small percentage of patients after thyroid hormone replacement.15
With Wernicke’s encephalopathy the oph- thalmoplegia, confusion, and ataxia usually respond rapidly to thiamine replacement.32,40 Some patients are left with a chronic memory disorder (Korsakoff’s syndrome) as well as mild ataxia due to midline cerebellar degeneration. In patients receiving thiamine replacement, vestibular function as measured by serial caloric testing slowly returns toward normal over sev- eral weeks, although in some cases recovery is asymmetric and incomplete. As suggested ear- lier, alcoholic cerebellar degeneration may also respond to thiamine replacement. Some patients with alcoholic cerebellar degeneration who stop drinking exhibit a significant and sometimes dramatic decrease in body sway as measured with posturography, compared with patients who continued drinking.51 This improvement after abstinence from alcohol may result from central plastic changes or from recovery of function where structural damage was not complete. The cortical shrinkage and
ventricular dilation seen on imaging of the cer- ebellum are partially reversible with abstinence from alcohol.52 Proton magnetic resonance spectography showed increases in the concen- tration of choline-containing compounds within the superior cerebellar vermis in chronic alco- holics after 3 to 4 weeks of abstinence, followed by a reduction in these compounds with relapse.53
METABOLIC DISORDERS OF THE TEMPORAL BONE
Otosclerosis
Otosclerosis is a metabolic disease of the bony labyrinth that usually manifests itself by immo- bilizing the stapes and thereby producing a conductive hearing loss.54,55 The disease, how- ever, can less commonly involve the entire otic capsule and be associated with sensorineural hearing loss and vestibular symptoms. Seventy percent of patients with clinical otosclerosis note hearing loss between the ages of 11 and
30. The disorder is most common in whites (prevalence about 0.2%–1%), is infrequent in blacks, and is almost nonexistent in Asians and American Indians.56 A positive family history for otosclerosis is reported in between 50% and 70% of cases. Otosclerosis is genetically hetero- geneous with rare autosomal dominant forms, but the majority are due to an interaction between genetic and environmental factors.57
So far in families with autosomal dominant transmission at least seven monogenic chromo- somal loci have been reported but no genetic mutations identified. Three susceptibility alleles have been identified and replicated in case control association studies: TGFB1, BMP2, and BMP4. All three are part of the transform- ing growth factor-beta pathway.57
Although otosclerosis is primarily a disorder of the auditory system, vestibular symptoms and signs are more common than generally appreciated. In one study,58 46% of 500 patients with nonsurgically treated otosclerosis com- plained of vestibular symptoms. The most common symptoms were recurrent attacks of vertigo (26%) and postural imbalance (22%). Those patients with more severe sensorineural hearing loss were more likely to have vestibular complaints. Abnormalities on vestibular func- tion testing have been found in as many as 50% of patients tested, with the most common abnormality being unilateral hypoexcitability to caloric stimulation.59,60 The vestibular abnor- malities are more common in patients with greater sensorineural hearing loss, but they are not necessarily seen in the poorer hearing ear.
The basic pathologic process of otosclerosis is a resorption of normal bone, particularly
around blood vessels, and its replacement by cellular fibrous connective tissue.55,61,62 With time, immature basophilic bone is produced in the resorption space; after several cycles of resorption and new bone formation a mature acidophilic bone with a laminated matrix is produced. Bilateral involvement is usual, but about a fourth of cases are unilateral. Areas of predilection for otosclerotic foci include the oval window region, the round window niche, the anterior wall of the internal auditory canal, and within the stapedial footplate (Fig. 17–3). Although conductive hearing loss is the hall- mark of otosclerosis, a combined conductive– sensorineural hearing loss pattern is frequent. The sensorineural component is perhaps caused by foci of otosclerosis next to the spiral ligament of the cochlea, producing atrophy of the spiral ligament. About 10% of patients with otosclerosis eventually develop a profound sen- sorineural hearing loss across all frequencies.63 The mechanism for the production of dizzi- ness in patients with otosclerosis is poorly under- stood. Direct mechanical deformation of the labyrinth or biochemical abnormalities of inner ear fluids are likely possibilities. Endolymphatic hydrops has been identified in a few temporal bones with multiple foci of otosclerosis.64
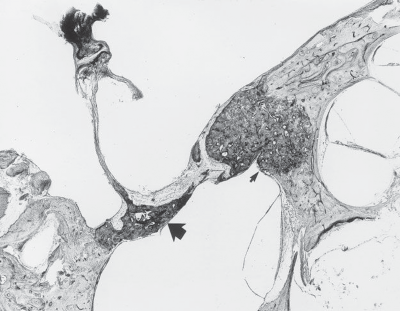
Figure 17–3. Histopathological section showing an otosclerotic lesion involving the bony labyrinth at the anterior margin of the oval window (small arrow) and the entire footplate of the stapes (larger arrow). The patient exhibited a combined conductive–sensorineural hearing loss. (Courtesy Harold Schuknecht, Boston, MA.)
Sando and colleagues65 studied four temporal bones of two patients with otosclerosis who complained of prominent vestibular symptoms and found otosclerotic foci in opposition to the superior vestibular nerve in each patient. Vestibular nerve degeneration distal to these foci was also present, and three of the four temporal bones exhibited a marked degenera- tion of the sensory epithelium of the cristae of the lateral semicircular canals.
Paget’s Disease
Paget’s disease is a metabolic disorder of bone marked by pronounced osteoclastic resorption of old, fully calcified bone and deposition of new osteoid layers that calcify normally.66–68 It usually becomes clinically manifest in the sixth decade, affecting men four times more commonly than women. The clinical picture varies from the classic one of an enlarged skull, progressive kyphosis, and short stature to the more common restricted forms confined to the skull, spine, pel- vis, and femur. Hearing loss is a common symp- tom, initially described by Paget in his early reports and subsequently studied in detail by numerous investigators.69,70 A progressive com- bined sensorineural and conducting hearing loss is usually found. The vestibular labyrinth may also be progressively destroyed, resulting in unsteadiness of gait and, in rare cases, episodic vertigo. In the late stages, complete destruction of the bony labyrinth may occur with invasion of the inner ears, fractures, and degeneration of the membranous labyrinth.
Genetic factors play an important role in the pathogenesis of Paget’s disease, and so far mutations have been identified in four differ- ent genes.71,72 By far the most common is SQSTM1, a gene that codes for a scaffold pro- tein in the nuclear factor KappaB signaling pathway. There is a mutation hot spot at the UBA domain of SQSTM1 with P392L being by far the most common mutation. Environmental factors must explain the varied clinical expres- sion within families with the mutation.
Other Disorders
Other less common metabolic disorders of the temporal bone that are associated with hearing loss and dizziness include osteogenesis
imperfecta,73 fibrous dysplasia,74 and osteopet- rosis.75 The clinical presentation of these disorders is often indistinguishable from that of otosclerosis and of Paget’s disease.
Diagnosis
The diagnosis of otosclerosis is based on find- ing a conductive hearing loss in a patient with the clinical picture outlined earlier. A flat tym- panogram with maximum compliance near zero pressure is characteristic (see Fig. 8–4C in Chapter 8). As the disease progresses, a mixed conductive, sensorineural hearing loss is com- mon. About 10% of patients exhibit hyperemia of the promontory mucosa of the middle ear, visible through the tympanic membrane (Schwartze’s sign). Computed tomography (CT) may show changes in the otic capsule (ranging from a small dehiscence in the nor- mal, crisp outline of the capsule to entire loss of anatomic details), but these changes are not specific for otosclerosis, as they are also found in osteogenesis imperfecta, fibrous dysplasia, and even some normal subjects. CT bone den- sity measurements at the fissula antefenestram may be useful for the diagnosis and as an indi- cator of disease progression.76
The diagnosis of Paget’s disease rests on finding the characteristic roentgenographic findings of increased density of bone with loss of the normal architecture, mingled with areas of decreased bone density.66 The skull is enlarged, with indistinct margins giving a “cotton wool” appearance. High-resolution CT scans of the temporal bone typically reveal poor definition of the cortical margins of the inner ear and internal auditory canals.
Management
In small observational studies, sodium fluoride has been associated with reduced progression of otosclerosis.77,78 A single controlled study from Denmark and France documented both biochemical and audiometric changes in a treated group of patients with otosclerosis com- pared with a control group.79 At 6 months, 7% (3/43) of treated patients had progressive hear- ing loss (>10 dB increase from baseline in pure- tone average) whereas 25% (13/52) of the control patients had progressive hearing loss.
Surgical treatment of otosclerosis is directed at improving the conductive hearing loss. Many different operations have been developed, and most have a high success rate. Although data on the results of surgical therapy in patients with vestibular symptoms are not available, there is little reason to expect improvement in such patients. In fact, surgical therapy may aggravate the vestibular symptoms. Smyth80 detected decreased caloric responses 3 months postoperatively in 30% of 26 ears with normal values preoperatively.
Bisphosphonate reduces bone turnover, and it has been shown to clear radiological lesions and restore normal histology in patients with Paget’s disease.71 Long-term follow-up on disease progression is lacking.
Patients who receive ototoxic drugs are often bedridden and suffer from multiple symptoms of systemic illness, so additional symptoms of audi- tory and vestibular dysfunction may be easily overlooked. Vestibular symptoms are particularly difficult to identify in this setting. Only after the patient begins to recover do the devastating effects of vestibular loss become apparent. By this time, the damage is irreversible. The examin- ing physician must be keenly aware of the poten- tial auditory and vestibular toxicity of any drug that is used, if ototoxicity is to be prevented.
Aminoglycosides
The commonly used aminoglycosides are listed in Table 17–1. Although each of these drugs can
Table 17–1 Relative Vestibular and Auditory Ototoxicities of Commonly Used Aminoglycosides
Vestibular | Auditory | |
Streptomycin | + + + | + |
Gentamicin | + + + | + |
Tobramycin | + + | + + |
Kanamycin | + | + + + |
Amikacin | + | + + + |
Dibekacin | + | + |
Netilmicin | + | + |
Sisomicin | + | + |
produce both auditory and vestibular damage, streptomycin and gentamicin are relatively spe- cific for the vestibular system, whereas kanamy- cin, tobramycin, and amikacin produce more damage to the auditory system.81,82 The newer aminoglycosides sisomicin, dibekacin, and netilmicin are overall less ototoxic than the older aminoglycosides.83 Although high blood levels and long durations of treatment are most likely to produce ototoxicity, some susceptible patients may develop ototoxicity after a brief low-dose course (see later discussion). The majority of patients with gentamicin vestibulotoxicity receive a total dose within the generally accepted safe range,84 and serum peak-and-trough gen- tamicin levels do not correlate with develop- ment of vestibulotoxicity.85 Also, both vestibular and cochlear toxicity have been observed with ototopic aminoglycoside ear drops.86
The pharmacologic and biochemical charac- teristics are similar for all of the aminoglyco- side antibiotics.87,88 They are excreted almost exclusively by glomerular filtration; they are not metabolized. Patients with renal impair- ment cannot excrete the drugs, so the amino- glycosides accumulate in the blood and inner ear tissues. The ototoxicity of the aminoglyco- sides has been shown convincingly to be due to hair cell damage in the inner ear. Unlike peni- cillin and other common antibiotics, aminogly- cosides are concentrated in the perilymph and endolymph. The earliest effect of the vestibu- lotoxic compounds, such as streptomycin and gentamicin, is a selective destruction of type I hair cells in the crista. Later, type II hair cells are destroyed, but the supporting cells remain unaffected. With the cochleotoxic agents, such as kanamycin and amikacin, there is first a selective destruction of the outer hair cells in
The mechanism by which aminoglycosides damage hair cells is only partially under- stood.82,83,88 Several studies suggest that amino- glycosides may interfere with mitochondrial energy metabolism by binding to the mitochon- drial membrane and increasing membrane permeability. Aminoglycosides have also been shown to reversibly block calcium-sensitive potassium channels, increase free-radical forma- tion, and have an excitotoxic effect on cochlear N-methyl-d-aspartate (NMDA) receptors.89,90 With the discovery of increased sensitivity to ototoxicity in families with mutations in the mitochondrial gene for the 12S ribosomal ribo- nucleic acid (rRNA), the possibility was raised that even normal human mitochondrial rRNA may be affected by the aminoglycosides.91,92 Interestingly, the region on the 12S rRNA that is mutated in families with ototoxin susceptibil- ity is morphologically similar to the area of bac- terial rRNA to which aminoglycosides bind in the course of their bacterial cytoaction. This mechanism could not explain the vestibular loss associated with gentamicin and streptomy- cin, however, since families with the predispos- ing mutations in the 12S rRNA gene have hearing loss but normal vestibular function.93 Furthermore, no mutations with confirmed pathogenicity were identified in the 12S rRNA gene in 66 patients with aminoglycoside ves- tibulotoxicity or in 15 patients with idiopathic bilateral vestibulopathy.92
As noted earlier, vestibular symptoms and signs due to aminoglycoside toxicity are often subtle. Although some patients complain of vertigo (presumably due to asymmetric involve- ment of the vestibular system), most complain of an unsteady gait, particularly at night or in a
darkened room. Head movement–induced oscillopsia is also common. The head-thrust test can be used at the bedside to identify ves- tibular impairment. Serial caloric and rotational examinations document a progressive bilateral loss of vestibular responsiveness. Because of their highly selective effect on the vestibular endorgan, streptomycin and gentamicin are used to produce a chemical vestibulectomy in patients with episodic vertigo from Meniere’s syndrome (see Chapter 11).
“Loop” Diuretics
The two main ototoxic diuretics, furosemide and ethacrynic acid, act by inhibiting active resorption of chloride in the loop of Henle, thereby preventing the renal resorption of sodium that passively follows chloride. The mechanism of their ototoxic effect is not com- pletely known, although these drugs clearly influence ion channels in the kidney and in the cochlear duct.94 They inhibit the Na-K-2Cl- cotransporter in the marginal and dark cells of the stria vascularis, resulting in a reduction in the endolymphatic potential and endolym- phatic K+ concentration. The ototoxic effects of the loop diuretics appear to be primarily confined to the cochlea, although vertigo has been rarely reported. About 6% of patients receiving furosemide develop a temporary hearing loss that is nearly always reversible. The newer loop-inhibiting diuretics, bume- tanide and piretanide, appear to have a much lower rate of cochleotoxic effects in both ani- mal and human studies.95,96 Loop diuretics may enhance aminoglycoside ototoxicity.97
Anti-inflammatory Drugs
Anti-inflammatory drugs known to be associ- ated with ototoxicity include salicylates, non- steroidal anti-inflammatory drugs (NSAIDs), and quinine.98 Salicylates and NSAIDs are among the most commonly used drugs for inflammation, fever, and pain. Quinine, origi- nally an antimalarial drug with properties simi- lar to those of the salicylates, is commonly used for treating nocturnal leg cramps. The main ototoxic effect of these drugs is a reversible, mild to moderate flat or high-frequency hearing loss along with a high-pitched tinnitus.
mg/ml.99 There is almost a linear relation- ship between serum unbound salicylate con- centration and ototoxic symptoms, although the threshold level for symptoms varies from patient to patient. These ototoxins cause a transient dysfunction of outer hair cells documented by both ultrastructural and electrophysiological studies. There is no evidence that the vestibular system is affected. All symptoms and signs are typically rapidly reversible after cessation of the drug ingestion (usually within 24 hr).
Platinum Compounds
Cis-platinum and carboplatin are the two most commonly used ototoxic chemotherapeutic agents. Both are associated with auditory toxicity, although carboplatin is less toxic than cis-platinum.88,100 By way of comparison, the incidence of aminoglycoside ototoxicity is about 10%, whereas the incidence of cis-platinum ototoxicity is in the range of 50%. Tinnitus and hearing loss are extremely common. Typically, the tinnitus is transient, lasting from a few hours to up to a week after therapy. The hear- ing loss is usually bilateral, beginning in the high frequencies and progressing to involve all frequencies; it may not appear until several days after treatment. The hearing loss usually has some degree of reversibility, although when it is severe and involves all frequencies it is often permanent. The critical cumulative oto- toxic dose of cis-platinum has been reported to be in the range of 3 to 4 mg/kg of body weight.101 The ototoxic effects can be decreased by using slow infusions and dividing the doses over sev- eral months.102 The vestibular system is relatively spared by these chemotherapeutic ototoxins.103 Morphologic studies in animals that have been given cis-platinum show hair cell damage similar to that seen with the amin- oglycosides. Free radical formation in the kidney and ear have been implicated in the pathogen- esis of toxicity with both compounds.94
Diagnosis
The clinician must be constantly on the alert for the early symptoms of ototoxic drugs.
This is particularly important in a patient who is seriously ill and confined to bed or in any patient who has renal impairment, particularly renal failure. Bedside audiometric assessment is available in most hospitals; bedridden patients can be tested with reproducible audi- tory stimuli. Although less satisfactory than conventional testing in a soundproof room, earphones help exclude ambient noise. Because the hearing loss due to ototoxic drugs usually begins in the high-frequency range, a screen of the high frequencies can be used to predict future low-frequency loss. By contrast, quanti- tative bedside vestibular testing (i.e., caloric and rotational testing) is not available in most hospitals so that the clinical examination is crit- ical for identifying early vestibular loss (see tests of vestibulo-ocular reflexes, Chapter 6). The head-thrust test can be performed even in critically ill patients and has consistently been shown to be a sensitive test for vestibulo- toxicity.104,105 Whenever possible the Romberg and tandem walking tests should be used to assess balance function. Ambulatory patients can undergo quantitative caloric and rotational testing. Rotational testing is ideally suited for identifying early vestibular ototoxic effects, because the normal response variability is much less than that seen with caloric testing (see Chapter 7). The earliest changes are short- ening of the dominant time constant and decreased gain at the lowest and highest frequencies.104
Management
The key to the management of the ototoxicity is prevention. Kidney function should be mea- sured prior to beginning any potentially oto- toxic drug. Patients in high-risk groups (Table 17–2) should be monitored with periodic audi- tory and vestibular testing. All patients should be questioned on a regular basis to identify early symptoms of auditory or vestibular loss. When the earliest effects of ototoxicity are identified, the drug should be stopped. Usually other drugs can be used that have less ototoxic potential.
Based on the hypothesis that aminoglycoside ototoxicity results from iron chelation and free- radical formation, use of iron chelators and radical scavengers was shown to prevent ototox- icity in guinea pigs.106 At the same time, these
Table 17–2 Major Risk Factors for Drug Ototoxicity
![]()
Impaired renal function High serum levels
Prior use of ototoxic drugs Course > 14 days
Preexisting sensorineural hearing loss Age > 65 years
![]()
drugs have no effect on serum levels of the aminoglycosides nor on its antibacterial effi- cacy. Assuming that the ototoxic effects of the aminoglycosides are at least in part due to exci- totoxic activation of cochlear NMDA receptors, Segal et al.90 showed that NMDA antagonists protected animals from aminoglycoside ototox- icity. Antioxidants may prevent both the free- radical formation and the excitotoxic effects on the NMDA receptors.107 A double-blind pla- cebo-controlled study in China found that aspi- rin (an antioxidant) given in combination with gentamicin was significantly more effective than placebo with gentamicin in preventing hearing loss.108,109 At 5–7 weeks after being treated with intravenous gentamicin for an acute infection, 3% (3/89) of patients treated with 3 g of aspirin per day had ototoxicity (defined as hearing loss of >15 dB from baseline at both 6 and 8 kHz) compared to 13% (14/106) of the control group. In another controlled study the antioxidant N-acetylcysteine (NAC) significantly decreased the incidence of hearing loss in hemodialysis patients receiving gentamicin for dialysis cathe- ter-related bacteremia.110 In this study at 6 weeks after gentamicin, 25% (5/20) patients treated with NAC had ototoxicity (defined as
>20 dB loss at any frequency or >10 dB loss at any two adjacent frequencies) compared to 60% (12/20) of the control group. Neither of these studies assessed vestibular or balance function as an outcome, despite the fact that gentamicin is more toxic to the vestibular sys- tem than to the auditory system.
As with other vestibular disorders, manage- ment of patients with permanent bilateral ves- tibular loss due to ototoxins should be directed at retraining the nervous system to use other sensory signals to replace the lost vestibular signals. Practical suggestions on how to avoid head movement–induced oscillopsia (stopping and holding the head still when attempting to
read a sign) and gait unsteadiness (always have a light on throughout the night) are useful, along with an active exercise program to force central compensation (see Chapter 20). Younger patients often will return to nearly normal activity over a period of years, but elderly patients are rarely able to compensate fully for the vestibular loss.
Heavy Metals
Both lead and mercury intoxication are known to produce auditory and vestibular symptoms, but the pathophysiologic mechanisms of these symptoms is poorly understood. In adults, acute lead poisoning typically leads to a motor neuropathy, whereas in children encephalopa- thy is more prominent. Chronic lead absorp- tion can lead to more subtle symptoms and signs, including nonspecific dizziness, imbal- ance, and cognitive deficits. Rats exposed to chronic low concentrations of lead acetate in their drinking water showed abnormalities in postrotatory nystagmus in the absence of clinical signs of lead intoxication.111 Segmental demyelination and axonal degeneration of the eighth nerve were identified in guinea pigs given weekly intraperitoneal injections of 1% lead acetate solution for 7 weeks.112 The end organs and ganglion cells showed no visible morphologic changes. By contrast, adult squirrel monkeys that were chronically poi- soned with lead showed minimal changes in the auditory and vestibular systems, both on clinical and histologic examination.113 The dif- ference in findings may be due to the well- known decreased susceptibility of the adult nervous system to lead toxicity. Hearing loss and vertigo have occasionally been reported in isolated cases of childhood and adult lead poisoning, but no detailed study has been undertaken of auditory and vestibular function in a large population of lead-poisoned patients.
Two large patient populations with organic mercury poisoning have undergone detailed neurotologic examination in Japan. Mizukoshi and colleagues114 studied 144 patients with organic mercury poisoning acquired from eat- ing contaminated fish caught in the Aganagawa
The site of vestibular and auditory dysfunction in mercury-poisoned patients is unclear. Oral administration of methyl mercury to rats rapidly leads to accumulation of mercury in the brain stem, particularly in the ventral cochlear nucleus and the superior vestibular nucleus.116 Mercury intoxication in guinea pigs resulted in changes in the labyrinth and cerebellum.117,118 Necropsy studies have not been performed on the tempo- ral bones and eighth nerves from patients dying of mercury intoxication. Examination of the brain in such patients reveals a selective sensitiv- ity of the granule cell layer of the neocerebellum, but cases of demyelination of the subcortical and brainstem white matter and peripheral nerves have also been reported.119,120
Organic Solvents
Acute intoxication with organic solvents leads to a well-described toxic encephalopathy mani- fested by symptoms of confusion, dizziness and imbalance, nausea, and generalized weakness. Whether a chronic toxic encephalopathy devel- ops from chronic low-level exposure to organic solvents is a more controversial issue.121,122 Some workers with long-term occupational exposure to organic solvents may become intol- erant even to low air concentrations so that they are no longer able to work in the industry.123 Studies in animals show direct effects of organic solvents on both vestibulo-ocular reflex gain
and the ability to suppress the vestibular-ocular reflex with fixation.124 Auditory and vestibular function testing in workers exposed to organic solvents identified a variety of nonspecific abnormalities, compared to controls, most being consistent with dysfunction within cen- tral auditory and vestibular pathways.125,126 However, these auditory–vestibular deficits found in workers with organic solvent exposure do not correlate with the degree of exposure or with the severity of symptoms. The effects of organic solvents on hearing may be synergistic with exposure to noise and other chemicals in the work environment.
Diagnosis
Auditory and vestibular symptoms and signs due to neurotoxins are typically part of a more generalized encephalopathic syndrome, with some unique features being associated with each of the different neurotoxins. Heavy metal intoxication leads to multisystem involvement with clear neurologic findings. With acute exposure, blood levels typically correlate with the severity of exposure and the severity of symptoms and signs. However, with chronic exposure, heavy metals are bound to bone and other hard tissues so that blood levels do not reflect the level of the body burden. Past expo- sure may be identified in hair and nail samples or by mobilizing bone stores with chelating agents. A 24-hr urine collection after exposure to a chelating agent provides a good means of assessment of body heavy-metal burden. There is no good way of assessing past exposure to organic solvents other than taking a careful history regarding types of exposure.
Management
The first step in managing exposure to neuro- toxins is to remove the exposure. With acute intoxication to heavy metals, residual symp- toms and signs are common, even with aggres- sive treatment. Chelating agents can be used to decrease the body burden of heavy metals, but neurologic damage is often irreversible. The chelating agents must be used with caution since they pull heavy metals from hard tissue into the blood, which can lead to further neurologic damage.
REFERENCES
Biurrun D, Ferrer JP, Lorente J, et al. Asymptomatic electronystagmographic abnormalities in patients with type 1 diabetes mellitus. ORL J Otorhionlaryngol Relat Spec. 1991;53:335.
Tay HL, Ray N, Ohri R, Frootko NJ. Diabetes mellitus and hearing loss. Clin Otolaryngol. 1995;20:130.
Gawron W, Pospiech L, Orendorz-Fraczkowska K, Noczynska A. Are there any disturbances in vestibular organ of children and young adults with Type I diabe- tes? Diabetologia. 2002;45(5):728. ePub ahead of print Apr 17, 2002.
Mitchell P, Gopinath B, McMahon CM, et al. Relationship of Type 2 diabetes to the prevalence, incidence and progression of age-related hearing loss. Diabet Med. 2009;26(5):483.
Austin DF, Konrad-Martin D, Griest S, McMillan GP, McDermott D, Fausti S. Diabetes-related changes in hearing. Laryngoscope. 2009;119(9):1788.
Makishilma K, Tanaka K. Pathological changes of the inner ear and central auditory pathways in diabetics. Ann Otol Rhinol Laryngol. 1971;80:218.
Wackym PA, Linthicum FH, Jr. Diabetes mellitus and hearing loss: clinical and histopathologic relationships. Am J Otol. 1986;7:176.
Myers SF, Ross MD, Jokelainen P, et al. Morphological evidence of vestibular pathology in long-term experi- mental diabetes mellitus. 1. Microvascular changes. Acta Otolaryngol (Stockh). 1985;100:351.
Aimoni C, Bianchini C, Borin M, et al. Diabetes, car- diovascular risk factors and idiopathic sudden sen- sorineural hearing loss: a case-control study. Audiol Neurootol. 2009;15(2):111.
Wu JS, Lu FH, Yang YC, Chang CJ. Postural hypotension and postural dizziness in patients with non–insulin-dependent diabetes. Arch Intern Med. 1999;159:1350.
Cohen G, Haag-Weber M, Horl WH. Immune dys- function in uremia. Kidney Int Suppl. 1997;62:S79.
Yassin A, Badry A, Fatt-Hi A. The relationship between electrolyte balance and cochlear disturbances in cases of renal failure. J Laryngol. 1970;84:429.
Oda M, Preciado MC, Quick CK, Paparella MM. Labyrinthine pathology of chronic renal failure patients treated with hemodialysis and kidney transplantation. Laryngoscope. 1974;84:1489.
Burn DJ, Bates D. Neurology and the kidney. J Neurol Neurosurg Psychiatry. 1998;65:810.
Meyerhoff WL. The thyroid and audition.
Laryngoscope. 1976;86:483.
Rubenstein N, Rubenstein C, Theodor R. Hearing dysfunction associated with congenital sporadic hypo- thyroidism. Ann Otol Rhinol Laryngol. 1974;83:814.
Rybak LP. Metabolic disorders of the vestibular sys- tem. Otolaryngol Head Neck Surg. 1995;112:128.
Brenner M, Hoistad DL, Hain TC. Prevalence of thyroid dysfunction in patients with Ménière’s dis- ease. Arch Otolaryngol Head Neck Surg. 2004;130(2): 226.
Meza G, Acuna D, Escobar C. Development of vestib- ular and auditory function: effects of hypothyroidism and thyroxine replacement therapy on nystagmus and auditory evoked potentials in the pigmented rat. Int J Dev Neurosci. 1996;14:515.
Wasniewska M, De Luca F, Siclari S, et al. Hearing loss in congenital hypothalamic hypothyroidism: a wide therapeutic window. Hear Res. 2002;172(1-2):87.
Withers BT, Reuter S, Janeke J. The effects of hypo- thyroidism on the ears of cats and squirrel monkeys: a pilot study. Laryngoscope. 1972;82:779.
Kashii S, Ito J, Matsuoka I, et al. Effects of etha- nol applied by electrosmosis on neurons in the lat- eral and medial vestibular nuclei. Jpn J Pharmacol. 1984;36:153.
Gilson RD, Schroeder DJ, Collins WE, Guedry FE. Effects of different alcohol dosages and display illu- mination on tracking performance during vestibular stimulation. Aerospace Med. 1972;4.3:656.
Guedry FE, Gilson RD, Schroeder DJ, Collins WE. Some effects of alcohol on various aspects of oculomo- tor control. Aviat Space Environ Med. 1975;46:1008.
Lehtinen I, Lang AH, Jantti V, Keskinen E. Acute effects of alcohol on saccadic eye movements. Psychopharmacology. 1979;63:17.
Lehtinen I, Nyrke T, Lang AH, et al. Quantitative effects of ethanol infusion on smooth pursuit eye move- ments in man. Psychopharmacology. 1982;77:74.
Wilkinson IMS, Kime R, Purnell M. Alcohol and human eye movement. Brain. 1974;97:785.
Coding CS, Dobie RA. Gaze nystagmus and blood alcohol. Laryngoscope. 1986;46:713.
Aschan C, Bergstedt M. Positional alcoholic nystag- mus (PAN) in man following repeated alcohol doses. Acta Otolaryngol Suppl (Stockh). 1975;330:15.
Money KE, Myles WS. Heavy water nystagmus and effects of alcohol. Nature. 1974;247:404.
Fetter M, Halswanter T, Bork M, Dichgans J. New insights into positional alcohol nystagmus using a three-dimensional eye-movement analysis. Ann Neurol. 1999;45:216.
Victor M, Adams RD, Collins CH. The Wernicke- Korsakoff Syndrome and Related Disorders Due to Alcoholism and Malnutrition. Philadelphia: FA Davis; 1989.
Kuo SH, Debnam JM, Fuller GN, de Groot J. Wernicke’s encephalopathy: an underrecognized and reversible cause of confusional state in cancer patients. Oncology. 2009;76(1):10. ePub ahead of print Nov 19, 2008.
Aasheim ET. Wernicke encephalopathy after bar- iatric surgery: a systematic review. Ann Surg. 2008;248(5):714.
Buscaglia J, Faris J. Unsteady, unfocused, and unable to hear. Am J Med. 2005;118:1215.
Ghez C. Vestibular paresis: a clinical feature of Wernicke’s disease. J Neurol Neurosurg Psychiatry. 1969;33:134.
Goor C, Endtz LJ, Muller Kobold MJR. Electro- nystagmography for the diagnosis of vestibular dys- function in Wernicke-Korsikoff syndrome. Clin Neurol Neurosurg. 1975;78:112.
Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol. 2009;192(2):501.
Tellez I, Terry RD. Fine structure of the early changes in the vestibular nuclei of the thiamine-deficient rat. Am J Pathol. 1968;52:777.
Cogan DG, Witt ED, Goldman-Rakic PS. Ocular signs in thiamine-deficient monkeys and in Wernicke’s dis- ease in humans. Ann Ophthalmol. 1985;103:1212.
Okeda R, Taki K, Ikari R, Funata N. Vascular changes in Wernicke’sencephalopathy. Acta Neuropathol (Berl). 1995;89:420.
Baker KG, Harding AJ, Halliday CM, Kril JJ, Harper CG. Neuronal loss in functional zones of the cerebel- lum of chronic alcoholics with and without Wernicke’s encephalopathy. Neuroscience. 1999;91:429.
Victor M, Adams RD, Mancall EL. A restricted form of cerebellar cortical degeneration occurring in alco- holic patients. Arch Neurol. 1959;1:579.
Yokota O, Tsuchiya K, Terada S, et al. Alcoholic cer- ebellar degeneration: a clinicopathological study of six Japanese autopsy cases and proposed potential progres- sion pattern in the cerebellar lesion. Neuropathology. 2007;27(2):99.
Diamond I, Messing RO. Neurologic effects of alco- holism. West J Med. 1994;161:279.
Seitz D, Widmann U, Seeger U, et al. Localized proton magnetic resonance spectroscopy of the cer- ebellum in detoxifying alcoholics. Alcohol Clin Exp Res. 1999;23:158.
Mancall E, McEntee WJ. Alterations of the cerebel- lar cortex in nutritional encephalopathy. Neurology. 1965;15:303.
Graham JR, Woodhouse D. Massive thiamine dosage in an alcoholic with cerebellar cortical degeneration. Lancet. 1971;2:107.
Ke ZJ, Wang X, Fan Z, Luo J. Ethanol promotes thia- mine deficiency-induced neuronal death: involvement of double-stranded RNA-activated protein kinase. Alcohol Clin Exp Res. 2009;33(6):1097.
Troni W, Carta Q, Cantello R, et al. Peripheral nerve function and metabolic control in diabetes mellitus. Ann Neurol. 1984;16:178.
Diener HC, DichgansJ, Bacher M, Guschlbauer
B. Improvement of ataxia in alcoholic cerebellar atrophy through alcohol abstinence. J Neurol. 1934;2.31:258.
Ron MA, Acker W, Shaw GK, Lishman WA. Computerized tomography of the brain in chronic alcoholism: a survey and follow-up study. Brain. 1982;105:497.
Martin PR, Gibbs SJ, Nimmerrichter AA, et al. Brain proton magnetic resonance spectroscopy studies in recently abstinent alcoholics. Alcohol Clin Exp Res. 1995;19:1078.
Larsson A. Otosclerosis: a genetic and clinical study.
Acta Otolaryngol Suppl (Stockh). 1960;154:6.
Markou K, Goudakos J. An overview of the etiol- ogy of otosclerosis. Eur Arch Otorhinolaryngol. 2009;266(1):25.
Sakihara Y, Christensen B, Parving A. Prevalence of hereditary hearing impairment in adults. Scand Audiol. 1999;28:39.
Thys M, Camp GV. Genetics of otosclerosis. Otol Neurotol. ePub ahead of print Jun 19, 2009.
Cody DTR, Baker HL. Otosclerosis: vestibular symp- toms and sensorineural hearing loss. Ann Otol Rhinol Laryngol. 1978;87:778.
Virolainen E. Vestibular disturbances in clini- cal otosclerosis. Acta Otolaryngol Suppl (Stockh). 1972;306:7.
Kujala J, Aalto H, Hirvonen TP. Video-oculography findings in patients with otosclerosis. Otol Neurotol. 2005;26(6):1134.
Youssef O, Rosen A, Chandrasekhar S, Lee HJ. Cochlear otosclerosis: the current understanding. Ann Otol Rhinol Laryngol. 1998;107:1076.
Thiers FA, Valvassori GE, Nadol JB, Jr. Pathology case of the month: otosclerosis of the cochlear capsule: correlation of computerized tomography and histopa- thology. Am J Otol. 1999;20:93.
Ramsay HAW, Linthicum FH. Mixed hearing loss in stapedial otosclerosis. Am J Otol. 1994;15:536.
Liston SL, Paparella MM, Mancall EL, Anderson JH. Otosclerosis and endolymphatic hydrops. Laryngoscope. 1984;94:1003.
Sando I, Hemenway WG, Miller DR, Black FO. Vestibular pathology in otosclerosis temporal bone histopathological report. Laryngoscope. 1974;84:593.
Proops D, Bayley D, Hawke M. Paget’s disease and the temporal bone—a clinical and histopathological review of six temporal bones. J Otolaryngol. 1985;14:20.
Siris ES. Paget’s disease of bone. J Bone Miner Res. 1998; 13:1061.
Seton M. Paget’s disease: epidemiology and pathophys- iology. Curr Osteoporos Rep. 2008;6(4):125.
Clemis J, Bayles J, Harford ER, Petasnick JP. The clin- ical diagnosis of Paget’s disease of the temporal bone. Ann Otol Rhinol Laryngol. 1967;76:611.
Davies D. Paget’s disease of the temporal bone: a clinical and histopathological survey. Acta Otolaryngol Suppl (Stockh). 1968;242:7.
Ralston SH, Langston AL, Reid IR. Pathogenesis and management of Paget’s disease of bone. Lancet. 2008;372(9633):155.
Falchetti A, Di Stefano M, Marini F, et al. Genetic epidemiology of Paget’s disease of bone in Italy: sequestosome1/p62 gene mutational test and haplo- type analysis at 5q35 in a large representative series of sporadic and familial Italian cases of Paget’s disease of bone. Calcif Tissue Int. 2009;84(1):20.
Tsipouras P, Barabas G, Matthews WS. Neurologic correlates of osteogenesis imperfecta. Arch Neurol. 1986;43:150.
Sharp M. Monostotic fibrous dysplasia of the temporal bone. J Laryngol. 1970;84:697.
Hamersma H. Osteopetrosis (marble bone disease) of the temporal bone. Laryngoscope. 1970;80:1518.
Grayeli AB, Yrieix CS, Imauchi Y, Cyna-Gorse F, Ferrary E, Sterkers O. Temporal bone density mea- surements using CT in otosclerosis. Acta Otolaryngol. 2004;124(10):1136.
Snow JB, Jr. Current status of fluoride therapy for oto- sclerosis. Am J Otol. 1985;6:56.
Naumann IC, Porcellini B, Fisch U. Otosclerosis: incidence of positive findings on high-resolution com- puted tomography and their correlation to audiologi- cal test data. Ann Otol Rhinol Laryngol. 2005;114(9): 709.
Bretlau P, Causse J, Causse JB, et al. Otospongiosis and sodium fluoride. Ann Otol Rhinol Laryngol. 1985;94:103.
Smyth GDL. Recent and future trends in the man- agement of otosclerotic conductive hearing loss. Clin Otolaryngol. 1982;7:153.
Smith CR, Lipsky JJ, Laskin OL, et al. Double blind comparison of the nephrotoxicity and auditory tox- icity of gentamicin and tobramycin. N Engl J Med. 1980;302:1106.
Selimoglu E. Aminoglycoside-induced ototoxicity.
Curr Pharm Des. 2007;13(1):119.
Roland JT, Jr, Cohen NL. Vestibular and auditory oto- toxicity. In: Cummings CW, Fredrickson JM, Harker LA, Krause CJ, Schuller DE, eds. Otolaryngology Head and Neck Surgery. 3rd ed. St. Louis, MO: CV Mosby; 1998.
Halmagyi GM, Fattore CM, Curthoys IS, Wade S. Gentamicin vestibulotoxicity. Otolaryngol Head Neck Surg. 1994;111:571.
Black FO, Pesznecker S, Stallings V. Permanent gentamicin vestibulotoxicity. Otol Neurotol. 2004;25(4):559.
Matz G, Rybak L, Roland PS, et al. Ototoxicity of oto- topical antibiotic drops in humans. Otolaryngol Head Neck Surg. 2004;130(suppl 3):S79.
Lerner SA, Matz GJ. Aminoglycoside ototoxicity. Am J Otolaryngol. 1980;1:169.
Rybak LP, Ramkumar V. Ototoxicity. Kidney Int. 2007;72(8):931.
Schacht J. Aminoglycoside ototoxicity: prevention in sight? Otolaryngol Head Neck Surg. 1998;118:674.
Segal JA, Harris BD, Kustova Y, Basile A, Skolnick
P. Aminoglycoside neurotoxicity involves NMDA receptor activation. Brain Res. 1999;815:270.
Casano RA, Johnson DF, Bykhovskaya Y, et al. Inherited susceptibility to aminoglycoside ototoxicity: genetic heterogeneity and clinical implications. Am J Otolaryngol. 1999;20:151.
Elstner M, Schmidt C, Zingler VC, et al. Mitochondrial 12S rRNA susceptibility mutations in aminoglycoside-associated and idiopathic bilat- eral vestibulopathy. Biochem Biophys Res Commun. 2008;377(2):379.
Braverman I, Jaber I, Levi H, et al. Audiovestibular findings in patients with deafness caused by a mito- chondrial susceptibility mutation and precipitated by an inherited nuclear mutation or aminoglycosides. Arch Otolaryngol Head Neck Surg. 1996;122:1001.
Humes HD. Insights into ototoxicity. Analogies to nephrotoxicity. Ann NY Acad Sci. 1999;884:15.
Rybak LP, Wellworth C. Comparative ototoxic- ity of furosemide and piretanide. Acta Otolaryngol (Stockh). 1986;101:59.
Tnzel IJ.Comparison of adverse reactions of bumetanide and furosemide. J Clin Pharmacol. 1981;21:615.
Bates DE, Beaumont SJ, Baylis BW. Ototoxicity induced by gentamicin and furosemide. Ann Pharmacother. 2002;36(3):446.
Jung TT, Rhee CK, Lee CS, et al. Ototoxicity of sali- cylate, nonsteroidal anti-inflammatory drugs and qui- nine. Otolaryngol Clin North Am. 1993;26:791.
Myers E, Bernstein J, Fostiropolous G. Salicylate ototoxicity: a clinical study. N Engl J Med. 1965; 273:587.
MacDonald MR, Harrison RV, Wake M, et al. Ototoxicity of carboplatin: comparing animal and clinical models at the Hospital for Sick Children. J Otolaryngol. 1994;23:151.
Hayes DM, Cvitkovic E, Golbey RB, et al. High dose cis-dichlorodiamineplatinum: amelioration by mannitol diuresis. Cancer. 1977;39:1372.
Schaefer SD, Post JD, Close LG, Wright CG. Ototoxicity of low- and moderate-dose cisplatinum. Cancer. 1985;56:1934.
Myers SE, Blakely BW, Schwan S. Is cis-platinum vestibulotoxic? Otolaryngol Head Neck Surg. 1993;108:322.
Ishiyama G, Ishiyama A, Kerber K, Baloh RW. Gentamicin ototoxicity: clinical features and the effect on the human vestibulo-ocular reflex. Acta Otolaryngol. 2006;126(10):1057.
Weber KP, Aw ST, Todd MJ, McGarvie LA, Curthoys IS, Halmagyi GM. Horizontal head impulse test detects gentamicin vestibulotoxicity. Neurology. 2009;72(16):1417.
Conlon BJ, Aran JM, Erre JP, Smith SW. Attenuation of aminoglycoside-induced cochlear damage with the metabolic antioxidant alpha-lipoic acid. Hear Res. 1999;128:40.
Sha SH, Schacht J. Are aminoglycoside antibiotics excitotoxic? Neuroreport. 1998;9:3893.
Chen Y, Huang W-G, Zha D-J, et al. Aspirin attenu- ates gentamicin ototoxicity: from laboratory to the clinic. Hearing Res. 2007;226:178.
Sha SH, Qiu JH, Schacht J. Aspirin to prevent gentamicin-induced hearing loss. N Engl J Med. 2006;354:1856.
Feldman L, Efrati S, Eviatar E, et al. Gentamicin- induced ototoxicity in hemodialysis patients is ameliorated by N-acetylcysteine. Kidney Int. 2007;72(3):359.
Mameli O, Caria MA, Melis F, et al. Neurotoxic effect of lead at low concentrations. Brain Res Bull. 2001;55(2):269.
Gozdzik-Zolnierkiewicz T, Moszynki B. VIIIth nerve in experimental lead poisoning. Acta Otolaryngol. 1969;68:85.
Wilpizeski D. Effects of lead on the vestibular system: preliminary findings. Laryngoscope. 1974;84:1585.
Mizukoshi K, Nagaba M, Ohno Y, et al. Neurotological studies upon intoxication by organic mercury com- pounds. ORL J Otorhinolanyngol Relat Spec. 1975;37:74.
Nosaka Y, Setoguti A, Suko H. Auditory and vestibu- lar disturbances in Minamata disease (in Japanese). Kumamoto Med J. 1958;32:1465.
Moller-Madsen B. Localization of mercury in CNS of the rat. III. Oral administration of methylmer- curic chloride (CH3HgCl). Fundam Appl Toxicol. 1991;16:172.
Anniko M, Sarkady L. Morphological changes of labyrinthine blood vessels following metal poisoning. Acta Otolaryngol Suppl (Stockh). 1977;330:15.
Young YH, Chuu JJ, Liu SH, Lin-Shiau SY. Neurotoxic mechanism of cinnabar and mercuric sulfide on the vestibulo-ocular reflex system of guinea pigs. Toxicol Sci. 2002;67(2):256.
Hunter D, Russell DS. Focal cerebral and cerebel- lar atrophy in a human subject due to organic mer- cury compounds. J Neurol Neurosurg Psychiatry. 1954;17:235.
Takeuchi T, Eto K, Kinjo Y, Tokunaga H. Human brain disturbance by methyhnercury poisoning, focusing on the long-term effect on brain weight. Neurotoxicology. 1996;17:187.
Niklasson M, Moller C, Odkvist LM, et al. Are defi- cits in the equilibrium system relevant to the clinical investigation of solvent-induced neurotoxicity? Scand J Work Environ Health. 1997;23:206.
Herpin G, Gargouri I, Gauchard GC, et al. Effect of chronic and subchronic organic solvents exposure on balance control of workers in plant manufactur- ing adhesive materials. Neurotox Res. 2009;15(2): 179.
Gyntelberg F, Vesterhauge S, Fog P, Isager H, Zillstorff K. Acquired tolerance to organic solvents and results of vestibular testing. Am J Ind Med. 1986;9:363.
Tham R, Bunnfors I, Eriksson B, et al. Vestibuloocular disturbances in rats exposed to organic solvents. Acta Pharmacol Toxicol (Copenh). 1984;54:58.
MorataTC, Nylen P, Johnson AC, Dunn DE. Auditory and vestibular functions after a single or combined exposure to toluene: a review. Arch Toxicol. 1995;69:431.
Niklasson M, Arlinger S, Ledin T, et al. Audio- logical disturbances caused by long-term exposure to industrial solvents. Relation to the diagnosis of toxic encephalopathy. Scand Audiol. 1998;27:131.
![]()
Acquired Disorders Hereditary Disorders Pathology
Diagnosis Management
DISORDERS OF THE CRANIAL VERTEBRAL JUNCTION
Basilar Impression Bony Fusions
Atlantoaxial Dislocation Chiari Malformation
Syringobulbia Diagnosis Management
INHERITED SPINOCEREBELLAR ATAXIA SYNDROMES
Autosomal Dominant Spinocerebellar Ataxia Syndromes
Autosomal Recessive Spinocerebellar Ataxia Syndromes
Episodic Ataxia and Vertigo Syndromes Diagnosis
Management
Permanent hearing loss occurs in about 1 to 3 out of every 1000 live births.1–3 In high-risk neonates, the prevalence is much higher, in the range of 1 in 50 live births. Although <10% of infants are treated in neonatal intensive care units, these newborns account for more than half of all cases of early-onset sensorineural hearing loss.1,4
Congenital deafness is usually recognized during infancy, but a congenital vestibular impairment is not, because the manifestations are more subtle. Children with a vestibular impairment typically learn to use other sensory information to compensate for vestibular loss and thus appear to be asymptomatic and often testnormalonstandardassessments. Congenital deafness has therefore received extensive study, whereas congenital vestibular loss has been relatively neglected. Given the large
number of children with congenital hearing loss, it seems likely that a large number of children also have congenital vestibular loss.
A consequence of congenital vestibular loss is that babies have difficulty learning to crawl and walk. Walking is often delayed to 15 to 18 months or later and the children are often con- sidered clumsy. They typically will participate in all children’s games and activities, although they will not perform well in tasks that require quick turns or excellent balance (for example, gymnastics). As they get older and develop good muscle control, their walking and running appear normal as long as they have good vision. However, if they begin to lose vision, the bal- ance problems become more and more notice- able, even becoming profound if a visual loss becomes severe.
Congenital disorders of the inner ear can be divided into two major categories: acquired and hereditary. Acquired disorders result from
383
development of a normal fertilized egg, and hereditary disorders result from abnormal genes or epigenetic factors.5 A large number of studies have looked at the relative frequency of hereditary and acquired hearing loss and most have found that each category accounts for about 30% to 40% of cases, with the remaining 20% to 40% of cases being due to unknown cause. It must be kept in mind, however, that these studies were all conducted before the recent rapid advances in the identification of genetic causes of hearing loss, so no doubt future studies will find a higher frequency of hereditary hearing loss.
Acquired Disorders
Currently, most cases of acquired severe hearing loss are associated with the perinatal complex of premature delivery, anoxia, and hyperbilirubinemia.1,4,6 A gestation <32 weeks, birth weight <1500 g, prolonged mechanical ventilation for >10 days, and an Apgar score after 5 or 10 min 6 are all significant risk fac- tors for developing permanent hearing loss. Hyperbilirubinemia at a level exceeding the indication for exchange transfusion is another major risk factor. Also, premature infants often receive potential ototoxic medications includ- ing but not limited to aminoglycosides.6
Pre-, peri-, or postnatal infections with toxo- plasmosis, herpes, cytomegalovirus, and rubella can all lead to permanent inner ear damage with hearing loss and vestibular loss. The inci- dence of congenital rubella and associated inner ear damage has markedly decreased since vaccines have been widely used.7–9 Infants born to mothers who acquire rubella in the first trimester of pregnancy may have multiple con- genital defects, including cataracts, patent duc- tus arteriosus, microcephaly, dental defects, and generally impaired growth and develop- ment. Hearing loss is more common than ves- tibular loss (apparently because of the longer critical developmental period). Although less common, the infant’s fully developed inner ear can be damaged by a maternal rubella infec- tion in the last two trimesters. Congenital cyto- megalovirus (CMV) infections are the leading cause of acquired hearing loss in developed countries. In the United States at least 1000 infants/year have hearing loss at birth due to CMV and another 3000–4000 have hearing loss
in childhood.10,11 Most infants with CMV- related hearing loss have no other symptoms or signs of CMV infection. Meningitis is another common cause of unilateral or bilateral auditory and vestibular loss in the postnatal period.4,6
The fetal alcohol syndrome is commonly associated with developmental abnormalities of the inner ear and other craniofacial struc- tures.12,13 Hearing loss, which can be traced to conductive, sensorineural, and central patholo- gies, contributes to delayed speech and language development. Postural imbalance results form both vestibular and cerebellar dysfunction.
Hereditary Disorders
INHERITED SYNDROMES
Although it is beyond the scope of this mono- graph to review all of the hereditary syndromes that may produce sensorineural deafness and vestibular loss, a few common disorders deserve mention (Table 18–1). About 3% to 6% of con- genitally deaf children have Usher’s syndrome, which is the leading cause of the combination of deafness and blindness.14,15 Usher’s syndrome was initially divided into three types on the basis of clinical features, with type 1 showing the most severe early-onset auditory and vestibular loss. Patients with type 2 Usher’s syndrome are born hard of hearing with a sloping sensorineural hearing loss, but vestibular function is normal. Type 3 is characterized by later-onset progres- sive hearing loss with some moderate loss of vestibular function. Visual loss due to retinitis pigmentosa begins in infancy or early childhood with type 1 but can be delayed until the teenage years with types 2 and 3. To date, seven differ- ent genetic loci have been linked to families with the clinical syndrome of Usher’s type 1 while only a single genetic locus has been linked to types 2 and 3 (for review see reference 16).
The protein products of the genes associated with all three Usher types are critical for devel- opment and maintenance of the inner ear ste- reocilia and kinocilia.16 Usher type I proteins cadherin 23 and protocadherin 15 are struc- tural components of the tip links (see Fig. 2-12 in Chapter 2), while Usher type II proteins are key components of the ankle link molecular complex. The actin-based motor protein myo- sin VIIA (Usher syndrome type 1B) is probably
![]()
Table 18–1 Genetic Syndromes with Audiovestibular Loss
Syndrome Mode of Inheritance
Unique Clinical Features Gene(s)
(Chromosome)
Gene Product
![]()
Usher 1B Recessive Retinitis pigmentosa MYO7A (11q) Myosin VIIa
Waardenberg type 1
Dominant White forelock, high nasal
root, hyperplastic eye brows, lateral displacement of medial canthi, heterochromia iridis
PAX3 (2q) Developmental protein
Jervell-Lange- Nielson (Long Q-T syndrome)
Pendred (enlarged vestibular aqueduct)
Dominant Cardiac arrhythmias KCNE1 (21q) K+ channel
Recessive Thyroid goiter PDS (7q) Pendrin
Alport’s X-linked Interstitial nephritis COL4A5 (Xq) Collagen Dominant COL4A4 (2q)
Dominant COL4A3 (2q)
![]()
the general transporter of the Usher proteins to their intended destination in the stereocilia. In the retina, Usher proteins have been local- ized to the cilia that connect the metabolically active inner segment to the photosensitive outer segment of the photoreceptor cell.17
The Dutch ophthalmologist Waardenberg described a syndrome characterized by con- genital hearing loss, white forelock (i.e., white hair that falls on the forehead), unusual eye color, and dystopia canthorum.18,19 The syn- drome has subsequently been expanded to include four subtypes, with type 1 representing Waardenburg’s initial families. With this syn- drome, vestibular function is usually impaired bilaterally and computed tomography (CT) studies of the temporal bone may reveal bony abnormalities of the inner ear. The gene for Waardenburg’s syndrome type 1 and type 3 is PAX-3, one of several genes that are important for controlling development of the face and inner ear. Other Waardenberg genes include MITF and SNA12 (type 2); and EDNRB, EDN, and SOX10 (type 4).19
The Jervell and Lange-Nielsen syndrome (also known as the long Q-T syndrome) is characterized by recessive congenital audiove- stibular loss and cardiac defects. The genes for this syndrome (KCNQ1 and KCNE1) code for potassium channels that allow potassium accumulating in the marginal cells to flow back into the endolymph, maintaining the high
potassium concentration in the endolymph required for endocochlear potentials (see Chapter 2).20–22 This is an important syndrome to recognize since children are at risk for sud- den infant death syndrome (from arrhythmias). Pendred syndrome is an autosomal recessive disorder characterized by the combination of congenital deafness and goiter (usually devel- oping in early puberty).23 It constitutes about 5% of all cases of childhood deafness. The inner ear is often malformed with a “Mondini- like” malformation, including an abnormal cochlea and an enlarged internal auditory canal and vestibular aqueduct. The degree of thyroid dysfunction is variable. The gene for Pendred’s syndrome, SLC26A4, codes for a chloride- iodide transporter (pendrin) critical for both inner ear and thyroid function.24 Mutations in SLC26A4 can also lead to a nonsyndromic autosomal recessive hearing loss associated with an enlarged vestibular aqueduct (see later
discussion).25,26
Alport’s syndrome has a combination of kid- ney and inner ear involvement.19,27 Hereditary nephritis (previously known as Bright’s disease) can have onset from childhood into adulthood and is the reason for about 2% of kidney trans- plants in this country. A bilateral progressive sensorineural hearing loss developing after the first decade is usually mild to moderate, although in rare patients the loss can be profound. Decreased vestibular responses have
also been identified in some patients.27 Mutations responsible for Alport’s syndrome have been found in three different collagen genes, the most common being inherited in an X-linked fashion (see Table 18–1).19,28
NONSYNDROMIC AUDIOVESTIBULAR LOSS
Most childhood deafness occurs in isolation without any other organ involvement and, as noted earlier, most of these cases are thought to be inherited.19,29 Of those with inherited hearing loss, the great majority are inherited in an autosomal recessive fashion. The number of genes associated with nonsyndromic inherited hearing loss is expanding at a rapid rate (Table 18–2). To date, there are more than 60 genetic loci linked to inherited hearing loss in different families. Many of the affected genes have already been identified and the rest will no doubt be identified within the next few years.30 Interestingly, different mutations in the same gene can lead to well-described syndromes or to nonsyndromic hearing loss with either a recessive or dominant pattern of inheritance (compare Tables 18–1 and 18–2). Correlation between the type of mutation and clinical
expression is just beginning to be investigated. As a general rule recessively inherited deafness is prelingual (e.g., present prior to language development) while dominantly inherited deaf- ness is postlingual (e.g, develops after language development). Vestibular loss occurs with many mutations, but most have not been ade- quately evaluated. A better understanding of how mutations in these genes cause audioves- tibular loss will lead not only to improved diag- nostic tests but also to improved understanding of pathophysiology and ultimately to the devel- opment of more effective treatments.
Recessively Inherited Hearing Loss
DFNB1 accounts for about two-thirds of Caucasians with recessively inherited hearing loss.31 It was initially linked to the long arm of chromosome 13 in several large families. The causative gene GJB2 codes for con- nexin-26, a component of a large family of pro- teins involved in gap junctions that are critical for transport of ions between cells in the inner ear.32,33 A frame-shift mutation in GJB2 (35delG) was found in most patients with DFNB1. This mutation has a carrier frequency of approximately 1 in 25 people in the
Table 18–2 Genes Associated with Nonsyndromic Hearing Loss
Disorder | Gene (Chromosome) | Protein | Location | Vestibular Lossa |
Dominant | ||||
DFNA1 | Diaphanous (5q) | Diaphanous protein | Hair cells | No |
DFNA2 | KCNQ4 (5q) | K+ channel | Hair cells | No |
DFNA3 | GJB2 (13q) | Connexin 26 | Gap junctions | No |
DFNA8/12 | TECTA (11q) | Tectorin | Tectorial membrane | No |
DFNA9 | COCH (14q) | Secreted protein | Extracellular matrix | Yes |
DFNA11 | MYO7A (11q) | Myosin VIIa | Hair cells | Yes |
DFNA15 | POU4F3 (5q) | Transcription factor | Inner ear | Yes |
Recessive | ||||
DFNB1 | GJB2 (13q) | Connexin 26 | Gap junctions | No |
DFNB2 | MYO7A (11q) | Myosin VIIa | Hair cells | Yes |
DFNB3 | MYO15 (17p) | Myosin XV | Hair cells | No |
DFNB4 | PDS (7q) | Pendrin | Inner ear | Yes |
DFNB21 | TECTA (11q) | Tectorin | Tectorial membrane | No |
X-linked | ||||
DFN3 | POV3F4 (Xq) | Transcription factor | Inner ear | Yes |
aBased on human or animal studies.
Source: Data from www.boystown.org/hhirr.
Mediterranean area and causes deafness in at least 1 in 2500 newborns in that area.34 Of 52 sequential probands with congenital sen- sorineural hearing loss referred for genetic analysis in the Midwest of the United States,
22 (42%) were found to have mutations in GJB2. The 35delG mutation was found in 29 of the 41 mutant alleles.37 Analysis of 560 controls for the carrier rate was determined to be 3%.
A wide range of auditory phenotypes has been associated with recessively inherited mutations in GJB2. In most, the deafness is congenital and nonprogressive. The typical audiogram is flat with a 50 to 60 dB loss. However, the hearing loss can be progressive and asymmetric with a sloping downward pat- tern.33 So far, only a few patients with the 35delG mutation in GJB2 have been tested for vestibular function and no abnormalities have been found.
Another recessively inherited hearing loss syndrome deserves comment because its com- bination of auditory and vestibular symptoms can mimic acquired disorders such as Meniere’s syndrome and perilymph fistula. As noted ear- lier, mutations in the SLC26A4 can result in Pendred’s syndrome, characterized by a com- bination of thyroid and inner ear dysfunction. Other mutations in the same gene can result in just audiovestibular symptoms associated with an enlarged vestibular aqueduct on imaging.25,35 The clinical picture is that of a childhood-onset bilateral sensorineural hearing loss that fluctu- ates in severity, with episodic worsening being triggered by head trauma or by coughing and sneezing. Vertigo spells can also be triggered by the same factors and there can be a gradual progressive deterioration in both auditory and vestibular function. The vertigo and vestibular dysfunction can be delayed into adulthood, even though the hearing loss was present since childhood. Most of the mutations causing this syndrome have been missense mutations, although deletions and insertions with frame shifts have also been found.
Dominantly Inherited Hearing Loss
Although less common than recessively inherited hearing loss, dominantly inherited hearing loss is more easy to recognize because multiple generations are typically involved.19,29
Depending on the type of mutation, several genes can produce either a recessive or domi- nant disorder (for example, GJB2, MYO7A, and tectorin) (Table 18–2). As a rule, the domi- nantly inherited sensorineural hearing loss dis- orders have a later onset (e.g., postlingual) and are less severe than the recessively inherited disorders. Some of them may even present later in life and be part of what is generally con- sidered presbycusis.
DFNA9 is of particular clinical interest because of the late onset of progressive bilat- eral auditory and vestibular loss.38 Hearing loss is typically noted in the fifth decade with an annual progression of about 3 dB eventually leading to profound bilateral hearing loss. There is a similar progression in vestibular loss, with a complete loss occurring in most patients by the sixth decade. Some patients will show fluctuating hearing loss and episodic vertigo suggestive of Meniere’s syndrome.39 A low- frequency hearing loss pattern can also be rem- iniscent of Meniere’s syndrome. Vestibular involvement is common although often clini- cally silent.36
The gene for DFNA9, COCH, located on 14q codes for a protein that is secreted into the extracellular matrix of the inner ear.40 The mechanism by which mutations in this gene lead to the delayed onset of progressive audio- vestibular loss is still unknown. Postmortem studies of temporal bones from patients with DFNA9 showed an acidophilic mucopolysac- charide-containing ground substance in the cochleas, macules, and cristae as well as some degeneration of vestibular and cochlear sensorineural elements.37,41 These deposits occurred in sites similar to those where COCH gene expression was seen in the normal inner ear.
Maternally Inherited Mitochondrial Disorders
Each mitochondrion contains 2 to 10 mito- chondrial chromosomes, so each cell contains thousands of mitochondrial chromosomes. Each of these mitochondrial DNA molecules is double stranded and forms a closed circle, and replication and transcription occur within the mitochondrion. Mitochondrial DNA encodes 13 messenger RNA genes, 2 ribosomal
RNA genes, and 22 organ-specific transfer RNA genes. The 13 messenger RNAs are translated on mitochondrion-specific ribo- somes with a mitochondrion-specific genetic code into 13 proteins. These mitochondrial- generated proteins interact with about 60 nuclear encoded proteins to form the five enzyme complexes required for oxidative phosphorylation. Mutations in mitochondrial DNA lead to both syndromic and nonsyndro- mic deafness.19,42 Hearing loss with mitochon- drial DNA mutations is typically delayed into adulthood, and there is often a great deal of variability in the degree of hearing loss among different family members. The charac- teristic maternal transmission suggests the pos- sibility of a mitochondrial DNA mutation. An A-to-G transition mutation at nucleotide 3243 in the mitochondrial gene for the leucine transfer RNA can produce a range of clinical syndromes, including mitochondrial encepha- lomyelopathy with lactic acidosis and stroke- like episodes (MELAS) and a combination of diabetes mellitus and hearing loss.43,44 This mutation was found in 2% to 6% of diabetic patients in Japan and in 3 out of 5 patients with diabetes and deafness.45 Twenty-seven of 44 patients with diabetes and the nucleotide 3243 mutation also had hearing loss. None of these cases had the other typical neurologic features of MELAS. In 37 adult patients identified with 3243A>G in Finland, first clini- cal manifestations appearing in childhood included sensorineural hearing loss, delayed maturation, migraine, learning difficulties, and exercise intolerance.46 A homoplasmic mutation at nucleotide 1555 in the mitochon- drial 12S RNA gene can produce a nonsyndro- mic deafness and an increased susceptibility to aminoglycoside-induced deafness in different families.47 Investigation of these families showed that mitochondrial mutations might lead to disease only in the presence of a specific nuclear genotype or some environ- mental factor. Mitochondrial DNA mutations presumably lead to hearing loss by interfering with the high energy requirements of the inner ear. Interestingly, although the 12S RNA gene mutations result in increased susceptibility to aminoglycoside-induce hear- ing loss, vestibular loss does not occur (see Chapter 17).
Inherited Vestibular Loss with Normal Hearing
Some patients presenting with a bilateral vestib- ular loss do not have hearing loss, and the clinical picture does not indicate a structural lesion, immune mediated disorder, or other etiology. In these presentations the most likely cause is an inherited vestibulopathy. In contrast to the many genetic causes of hearing loss, only a few familial bilateral vestibulopathy families have been described.48–50 As mentioned earlier, this dispar- ity is likely explained by difficulty in recognizing the symptoms and signs of a bilateral vestibul- opathy and because quantitative rotational ves- tibular function testing is not widely available. In addition, many patients with a bilateral vestibul- opathy may not present to a physician if they have achieved an acceptable level of compensa- tion or substitution. Patients with inherited bilat- eral vestibulopathy typically have recurrent attacks of isolated vertigo and a high prevalence of migraine headaches.48 Age of onset ranges from the first to sixth decade. Vertigo attacks are often brief in duration (seconds to minutes). Progression of peripheral vestibular function loss will eventually cause imbalance and oscillopsia, typically by the fifth decade. The bedside head- thrust test may show bilateral corrective saccades when vestibulopathy is severe enough; otherwise the rotational chair test is used to identify these patients. As the vestibulopathy becomes more severe, attacks of vertigo become less frequent and eventually cease. Linkage analysis performed in four families with familial bilateral vestibulop- athy maps to a chromosomal locus on 6q.49 This region does not overlap any known autosomal- dominant deafness or migraine syndromes. One additional familial bilateral vestibulopathy family did not link to the region on chromosome 6, thus supporting genetic heterogeneity. A candidate gene, OPRM1, in the region of interest on chro- mosome 6 was sequenced in probands, but no mutations were found.49 A survey of gene expres- sion within the vestibular labyrinth would enhance the ability to identify genetic causes of familial bilateral vestibulopathy.
Pathology
The first pathologic study of an inner ear con- genital malformation was described by Mondini
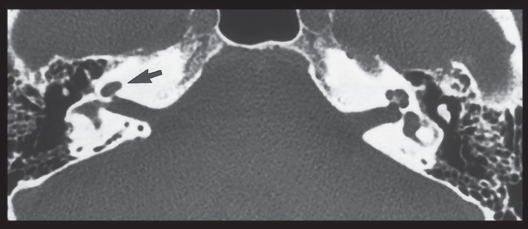
Figure 18–1. Computed tomography scan of the temporal bones showing a Mondini malformation on the right side. Only the basal turn of the cochlea is fully developed (arrow). Cochlea on the left is normal.
in 1791. The Mondini malformation consists of subtotal development of the osseous and membranous labyrinth with only the basal turn of the cochlea being completely formed (Fig. 18–1).51,52 The endolymphatic duct sys- tem is dilated and the vestibular labyrinth is underdeveloped. This deformity occurs with many different syndromes, both hereditary and acquired, and is invariably associated with some (and often complete) loss of auditory and ves- tibular function. Cochleosaccular dysgenesis initially described by Scheibe consists of dys- plasia of the pars inferior (cochlea and saccule) with a fully developed bony labyrinth and normal pars superior (semicircular canals and utricle).53 The Scheibe deformity has classically been associated with congenital rubella, accounting for the relative sparing of vestibular function in many of these children. A rare deformity characterized by complete failure of development of the inner ear (Michel’s defor- mity) is associated total loss of auditory and vestibular function. This deformity has been found in several patients with thalidomide anomalies of the ear.54
Diagnosis
As noted earlier, although inherited loss of auditory and/or vestibular function may be part of a well-described multiorgan syndrome, most cases occur in isolation with an autosomal
recessive mode of inheritance. Even with the well-defined syndromes, variability in gene penetrance can complicate the clinical picture. For example, with Waardenburg’s syndrome, penetrance for deafness is only 20%. Thus, although this dominant disorder is passed to 50%, only 20% of the 50% will be deaf. For nonsyndromic audiovestibular loss, allelism and modifier genes are important for pheno- type expression.55 In the initial assessment of any patient with early-onset hearing loss, one should obtain a detailed family history and search for other family members with possible hearing loss, vestibular loss, or other features characteristic of the syndrome.56
LABORATORY TESTS
Identification of hearing loss or vestibular loss in an infant requires objective measurements, as behavioral testing is usually impractical. Brainstem auditory-evoked responses (BAERs) reflect the electrical activity of the auditory pathways in the brain stem and therefore pro- vide an objective measure of whether the end organ and peripheral nerve generate signals to transmit through the brainstem pathways.57 One must keep in mind, however, that the presence of BAER does not mean that the infant is able to discriminate sounds. Measurement of otoacoustic emissions pro- vides an objective measurement of outer hair cell function. Vestibular evoked myogenic
potentials (VEMPs) provide a rapid objective measure of vestibular function in infants.58 Rotational testing can be performed on infants by rotating the child on the mother’s lap while eye movements are recorded with electro- or videonystagmography. Caloric testing is more difficult to perform but also can be obtained in most infants.58
Most genetic disorders of the inner ear do not affect the otic capsule and therefore the inner ear appears normal both on high-resolu- tion CT scans and on magnetic resonance imag- ing (MRI). Malformations are readily identified both with CT and MR scans of the temporal bone.59 An enlarged vestibular aqueduct in a patient with auditory and vestibular symptoms suggests the likelihood of a mutation in SLC26A3. In patients with fetal alcohol syn- drome, MRI will often show abnormal develop- ment of the cerebellar vermis (lobules I–V).60
GENETIC TESTING
At the present time, the only genetic test for congenital hearing loss that is widely available is for the 35delG mutation in GJB2. Since the carrier rate for this recessive deafness-causing mutation is approximately 3%, screening for the mutation could lead to an early diagnosis in a large percentage of patients with recessively inherited deafness. None of the other muta- tions in GJB2 or in any of the other genes associated with either recessively or dominantly inherited deafness occur with a frequency rate high enough to warrant routine screening. However, specific genes can be selected for based on the following: (1) a characteristic clinical syndrome (e.g., Usher syndrome type IB, MYO7A), (2) recognizable imaging features (e.g., enlarged vestibular aquaduct, SLC26A3); (3) recognizable audioprofile (e.g., DFNA6, WFS1); or (4) prominent vestibular symptoms (e.g., DFNA9, COCH).30 In the future, with rapidly developing technology, “gene chips” will be available to screen a large number of mutations in several genes at the same time.
Management
Congenital hearing loss is obviously important to identify as early as possible because of the rami- fications regarding early language development
and learning abilities. Congenital vestibular loss might explain delayed developmental milestones and early problems with coordination. It is also critical to identify such patients since they need to be counseled about the dangers of drowning when diving into deep water. Infants with sib- lings or parents with known hereditary deafness should be screened for hearing loss. Genetic counseling is an important part of management in these families. Many but not all are candidates for cochlear implants.61
DISORDERS OF THE CRANIAL VERTEBRAL JUNCTION
Cranial vertebral junction disorders are often congenital disorders and many have familial patterns of inheritance. Patients with disorders of the cranial vertebral junction present with a range of brainstem and lower cranial nerve symptoms, including tinnitus, vertigo, hearing loss, pharyngeal dysfunction, hoarseness, or even airway obstruction. The basic pathophysi- ologic mechanism for these symptoms is com- pression of the nervous system at the upper spinal cord and medulla. The rostrocaudal extent of the compression is variable, and the impingement can be ventral, dorsal, or (rarely) both. A second, less common, cause of symp- toms is vascular insufficiency due to angulation, stretching, or extrinsic compression of the anterior spinal or vertebral arteries.
Basilar Impression
Basilar impression is an upward indentation or invagination of the ridged cervical spine into the normally convex skull base.62 The odontoid projects intracranially to compress the ventral aspect of the medulla: the cerebellum is compressed posteriorly by the first and second cervical vertebrae. Disorders known to cause basilar impression include Paget’s disease, rheumatoid arthritis, osteomalacia, osteogenesis imperfecta, cretin- ism, and rickets.63 The term platybasia has been used synonymously with basilar impres- sion by some authors. Technically, it is not a measure of basilar impression and although the two often coexist, platybasia by itself causes no symptoms.
Assimilation of the atlas (also called occipital- ization of the atlas) is a bony union between the first cervical vertebra and the skull.64 The amount of union varies, but motion between the two structures does not occur. As a result, the odontoid impinges on the effective anterior-posterior diameter of the foramen. Frequently, there is associated fusion of the axis to the third cervical vertebra. Many differ- ent varieties of cervicovertebral fusion have been reported. Klippel and Feil initially described a patient with only four cervical ver- tebrae that were fused into a single column of bone. These anatomic features were associated with a clinical triad of short neck, low hair line, and limitation of neck movements. Although partial coalescence of two or more cervical ver- tebrae occurs in many patients, few develop the syndrome originally described by Klippel and Feil. Probably fusion of cervical vertebrae should be called congenital cervical synostosis and the term Klippel-Feil should be used to describe only typical clinical syndromes associ- ated with either complete fusion of the cervical spine or reduction in the number of cervical vertebrae.65 Most cases of Klippel-Feil syn- drome are sporadic, but both dominant and recessive inheritance has been described. Recently mutations in bone morphogenetic protein 13 (BMP13) have been associated with numerous skeletal and developmental defects, including spinal fusion.66 Associated develop- mental anomalies such as split cervical spinal cord and cleft palate are common.67 Hearing loss is common.68 The temporal bone of one such patient showed a vestigial inner ear hav- ing a rudimentary cystic cavity for a cochlea and only one semicircular canal incompletely formed.69 The inner ear abnormalities with Klippel-Feil syndrome may be unilateral or bilateral and may be evident on high-resolution CT scans of the temporal bone.
Atlantoaxial Dislocation
During flexion and extension of the neck, con- genital fusion of the occiput to the atlas increases the strain on the structures that nor- mally restrict the motion of the atlas on the axis, especially if there is fusion of other cervi- cal vertebrae as well.70 The transverse ligament
that normally secures the odontoid against the anterior aspect of the arch of the atlas may weaken because of this repeated strain, and the resultant laxity allows the odontoid to move posterior into the lumen of the foramen mag- num. Flexion or extension of the neck may then produce symptoms, depending on whether the predominant neural compression is anterior from the odontoid or posterior from the poste- rior arch of C1. When the atlas or congenital cervical fusion has been assimilated, the transverse odontoid ligament is sometimes hypoplastic, which makes laxity and atlantoaxial dislocation even more likely. Atlantoaxial insta- bility is also known to be associated with a number of congenital and acquired disease processes. It occurs in 18% to 30% of individu- als with Down’s syndrome and is frequently seen with spondyloepiphyseal dysplasia, Hurler’s syndrome, Morquio’s syndrome, and in achondroplastic dwarfs.70,71 Of patients with rheumatoid arthritis, 25% have atlantoaxial instability secondary to destruction of normal stabilizing mechanisms by inflammatory rheu- matoid tissue in the synovial membrane.72 Similarly, ligamentous laxity can result from inflammatory conditions affecting retropharyn- geal soft tissue or cervical bony structures, such as tuberculosis (or other bacterial) osteitis, retropharyngeal abscess, or lymphadenitis.
Chiari Malformation
In 1895, Chiari described a congenital malfor- mation of the hindbrain in which the brain stem and cerebellum were elongated downward into the cervical canal. Most frequently, the defor- mity manifests itself in the first few months of life and is associated with hydrocephalus and other nervous system malformations (Chiari type II malformation). Less frequent but more important to the neurotologist are those cases in which the onset of symptoms and signs is delayed until adult life (Chiari type I malforma- tion). These cases often present with subtle neurotologic symptoms and signs and are usu- ally not associated with other developmental defects.73–75 The most common neurologic symptom is slowly progressive unsteadiness of gait, which the patient frequently describes as dizziness. Vertigo (particularly positional vertigo), tinnitus, hearing loss, and recurrent facial paresis occur in a small percentage
of patients.75,76 Some patients have fluctuating hearing loss and vertigo suggestive of Meniere’s syndrome.74 Abnormalities on the neurologic exam are generally required before considering low-lying cerebellar tonsils to be a pathological finding. On neurologic examination the patient is ataxic, suggesting midline cerebellar involve- ment. Pathologic nystagmus is nearly always present. Spontaneous and positional downbeat nystagmus are particularly common, but other forms of central spontaneous nystagmus and rebound nystagmus also occur. Oscillopsia is nearly always associated with the spontaneous nystagmus. Dysphagia, hoarseness, and dysar- thria result from stretching of the lower cranial nerves, and obstructive hydrocephalus results from occlusion of the basilar cisterns. Some patients experience episodes of dizziness or even syncope triggered by coughing or sneez- ing. The Chiari type 1 malformation is a devel- opmental disorder of para-axial mesoderm leading to underdevelopment of the posterior cranial fossa and overcrowding of the normally developed hindbrain.73 Although usually spo- radic, both autosomal dominant and recessive families have been described.
Syringobulbia
Syrinx formation in the medulla (syringobul- bia) damages any of the lower cranial nerve nuclei but most often involves the twelfth nuclei and the descending tract and nucleus of the fifth nerve, producing atrophy and fascicu- lations of the tongue and loss of pain and tem- perature sensation on one or both sides of the face. Dysphonia and dysphagia are also preva- lent because of the damage to the ninth and tenth nuclei. As with Chiari malformation, pathologic nystagmus is a common finding in nearly all reported series, and occasionally it is the only abnormal neurologic sign.77 A pure torsional nystagmus either in the primary posi- tion or on lateral gaze is particularly character- istic of syringobulbia. Any adult presenting with oscillopsia and central spontaneous nys- tagmus should be suspected of having either syringobulbia or a Chiari type I malformation.
Diagnosis
Patients with congenital abnormalities of the craniovertebral junction often exhibit
associated morphologic abnormalities of the neck, such as low hairline, short neck, abnor- mal head position, limitation of motion, and painful torticollis. Accentuation of symptoms by coughing, straining, or change in neck posi- tion is common. Clinical manifestations of cervi- comedullary compression are usually relentless and severe, progressing over months to years. Occipital pain with radiation toward the vertex is a common presenting symptom. Other symp- toms can be related to brainstem and cranial nerve dysfunction from compression.
The diagnosis of basilar impression is con- firmed when lateral radiographs of the skull demonstrate that the tip of the odontoid either extends above Chamberlain’s line (a line drawn from the posterior edge of the hard palate to the posterior lip of the foramen magnum) or projects posterior to Wackenheim’s clivus– canal line.70 With assimilation of the atlas or atlantoaxial dislocation, the critical assessment is whether the abnormality is reducible and the direction of encroachment on the cervical medullary junction. High-resolution CT scan- ning is performed in the frontal and lateral pro- jections with the patient’s head in both neutral and extended positions (with the attending neurosurgeon supervising the procedure).78 Magnetic resonance imaging is the procedure of choice for assessing the degree of soft tissue compression and for identifying Chiari malfor- mations and syrinx formation.73,57 Midline saggital sections are ideal for identifying the level of the cerebellar tonsils (Fig. 18–2) and syrinx formation in the medulla and high cervical cord. Vertical descent of the brain due to a cerebrospinal (CSF) leak can mimic a Chiari malformation, but usually there is an associated dural enhancement on MRI after contrast.80
Management
A series of operations have been developed to correct bony deformities at the craniovertebral junction, to eliminate the cervical medullary compression, and to prevent its recurrence. They are designed to reduce the odontoid from its cranial position, to remove any bony liga- mentous or inflammatory soft tissue compres- sion of the cervicomedullary junction, and to fix the skull to the cervicovertebral column in the reduced position when necessary. In patients with Chiari type I malformations,
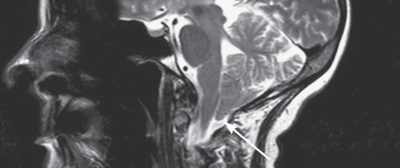
Figure 18–2. Chiari type I malformation. Magnetic resonance image shows a Chiari type I malformation. Arrow points to the tip of the cerebellar tonsils.
suboccipital decompression of the foramen magnum region can stop the progression and occasionally lead to improvement in neurologic symptoms and signs.73,81,83 Special emphasis should be given to patients with rheumatoid arthritis because up to 25% of them have sig- nificant cranial vertebral abnormalities. In a series of 45 patients surgically treated for cervi- cal medullary compression secondary to rheu- matoid arthritis and cranial settling, there were no operative deaths and no infections.83 All the patients improved to a functional class two grades above the preoperative level, and some improvement in cranial nerve function occurred in patients who had preoperative deficits.
INHERITED SPINOCEREBELLAR ATAXIA SYNDROMES
Early classifications of the inherited spinocer- ebellar ataxias (SCAs) were based on a confus- ing array of eponyms and clinical–pathological terms (e.g., Marie ataxia, Holmes ataxia, olivo- pontocerebellar ataxia, parenchymal cerebellar ataxia, etc.). With the rapid advances in genetic analysis at the turn of the century, classifica- tions based on phenotypic descriptions have been replaced with a classification based on the genotype.84 The prevalence of inherited SCA in the general population has not been well studied, although it is probably no more than 1 to 5 per 100,000 in unselected populations. In some isolated populations, however, the preva- lence can be as high as 20 to 25 per 100,000.85
Auditory and vestibular symptoms and signs occur with several of the hereditary SCA syn- dromes. In addition, isolated families with
atypical ataxia syndromes associated with hear- ing loss and abnormal vestibular function have been reported.86–88 Clinically, however, cere- bellar findings usually overshadow the loss of vestibular function; patients present with ataxia and incoordination. In most, the symptoms are slowly progressive, although in some they are episodic. Head movement–induced oscillopsia and dizziness are common, usually because of the patient’s inability to suppress the vestibulo- ocular reflex with fixation, but in some cases, also because of bilateral vestibular loss. Often, only after performing caloric or rotational test- ing is the loss of vestibular function identified. Vertigo is usually not present because the vestibular loss occurs gradually in a bilateral symmetrical fashion, but vertigo is a common feature of several of the episodic ataxia syn- dromes. Many types of pathological nystagmus are encountered, including gaze-evoked, cen- tral spontaneous (particularly upbeat and downbeat), rebound, and central paroxysmal positional. Bedside and laboratory assessment of eye movements can be helpful in identifying the phenotype prior to genetic testing (see later discussion).
Autosomal Dominant Spinocerebellar Ataxia Syndromes
About a third of the inherited SCA syndromes are inherited in an autosomal dominant fashion. Classification of the autosomal domi- nant SCA syndromes has long been a source of confusion and controversy. Harding89 sepa- rated these disorders into types 1 through 3. The most common, type 1, manifested ataxia, pyramidal and extrapyramidal signs, and
Table 18–3 Mutations associated with some common Spinocerebellar Ataxias
![]()
Classification Chromosomal Mutational Mechanisms Characteristic Features Location
![]()
SCA-1 6q CAG: nl 16–37,a abnl 39–81 Dysphagia, pyramidal signs,
vibratory loss
SCA-2 12q CAG: nl 15–32, abnl 34–64 Slow saccades, loss of reflexes,
dementia
SCA-3/MJD 14q CAG: nl 12–40, abnl 66–84 Pyramidal and extrapyramidal
signs, peripheral motorsensory neuropathy
SCA-5 11cent Coding mutations in beta-III
spectrin
Relatively benign course, pyramidal signs in early onset
SCA-6 19p CAG: nl 4–19, abnl 21–29 Predominant cerebellar signs SCA-7 3p CAG: nl 7–17, abnl 38–130 Retinal degeneration
SCA-8 13q CTG: nl < 50, abnl 80–250 Predominant cerebellar syndrome SCA-10 22q ATTCT: nl<280, abnl 850–4,500 Predominant cerebellar syndrome
with epilepsy
![]()
aThere are longer normal SCA-1 alleles, but they carry a CAT interruption. Number of repeats, nl-normal, abnl-abnormal
ophthalmoplegia. Type 2 was similar to type 1 but also included retinal degeneration, while type 3 was a relatively pure cerebellar syn- drome. This classification has been shown to be genetically heterogeneous, composed of a vari- ety of distinct SCA subtypes (Table 18–3).67 To date, 28 autosomal dominant SCA syn- dromes have been linked to chromosomal loci, and 17 causative genes have been identified.90 Spinocerebellar ataxia types 1, 2, 3, and 6 are by far the most common of the SCA syndromes, accounting for more than half of all cases.91 Each of these disorders is caused by an expanded CAG triplet repeat in the open read- ing frame of the gene, leading to an expanded polyglutamine tract in the predicted protein.92,93 The size range of the repeat expansion for each is roughly similar, with less than about 30 repeats being asymptomatic and more than about 40 being symptomatic. The size of the repeat correlates with disease severity and age of onset. Repeat expansion constitutes the molecular basis of anticipation, which typically occurs with paternal transmission. With each generation, the disease tends to come on ear- lier and be more severe. Spinocerebellar ataxia type 6 is the lone exception to this rule, having a smaller stable repeat expansion, although disease severity is correlated with the length of the expansion.94
All of the SCA syndromes share the main clinical features of gait ataxia, dysarthria, dysphagia, dysmetria, and intention tremor due to involvement of the afferent and efferent cerebellar pathways.95 Involvement of other structures, including brain stem and spinal cord, basal ganglia, peripheral nerves, optic nerve and retina, and cerebrum, occurs in indi- vidual syndromes. Disorders of oculomotor control are common with all of the SCA syn- dromes, and the particular pattern can help define the phenotype (Table 18–4).96 The ves- tibular-ocular reflex gain is typically decreased in SCA-3, presumably because of involvement of brainstem vestibulo-ocular reflex pathways. Slowing of both voluntary and involuntary sac- cades is prominent with SCA-1, SCA-2, and SCA-7, so that even though the vestibulo- ocular reflex remains intact, vestibular nystag- mus may not be generated and vestibular test- ing results in a slow deviation of the eyes and pinning in an extreme lateral position because of the absence of fast components.
Autosomal Recessive Spinocerebellar Ataxia Syndromes
Friedreich’s ataxia (FA) is by far the most com- mon of the hereditary ataxias, accounting for
Table 18–4 Summary of Type and Degree of Horizontal Eye Movement Abnormalities with Different Spinocerebellar Ataxia Syndromes
Saccade Velocity | Pursuit/OKN Gain | VOR Gain | VOR-Fix Gain | |
SCA-1 | Moderate | Moderate | Normal | Moderate |
SCA-2 | Severe | Mild | Normal | Mild |
SCA-3 | Mild | Moderate | Moderate | Mild |
SCA-6 | Normal | Severe | Normal | Severe |
SCA-7 | Severe | Severe | Mild | Severe |
OKN, optokinetic nystagmus; VOR, vestibular-ocular reflex; VOR-Fix, fixation suppression of VOR.
Source: Adapted from Buttner et al.96
the majority of the recessively inherited ataxias.97–99 Its prevalence of about 2 per 100,000 equals nearly that of all of the domi- nant ataxia syndromes combined. It was first described by Nicholas Friedreich in 1863.100 He emphasized the progressive ataxia, sensory loss, and muscle weakness often associated with scoliosis, foot deformity, and heart dis- ease. The currently accepted clinical criteria include (1) autosomal recessive inheritance,
onset before age 25 years, (3) progressive limb and gait ataxia, (4) distal loss of position and vibration sense, and (5) absent tendon reflexes in the legs. Cardiomyopathy, kyphosco- liosis, pes cavus, optic atrophy, hearing and vestibular loss, and diabetes mellitus occur in some patients. With the discovery of the gene for FA, it has become clear that the disease can show remarkable clinical variability, sometimes even within the same family.99,101 Age of onset, presence or absence of deep tendon reflexes, and presence or absence of associated features can all be variable according to the type of genetic defect.
Friedreich’s ataxia results from a large expan- sion of a GAA repeat in intron 1 of the gene frataxin.102,103 This gene has 5 exons spread over 40,000 base pairs encoding a 210 amino acid protein designated frataxin. Most patients have the expanded GAA repeat in both alleles, but some have an expanded GAA repeat in one allele and a point mutation in the other, and rare patients have point mutations in both alleles. Frataxin is a mitochondrial protein that is important in iron metabolism.104 Yeast organ- isms deficient in the frataxin homologue accu- mulate iron in mitochondria and have increased sensitivity to oxidative stress. From this observation, it has been suggested that the
neurological degeneration seen with FA results from free-radical toxicity due to mitochondrial damage. Selective cell vulnerability may be due to the levels of respiratory activity (which is high in the brain and heart) and to levels of iron metabolism.
Both auditory and vestibular loss commonly occur with FA, particularly in the later stages of the disease.105,106 Two sisters with FA showed extensive degeneration of the neurons of the eighth nerve (both auditory and vestibular), with preservation of the peripheral receptor organs.107 These changes correlated with the clinical findings of progressive bilateral deaf- ness and caloric hypoexcitability for several years prior to death. Oculomotor testing typi- cally shows prominent saccade dysmetria and ocular flutter superimposed on voluntary and involuntary eye movements.105,108
As with the other trinucleide repeat syn- dromes, larger expansions are correlated with earlier disease onset and more rapid progres- sion.103 The best correlation is seen with the size of the smaller allele, indicating that smaller expansions are consistent with some residual function. The frequencies of cardiomyopathy, flexor plantar responses, and skeletal deformi- ties all increase with increasing GAA repeat number.103
Several FA-like syndromes can result from metabolic disorders that lead to vitamin E defi- ciency. Mutations in the alpha-tocopherol transfer protein (-TTP) on chromosome 8q and mutations in the microsomal triglyceride transfer protein (MTP) on chromosome 4q result in vitamin E deficiency and a clinical syndrome similar to FA.99,109 These syndromes are important to recognize because they can be treated with vitamin E replacement.
Refsum’s syndrome is characterized by a combination of retinitis pigmentosa, bilateral sensorineural hearing loss, cerebellar ataxia, and peripheral neuropathy. Patients with Refsum’s disease have an elevated serum phy- tanic acid, the result of a defect in lipid alpha oxidase.110 The disorder typically begins in the first decade, is slowly progressive, and like FA can be associated with cardiac conduction defects and cardiomyopathy. Refsum’s syn- drome results from missense mutations in the phytanoyl-CoA hydroxylase (PAHX) gene located on chromosome 10.111
Several recessively inherited ataxia syn- dromes are associated with oculomotor apraxia: ataxia telangectasia, ATM gene; ataxia with oculomotor apraxia I, APTX gene; ataxia with oculomotor apraxia II, SETX gene.112–114 Children with these disorders have impaired voluntary and reflexive saccades so that vestib- ular stimulation causes a slow deviation of the eyes without corrective fast components. Refixations are made with head thrusts rather than saccades. How these genes that are pri- marily involved in DNA repair lead to ataxia and oculomotor apraxia is poorly understood.
Episodic Ataxia and Vertigo Syndromes
The episodic ataxias are a group of rare disor- ders characterized by attacks of generalized ataxia with normal or near-normal neurologic function in between.115,116 The attacks typically begin in early childhood or early adulthood. Although a large number of recessive and X-linked causes of episodic ataxia are known, most cases are autosomal dominant. The genes for several dominant forms of episodic ataxia have recently been discovered; this may pro- vide insight into the pathophysiology of other episodic vertigo and ataxia syndromes.116
EPISODIC ATAXIA TYPE 1
Episodic ataxia type 1 (EA-1) is characterized by brief episodes of ataxia (minutes) and interictal myokymia.117 The myokymia (muscle rippling) is typically most evident in the eyelids and fingers. In some cases it can only be identi- fied with electromyography. The gene for EA-1 is a voltage-gated potassium channel gene (KCNA1) located on chromosome 12q.118
A large variety of missense mutations in KCNA1 have been found in families with EA-1 with no mutation being predominant.
EPISODIC ATAXIA TYPE 2
Episodic ataxia type 2 (EA-2) is characterized by more prolonged episodes of ataxia (lasting hours) and interictal nystagmus.119 Often the episodes of ataxia are associated with vertigo and nausea and vomiting. The ataxia can be confined to just the trunk or may involve the upper extremities as well. The interictal nys- tagmus can be gaze-evoked, rebound, or spon- taneous vertical (particularly downbeat). Some patients develop a slowly progressive ataxia later in life. The gene for EA-2 is a calcium channel gene, CACNA1A, located on chromo- some 19p.120 Missense mutations in CACNA1A typically lead to a variety of familial hemiplegic migraine, whereas nonsense mutations result in EA-2.
EPISODIC ATAXIA TYPES 3–7
At least five other autosomal dominant EA syn- dromes have been described in one or two families.116 Genetic analysis ruled out linkage to the EA1 and EA2 loci, and two additional genes have been identified: EA-5, CACNB4, another calcium channel gene and EA-6, SLC1A3, a glutamate transporter gene.121,122 All of the genes identified so far with autosomal dominant episodic ataxia code for ion channels or transporters. Similar genes are good candi- dates for other EA syndromes and for the more common episodic vertigo syndromes, including migrainous vertigo.
Diagnosis
The first step in the diagnosis of one of the inherited SCA syndromes is to recognize the phenotype. This will define the diagnostic workup and the type of DNA testing.95,99 The clinical examination will indicate whether the phenotype is a pure cerebellar syndrome or whether there are combined brainstem, cerebellar, and spinal cord findings. Most patients with an inherited disorder will have a positive family history. It must be kept in mind, however, that a negative family history based on the patient’s report can be deceptive.
Neuroimaging studies, particularly MRI of the brain and upper cervical cord, can help confirm which anatomical structures are involved. Of the common dominant SCA syndromes, SCA-1, -2, -3, and -7 show com- bined cerebellar and brainstem atrophy at least by the mid to late stages, whereas SCA-6 shows a selective cerebellar atrophy, which is most prominent in the vermis.123 With FA, the cerebellum is typically spared, but the spinal cord is atrophic. Cerebellar vermian atrophy is also seen with EA-2 (Fig. 18–3)124 but not with EA-1.
Specific DNA tests are now available for all of the trinucleotide repeat syndromes. The battery of tests can be ordered on any patient with a dominantly inherited SCA syndrome. The GAA expansion with FA should be consid- ered in just about any patient with a recessively inherited ataxia syndrome. Unfortunately at the present time, none of the missense muta- tions, either in the frataxin gene or in the epi- sodic ataxia genes, can be routinely ordered since no single dominant missense mutation has been identified. However, as in the case with the many missense mutations causing nonsyndromic hearing loss, “gene chips” should be available in the near future to screen for the large number of mutations that have been
identified. The genes can be sequenced, but this is expensive especially for a large gene such as CACNA1A.
Management
With a few exceptions, management of patients with inherited ataxia is symptomatic. Patients are encouraged to use a cane or walker to improve sensory input and to avoid falls. Regular physical therapy to maintain range of motion for all joints is critical to avoid painful contractures. A special diet low in long-chain fatty acids can be effective in controlling the progression of symptoms and signs in patients with Refsum’s syndrome.110 Acetazolamide (Diamox) is remarkably effective for relieving the episodic symptoms in patients with EA-2 and, to a lesser degree, in patients with EA-1.115 Acetazolamide is not very effective in patients with other EA syndromes. Acetazolamide pre- sumably works by altering the pH within the cerebellum, thus stabilizing the mutated ion channels. One typically begins with a low dos- age (125 mg/day) and then works up to an aver- age effective dosage of between 500 and 750 mg/day. Most patients will experience par- esthesias of the extremities after taking the drug, but these symptoms typically decrease over time. The main long-term side effect is the development of kidney stones, which may
Figure 18–3. Magnetic resonance image of brain in a patient with episodic ataxia type II (EA-2) showing atrophy of the cerebellar vermis. T1-weighted sagittal section.
markedly decrease if the patient regularly drinks citrus juices. Patients with SCA-6 who have episodic features may also respond to acetazolamide.125 It is unknown whether the long-term slowly progressive ataxia is affected by the regular use of acetazolamide. A thera- peutic trial of acetazolamide should be consid- ered in any patient with early-onset episodic
Smith RJH, Bale JF, White KR. Sensorineural hearing loss in children. Lancet. 2005;365:879.
Colugnati FA, Staras SA, Dollard SC, Cannon MJ. Incidence of cytomegalovirus infection among the general population and pregnant women in the United States. BMC Infect Dis. 2007;7:71.
Church MW, Abel EL. Fetal alcohol syndrome. Hearing, speech, language, and vestibular disorders. Obstet Gynecol Clin North Am. 1998;25:85.
Roebuck TM, Simmons RW, Mattson SN, Riley
vertigo and/or ataxia who has a family history of similar episodes.
Although identifying the genes responsible for the other SCA syndromes is the first step in
EP. Prenatal exposure to alcohol affects the ability to maintain postural balance. Alcohol Clin Exp Res. 1998;22:252.
Kumar A, Fishman G, Torok N. Vestibular and audi-
finding effective treatment, at present no effective treatment exists. Therefore, genetic counseling is critical for patients both before and after genetic tests are ordered.123 Presymptomatic testing should probably be
tory function in Usher’s syndrome. Ann Otol Rhinol
Laryngol. 1984;93:600.
Nikolopoulos TP, Lioumi D, Stamataki S, O’Donoghue GM. Evidence-based overview of ophthalmic disorders in deaf children: a literature update. Otol Neurotol. 2006;27(2 suppl 1):S1.
Saihan Z, Webster AR, Luxon L, Bitner-Glindzicz
restricted to adults, as informed decision mak- ing and psychological counseling can be more effectively employed and the risk of stigmatiza- tion is lessened.
REFERENCES
Vartiainen E, Kemppinen P, Karjalainen S. Prevalence and etiology of bilateral sensorineural hearing impair- ment in Finnish childhood population. Int J Ped Otorhinolaryngol. 1997;41:175.
Hess M, Finckh-Krämer U, Bartsch M, et al. Hearing screening in at-risk neonate cohort. Int J Ped Otorhinolaryngol. 1998;46:81.
Mehra S, Eavey RD, Keamy DG, Jr. The epidemiology of hearing impairment in the United States: newborns, children, and adolescents. Otolaryngol Head Neck Surg. 2009;140(4):461.
Robertson CM, Howarth TM, Bork DL, Dinu IA. Permanent bilateral sensory and neural hearing loss of children after neonatal intensive care because of extreme prematurity: a thirty-year study. Pediatrics. 2009;123(5):e797.
Provenzano MJ, Domann FE. A role for epigenetics in hearing: establishment and maintenance of auditory specific gene expression patterns. Hear Res. 2007;233(1- 2):1.
Streppel M, Richling F, Roth B, et al. Epidemiology of acquired hearing disorders in childhood in the Cologne area. Int J Ped Otorhinolaryngol. 1998;44:235.
Cochi SL, Edmonds LE, Dyer K, et al. Congenitalrubella syndrome in the United States, 1970–1985: on the verge of elimination. Am J Epidemiol. 1989;129:349.
Kadoya R, Ueda K, Miyazaki C, Hidaka Y, Tokugawa
Incidence of congenital rubella syndrome and influence of the rubella vaccination program for schoolgirls in Japan, 1981–1989. Am J Epidemiol. 1998;148:263.
Morice A, Ulloa-Gutierrez R, Avila-Agüero ML. Congenital rubella syndrome: progress and future chal- lenges. Expert Rev Vaccines. 2009;8(3):323.
M. Update on Usher syndrome.Curr Opin Neurol. 2009;22(1):19.
Maerker T, van Wijk E, Overlack N, et al. A novel Usher protein network at the periciliary reload- ing point between molecular transport machineries in vertebrate photoreceptor cells. Hum Mol Genet. 2008;17:71.
Newton VE. Clinical features of the Waardenburg syndromes. Adv Otorhinolaryngol. 2002;61:201.
Kochhar A, Hildebrand MS, Smith RJ. Clinical aspects of hereditary hearing loss. Genet Med. 2007;9(7): 393.
Neyroud N, Tesson F, Denjoy I, et al. A novel muta- tion in the potassium channel gene KVLTQT1 causes the Jervell and Lange-Nielsen cardioauditory syn- drome. Nat Genet. 1997;5:186.
Neyroud N, Tesson F, Denjoy I, Leibovici M, et al. A novel mutation in the potassium channel gene KVLTQ1 causes Jervell Lange-Nielsen cardioauditory syndrome. Nat Genet. 1997;15:186.
Steel KP: The benefits of recycling. Science. 1999;285:1363.
Cremers CWRJ, Bolder C, Admiraal RJC, et al. Progressive sensorineural hearing loss and a wid- ened vestibular aqueduct in Pendred syndrome. Arch Otolaryngol Head Neck Surg. 1998;124:501.
Scott DA, Wang R, Kreman TM, Sheffield VC, Karnishki LP. The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet. 1999;21:440.
Usami S-I, Abe, S, Weston MD, et al. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is causes by PDS mutations. Hum Genet. 1999;104:188.
Maciaszczyk K, Lewin´ski A. Pendred syndrome and hypoacusis with enlarged vestibular aqueduct. Neuro Endocrinol Lett. 2008;29(1):29.
Miller GW, Joseph DJ, Cozad RL, McCabe BE. Alport’s syndrome. Arch Otolaryngol. 1970;92:419.
Barker DF, Hostikka SL, Zhou J, et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science. 1990;248:1224.
Kunst K, Marres H, Van Camp G, Cremers C. Non- syndromic autosomal dominant sensorineural hearing
loss: a new field of research. Clin Otolaryngol. 1998; 23:9.
Hilgert N, Smith RJ, Van Camp G. Forty-six genes causing nonsyndromic hearing impairment: which one should be analyzed in DNA diagnostics? Mutat Res. 2009;681(2-3):189.
Snoeckx RL, Huygen PL, Feldmann D, et al. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet. 2005;77:945.
Kelsell DP, Dunlop J, Stevens HP, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural hearing loss (letter). Nature. 1997;387:80.
Wilcox SA, Osborn AH, Allen-Powell DR, et al. Connexin26 deafness in several interconnected fami- lies. J Med Genet. 1999;36:383.
Zelante L, Gasparini P, Estivill X, et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum Mol Genet. 1997;6:1605.
Das V, Smith R, Choo D, Greinwald J. The influence of mutations in the SLC26A4 gene on the temporal bone in a population with enlarged vestibular aque- duct. Arch Otolaryngol Head Neck Surg. 2007;133(2): 162.
Street VA, Kallman JC, Strombom PD, Bramhall NF, Phillips JO. Vestibular function in families with inher- ited autosomal dominant hearing loss. J Vestib Res. 2008;18(1):51.
Green GE, Scott DA, McDonald JM, et al. Carrier rates in the mid western United States for GJB2 mutations causing inherited deafness. JAMA. 1999; 281:2211.
Robertson NG, Lu L, Heller S, et al. Mutations in a novel cochlear gene COCH cause DFNA9, a human nonsyndromic sensorineural dysfunction. Nat Genet. 1998;20:299.
Fransen E, Verstreken M, Verhagen WI, et al. High prevalence of symptoms of Meniere’s disease in three families with a mutation in the COCH gene. Hum Mol Genet. 1999;8:1425.
Bon SJH, Kemperman MH, De Kok YJM, et al. Progressive cochleovestibular impairment caused by a point mutation in the COCH gene at DFNA9. Laryngoscope. 1999;109:1525.
Khetarpal U, Schuknecht HF, Gacek RR, Holmes LB. Autosomal dominant sensorineural hearing loss: pedi- grees, audiologic findings, and temporal bone findings in two kindreds. Arch Otolaryngol Head Neck Surg. 1991;117:1032.
Fischel-Ghodsian N. Mitochondrial genetics and hear- ing loss. Bull NIDCD-HHIRR. 1996;2:1.
van den Ouweland JM, Lemkes HH, Ruitenbeek W, et al. Mutation in mitochondrial tRNA (Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat Genet. 1992;1:368.
Oka Y, Katagiri H, Yazaki Y, Murase T, Kobayashi T. Mitochondrial gene mutation in islet-cellantibody– positive patients who were initially non–insulin- dependent diabetics. Lancet. 1993;342:527.
Kadowaki T, Kadoaki H, Mori Y, et al. A subtype of diabetes mellitus associated with a mutation of mito- chondrial DNA. N Engl J Med. 1994;330:962.
Uusimaa J, Moilanen JS, Vainionpää L, et al. Prevalence, segregation, and phenotype of the
mitochondrial DNA 3243A>G mutation in children.
Ann Neurol. 2007;62(3):278.
Prezant TR, Agapian JV, Bohlman MC, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deaf- ness. Nat Genet. 1993;4:289.
Baloh RW, Jacobson K, Fife T. Familial vestibulopa- thy: a new dominantly inherited syndrome. Neurology. 1994;44:20.
Jen JC, Wang H, Lee H, et al. Suggestive linkage to chromosome 6q in families with bilateral vestibulopa- thy. Neurology. 2004;63:2376.
Brantberg K. Familial early-onset progressive vestibul- opathy without hearing impairment. Acta Otolaryngol. 2003;123:713.
Holden PK, Linthicum FH, Jr. Mondini dysplasia of the bony and membranous labyrinth. Otol Neurotol. 2005;26(1):133.
Zheng Y, Schachern PA, Cureoglu S, Mutlu C, Dijalilian H, Paparella MM. The shortened cochlea: its classification and histopathologic features. Int J Pediatr Otorhinolaryngol. 2002;63(1):29.
Schuknecht HF. Pathology of the Ear. 2nd ed. Philadelphia: Lea & Febiger; 1993.
Takemori S, Tanaka Y, Suzuki J. Thalidomide anoma- lies of the ear. Arch Otolaryngol. 1976;10:425.
McHugh RK, Friedman RA. Genetics of hearing loss: Allelism and modifier genes produce a phenotypic continuum. Anat Rec A Discov Mol Cell Evol Biol. 2006;288(4):370.
Morzaria S, Westerberg BD, Kozak FK. Evidence- based algorithm for the evaluation of a child with bilateral sensorineural hearing loss. J Otolaryngol. 2005;34(5):297.
Riko K, Hyde ML, Alberti PW. Hearing loss in early infancy: incidence, detection, and assessment. Laryngoscope. 1985;95:137.
Zagólski O. Vestibular-evoked myogenic potentials and caloric tests in infants with congenital rubella. B-ENT. 2009;5(1):7.
Robson CD. Congenital hearing impairment. Pediatr Radiol. 2006;36(4):309.
Sowell ER, Jernigan TL, Mattson SN, et al. Abnormal development of the cerebellar vermis in children pre- natally exposed to alcohol: size reduction in lobes I-V. Alcohol Clin Exp Res. 1996;20:31.
Pau H, Gibson WP. Cochlear implantations in chil- dren with Waardenburg syndrome: an electrophysi- ological and psychophysical review. Cochlear Implants Int. 2006;7(4):202.
Goel A, Bhatjiwale M, Desai K. Basilar invagination: a study based on 190 surgically treated patients. J Neurosurg. 1998;88:962.
Menezes AH. Specific entities affecting the craniocervical region: osteogenesis imperfecta and related osteochon- drodysplasias: medical and surgical management of basi- lar impression. Childs Nerv Syst. 2008;24(10):1169.
Klimo P Jr, Rao G, Brockmeyer D. Congenital anom- alies of the cervical spine. Neurosurg Clin N Am. 2007;18(3):463.
Spillane JD, Pallis C, Jones AM. Developmental abnormalities in the region of the foramen magnum. Brain. 1957;80:11.
Williams LA, Bhargav D, Diwan AD. Unveiling the bmp13 enigma: redundant morphogen or crucial regu- lator? Int J Biol Sci. 2008;4(5):318.
David KM, Copp AJ, Stevens JM, Hayward RD, Crockard HA. Split cervical spinal cord with Klippel- Feil syndrome: seven cases. Brain. 1996;119:1859.
McGaughran JM, Kuna P, Das V. Audiological abnor- malities in the Klippel–Feil syndrome. Arch Dis Child. 1998;79:352.
McLay K, Maran A. Deafness and Klippel-Feil syn- drome. J Laryngol. 1969;83:175.
Smoker WR, Khanna G. Imaging the craniocervical junction. Childs Nerv Syst. 2008;24(10):1123.
Menezes AH. Specific entities affecting the cranio- cervical region: Down’s syndrome. Childs Nerv Syst. 2008;24(10):1165.
Nakano KK, Schoene WC, Baker RA, Dawson DM. The cervical myelopathy associated with rheumatoid arthritis: analysis of 32 patients, with 2 postmortem cases. Ann Neurol. 1978;3:144.
Milhorat TH, Chou MW, Trinidad EM, et al. Chiari I malformation redefined: clinical and radiographic findings for 364 symptomatic patients. Neurosurgery. 1999;44:1005.
Ahmmed AU, Mackenzie I, Das VK, Chatterjee S, Lye RH. Audio-vestibular manifestations of Chiari malfor- mation and outcome of surgical decompression: a case report. J Laryngol Otol. 1996;110:1060.
Levo H, Tapani E, Karppinen A, Kentala E. Chiari malformation in otology practice. Auris Nasus Larynx. ePub ahead of print Apr 29, 2009.
Heuer GG, Gabel B, Lemberg PS, Sutton LN. Chiari I malformation presenting with hearing loss: surgical treatment and literature review. Childs Nerv Syst. 2008;24(9):1063.
Thrush DC, Foster JB. An analysis of nystagmus in 100 consecutive patients with communicating syringo- myelia. J Neurol Sci. 1973;20:381.
Kumar A, Jafar J, Mafu M, Glick R. Diagnosis and management of anomalies of the craniovertebral junc- tion. Ann Otol Rhinol Laryngol. 1986;95:487.
Amer TA, el-Shmam OM. Chiari malformation type I: a new MRI classification. Magn Reson Imaging. 1997;15:397.
Samii C, Mobius E, Weber W, Heienbrok HW, Berlit
P. Pseudo Chiari type I malformation secondary to cerebrospinal fluid leakage. J Neurol. 1999;246:162.
Spooner JW, Baloh RW. Arnold-Chiari malformation: improvement in eye movements after surgical treat- ment. Brain. 1981;104:51.
Menezes AH, Van Gilder JC, Clark CR, el-Khoury
G. Odontoid upward migration in rheumatoid arthri- tis or “cranial settling”: an analysis of 45 patients. J Neurosurg. 1985;63:500.
Sindou M, Gimbert E. Decompression for Chiari type I-malformation (with or without syringomyelia) by extreme lateral foramen magnum opening and expan- sile duraplasty with arachnoid preservation: com- parison with other technical modalities (Literature review). Adv Tech Stand Neurosurg. 2009;34:85.
van de Warrenburg BP, Sinke RJ, Kremer B. Recent advances in hereditary spinocerebellar ataxias. J Neuropathol Exp Neurol. 2005;64(3):171.
Gudmundsson K. The prevalence and occurrence of some rare neurological diseases in Iceland. Acta Neurol Scand. 1969;45:114.
Bogaert L, Van Martin L. Optic and cochleovestibu- lar degenerations in the hereditary ataxias. I. Clinico- pathological and genetic aspects. Brain. 1974;97:15.
Schmidley JW, Levinsohn MW, Manetto V. Infantile X-linked ataxia and deafness: a new clinicopathologic entity? Neurology. 1987;37:1344.
Melberg A, Dahl N, Hetta J, et al. Neuro imagingstudyinautosomaldominantcerebellarataxia, deafness, and narcolepsy. Neurology. 1999; 53:2190.
Harding AE. The clinical features and classification of the late onset autosomal dominant cerebellar ataxia: a study of 11 families, including descendants of the Drew family of Walworth. Brain. 1982:105:1.
Carlson KM, Andresen JM, Orr HT. Emerging pathogenic pathways in the spinocerebellar ataxias. Curr Opin Genet Dev. 2009;19(3):247.
Moseley ML, Benzow KA, Schut LJ, et al. Incidence of dominant spinocerebellar and Friedreich trip- let repeats among 361 ataxia families. Neurology. 1998;51:1666.
Timchenko LT, Caskey CT. Triplet repeat disorders: discussion of molecular mechanisms. Cell Mol Life Sci. 1999;55:1432.
Brouwer JR, Willemsen R, Oostra BA. Microsatellite repeat instability and neurological disease. Bioessays. 2009;31(1):71.
Baloh RW, Jen JC. Episodic ataxia type 2/spi- nocerebellar ataxia type 6. In: Klockgether T, ed. Neurological Ataxia. New York: Marcel Dekker; 2000.
Schöls L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical fea- tures, genetics, and pathogenesis. Lancet Neurol. 2004;3(5):291.
Buttner N, Geschwind D, Jen JC, et al. Oculomotor phenotypes in autosomal dominant ataxias. Arch Neurol. 1998;55:1353.
Harding AE. Friedreich’s ataxia: a clinical and genetic study of 90 families with an analysis of early diagno- sis criteria and intrafamilial clustering of clinical fea- tures. Brain. 1981;104:589.
Leone M, Brignolio F, Rosso MG, et al. Friedreich’s ataxia: a descriptive epidemiological study in an Italian population. Clin Genet. 1990;38:161.
Fogel BL, Perlman S. Clinical features and molecu- lar genetics of autosomal recessive cerebellar ataxias. Lancet Neurol. 2007;6(3):245.
Friedreich N. Über degenerative Atrophie der spi- nalen Hinterstränge. Virchows Arch Pathol Anat. 1863;27:1.
Montermini L, Richter A, Morgan K, et al. Phenotypic variability in Friedreich ataxia: role of the associated GAA triplet repeat expansion. Ann Neurol. 1997;41:675.
Campuzano V, Montermini L, Moltó MD, et al. Friedreich ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423.
Pandolfo M. Molecular pathogenesis of Friedreich ataxia. Arch Neurol. 1999;56:1201.
Cavadini P, O’Neill HA, Benada O, Isaya G. Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia, Hum Mol Genet. 2002;11: 217.
Fahey MC, Cremer PD, Aw ST, et al. Vestibular, saccadic and fixation abnormalities in genetically confirmed Friedreich ataxia. Brain. 2008;131(pt 4):1035.
López-Díaz-de-León E, Silva-Rojas A, Ysunza A, Amavisca R, Rivera R. Auditory neuropathy in Friedreich ataxia. A report of two cases. Int J Pediatr Otorhinolaryngol. 2003;67(6):641.
Spoendlin H. Optic and cochleovestibular degenera- tions in the hereditary ataxias. II. Temporal bone his- topathology in two cases of Friedreich’s ataxia with vestibulo-cochlear disorders. Brain. 1974;97:41.
Furman JM, Perlman S, Baloh RW. Eye move- ments in Friedreich’s ataxia. Arch Neurol. 1983;40: 343.
Ouahchi K, Arita M, Kayden H, et al. Ataxia with isolated vitamin E deficiency is caused by mutations in alpha-tocopherol transfer protein. Nat Genet. 1995;9:141.
Herndon JH, Steinberg D, Vhlendorf BW. Refsum’s disease: defective oxidation of phytanic acid in tissue cutures derived from homozygotes and heterozy- gotes. N Engl J Med. 1969;281:1034.
Jansen GA, Waterham HR, Wanders RJA. Molecular basis of Refsum disease: sequence variations in phytanoyl-CoA hydroxylase (PHYH) and the PTS2 receptor (PEX7). Hum Mutat. 2004;23:209.
McKinnon PJ. ATM and ataxia telangiectasia. EMBO Rep. 2004;5:772.
Le Ber I, Moreira MC, Rivaud-Pechoux S, et al. Cerebellar ataxia with oculomotor apraxia type 1: clinical and genetic studies, Brain. 2003;126:2761.
Le Ber I, Bouslam N, Rivaud-Pechoux S, et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain. 2004;127:759.
Baloh RW. Periodic and progressive ataxias. In: Rose MR, Griggs R, eds. Channelopathies of the Nervous System. London: Butterworth-Heinamann; 2001.
Jen JC, Baloh RW. Familial episodic ataxia: a model for migrainous vertigo. Ann NY Acad Sci. 2009;1164:252.
Brunt ER, van Weerden TW. Familial paroxysmal kinesigenic ataxia and continuous myokymia. Brain. 1990;113:1361.
Browne DL, Gancher ST, Nutt JG, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136.
Jen JC, Kim GW, Baloh RW. Clinical spectrum of episodic ataxia type 2. Neurology. 2004;62:17.
Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hémiplégie migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNA1A. Cell. 1996;87:543.
Escayg A, MacDonald BT, Meisler MH, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. 2000;24:343.
Jen JC, Wan J, Palos TP, et al. Mutation in the glu- tamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. 2005;65:529.
Pulst S-M, Perlman S. Hereditary ataxias. In: Pulst S- M, ed. Neurogenetics. New York: Oxford University Press; 2000: 231.
Baloh RW, Yue Q, Furman JM, Nelson SE. Familial episodic ataxia: clinical heterogeneity in four families linked to chromosome 19p. Ann Neurol. 1997;41:8.
Jen JC, Yue Q, Karrim J, et al. Spinocerebellar ataxia type 6 with positional vertigo and acetazolamide- responsive episodic ataxia. J Neurol Neurosurg Psychiatry. 1998;65:565.
This page intentionally left blank
![]()
![]()
This page intentionally left blank
![]()
VESTIBULAR SUPPRESSANTS
How Do They Work? How to Use Them Indications Precautions
What to Tell the Patient
ANTIEMETIC DRUGS
How Do They Work? How to Use Them Precautions
What to Tell the Patient
SPECIFIC DRUGS
Scopolamine (Transderm Scop) Buclizine Hydrochloride (Bucladin-S)
Diphenhydramine Hydrochloride (Benadryl) Meclizine (Antivert, Bonine) Dimenhydrinate (Dramamine) Promethazine Hydrochloride (Phenergan) Betahistine (Serc)
Metoclopramide (Reglan)
Benzquinamide Hydrochloride (Emete-con) Trimethobenzamide Hydrochloride (Tigan) Diazepam (Valium)
Droperidol (Inapsine) Diphenidol (Vontrol) Prochlorperazine (Compazine) Dronabinol (Marinol)
Vertigo is an unpleasant sensation that is frequently accompanied by nausea and vomiting. These symptoms can result from a variety of diseases affecting the vestibular system (see Chapters 9–18). The best therapy is to elimi- nate vertigo with specific treatment of the underlying disease. However, in cases in which the disease is not readily treatable, when treat- ment must be continued for a long period before improvement occurs or when vertigo is prolonged and severe, there is a need for symp- tomatic therapy.
The ideal symptomatic medication should suppress the sensation of vertigo, help restore normal balance, and prevent vomiting. There should be minimal side effects, and the treat- ment should not impede the normal process of recovery from the vestibular lesion. At the present time, there is no medication available that would meet these objectives. In the absence of the ideal drug, the choice of thera- pies should take into account the patient’s
underlying disease, the expected course of the disease, and the patient’s need for mobility during recovery.
Two general classes of drugs are used to treat vertigo, nausea, and vomiting: vestibular suppressants and antiemetics.1 Vestibular sup- pressants treat a variety of symptoms associ- ated with vestibular illness, usually reducing the sensation of vertigo and associated nausea and vomiting. Antiemetics are more selective in action; they are used primarily to reduce nausea and vomiting associated with many illnesses and can also be used in combination with vestibular suppressants for treating vertigo.
The main types of vestibular suppressants include antihistamines, anticholinergics, and benzodiazepines, although a variety of other drugs have also been used (Table 19–1). Abundant empirical evidence demonstrates the efficacy of these medications in the treat- ment of vertigo. Although the exact mechanism
405
406
406
Clinical Neurophysiology of the Vestibular System
Clinical Neurophysiology of the Vestibular System
Table 19–1 Main Actions of Commonly Used Antivertiginous and Antiemetic Drugs
Class | Drug | Histamine Antagonist | Acetylcholine Antagonist | Dopamine Antagonist | Serotonin GABA Agonist Opiate Agonist Antagonist | |
Antihistamine | Diphenhydramine | + | + | |||
Meclizine | + | + | ||||
Cyclizine | + | + | ||||
Buclizine | + | + | ||||
Promethazine | + | + | + | |||
Anticholinergic | Scopolamine | + | ||||
Benzamide derivative | Trimethobenzamide | + | + | |||
Benzquinamide | + | + | ||||
Metoclopramide | + | + | + | |||
Phenothiazine | Chlorpromazine | + | + | |||
Perphenazine | + | + | ||||
Prochlorperazine | + | + | ||||
Butyrophenone | Droperidol | + | + | |||
Antiserotonergic | Ondansetron | + | ||||
Benzodiazepine | Diazepam | + | ||||
Lorazepam | + | |||||
Cannabinoid | Dronabinol | + | ||||
Other | Diphenidol | + | + | |||
Domperidone | + | + | ||||
![]()
(Cannabinoids) (Benzodiazepines)
![]()
Chemoreceptor trigger zone
(Antidopaminergics)
![]()
G.I. tract
Emetic center
Vestibular nuclei
![]()
(Antiseritonergics) (Antihistamines) (Anticholinergics)
(Antihistamines) (Anticholinergics) (Benzodiazepines) (Antidopaminergics?)
Figure 19–1. Schematic drawing of the major inputs to the emetic center and probable site of action of different classes of drugs. G.I., gastrointestinal.
of action of many of these drugs is unclear, most appear to act at the level of the neu- rotransmitters involved in propagation of impulses from primary to secondary vestibular neurons and in maintenance of tone in the ves- tibular nuclei. Antiemetic drugs are directed against the areas of the nervous system control- ling vomiting (Fig. 19–1).2 This system of neu- rons contains central components (loosely described as the emetic center) and peripheral components in the gastrointestinal tract.3 Although the exact pathways mediating vestib- ular-induced vomiting are not completely known, patients who have a baseline sensitivity to motion (i.e., low threshold for developing nausea and vomiting) are often more affected by nausea and vomiting in the setting of a ves- tibular disorder.4 Many of the vestibular suppressants also have antiemetic action and vice versa, and many of the antiemetic drugs produce vestibular suppression.
The study and understanding of neurotrans- mitters within the vestibular nuclei is in its infancy (see Chapter 3). New information on the neurotransmitters will lead to new and bet- ter vestibular suppressants and a better under- standing of how vestibular suppressants work. For example, substantial evidence shows that the main neurotransmitter between primary and secondary vestibular neurons is an excit- atory amino acid, either glutamate or aspartate, acting on kainate receptors.5,6 Despite this information, the therapeutic potential of drugs
that block or otherwise modulate excitatory amino acid receptors has been limited. Possibly because of the widespread distribution of excit- atory amino acid receptors in the central ner- vous system (CNS), therapeutic benefits that could be derived from systemic administration of antagonists of the excitatory amino acid receptor are offset by generalized side effects.
How Do They Work?
As noted earlier, vestibular suppressants are thought to work by depressing transmission of vestibular signals through the vestibular nuclei to the emetic center and other brainstem auto- nomic centers. As shown in Table 19–1, these drugs typically have multiple actions. The transmitters that they affect are not specific to the vestibular system but are present through- out the CNS. This may account for the many associated side effects. Acetylcholine is an excitatory neurotransmitter in the vestibular system; muscarinic receptors in the vestibular nuclei are the presumed site of action for anti- cholinergic vestibular suppressants.6,7 A major histaminergic system projects to the vestibular nuclei and brainstem autonomic centers.8,9 All three histamine receptors are expressed in the vestibular nuclei and in the vestibular periphery.10,11,12 H1and H2 receptors are post-
synaptic, while H3 receptors are presynaptic. Most antihistamines used in the treatment of
vertigo are H1 blockers, but they also have anti- cholinergic actions. The benzodiazepines are agonists of -aminobutyric acid (GABA), which
is the primary inhibitory neurotransmitter for vestibular neurons.13 Furthermore, H3 receptors
modulate GABAergic transmission in the ves- tibular nuclei.14 Dopamine receptors have also been identified within the vestibular nucleus complex,15 but it is unclear whether antidop- aminergic drugs have a direct effect on the vestibular nuclei or affect other centers that influence the vestibular nuclei. Finally, sero- tonergic neurons in the dorsal raphe nucleus project directly to the vestibular nuclei.16
How to Use Them
Vestibular suppressants can be used acutely to treat a discrete attack or chronically as prophy- laxis against future attacks. To decrease the risks of unacceptable side effects, the choice of drug and administration must be determined by the underlying vestibular problem. Acute severe attacks of vertigo accompanied by severe nausea and vomiting are the most distressing form of vestibular illness. Vestibular suppres- sants are useful only for attacks that last long enough for the drug to reach an effective blood level before the attack ends. This limits acute therapy to attacks lasting at least an hour. Because maximum vestibular suppression may not be reached for 2 or more hours after dos- ing, treatment is most useful for attacks that last several hours. In general, the stronger sup- pressants are more sedating and should be reserved for acute treatment. Brief vertigo spells, such as those associated with benign positional vertigo or vertebrobasilar insuffi- ciency, cannot be controlled by ingestion of a suppressant at the time of the attack.
Chronic prophylactic treatment with vestib- ular suppressants should be avoided because of potential problems with tolerance and depen- dency. Withdraw symptoms such as motion sensitivity and nausea commonly occur and may be misinterpreted as due to the underly- ing disorder. The need to control vertigo must be weighed against the need for the patient to maintain full mobility and function. Patients who complain of almost continuous weeks to months of dizziness should not be managed with vestibular suppressants. Such a complaint suggests a diagnosis other than a specific ves- tibular disorder. Vestibular suppressants are not useful in treating chronic dizziness of non- vestibular origin.
A sudden loss of peripheral vestibular func- tion on one side is a common disorder with
many different causes, but regardless of the cause, central compensation will occur and the patient will recover. Vestibular suppressants can impair the process of central compensation.17,18 Patients are often extremely vertiginous with severe nausea and vomiting for the first few days after the vestibular lesion, and it is appropriate to use strong vestibular suppressants in that time frame. However, as soon as the vomiting ceases, vestibular suppressants should be with- drawn gradually to allow compensation.
The commonly used antihistamine vestibular suppressants require 20 to 30 min to initiate action, reach a peak plasma level in 1 to 2 hr, and have a half-life of about 8 hr. Therefore, dosing can be as infrequent as twice a day. Transdermal scopolamine is programmed to deliver through the systemic circulation 0.5 mg of scopolamine at a constant rate each day over the 3-day life- time of the system. When used as a prophylaxis for motion sickness, the patch should be in place several hours before exposure to motion.
Indications
The less sedating drugs are milder vestibular suppressants than the more sedating drugs (Table 19–2). The less sedating medications, such as scopolamine and meclizine, are partic- ularly useful for treating chronic recurrent attacks of vertigo during which the patient is attempting to carry on normal activities, includ- ing work. The more sedating medications, such as promethazine, the benzodiazepines, and droperidol, are particularly effective for an acute severe attack of vertigo, nausea, and vomiting when the patient desires sedation and rest. For similar reasons, the less sedating medications are used in the prophylaxis of motion sickness.
Precautions
Because drowsiness and disorientation can occur with all of these drugs, patients should be warned about engaging in tasks requiring mental alertness. All medications with anticho- linergic properties should be used with caution in patients with glaucoma, pyloric obstruction, or urinary bladder neck obstruction and who are suspected of having intestinal obstruction. These drugs should be used with caution in
Table 19–2 Indications for Use of Common Vestibular Suppressants
![]()
![]()
19 Antiemetic and Antivertigo Drugs
19 Antiemetic and Antivertigo Drugs
LESS SEDATING MORE SEDATING
![]()
Type of Vertigo
Acute periph- eral vertigo
Chronic recurrent peripheral vertigo
Central
vertigo Prevent
409
409
motion sickness
Scopolamine Meclizine Cyclizine Buclizine Diphenhydramine Promethazine Diazepam Lorazepam Droperidol
+ + + +
+ + + + + +
+ + +
+ + + + + +
![]()
patients using CNS depressant drugs or con- suming alcohol because the additive effects on the CNS may cause adverse reactions. As noted earlier, withdrawal symptoms can occur after discontinuing any of these medications but are particularly prominent with the benzodiaz- epines and scopolamine.19 These symptoms include dizziness, nausea and vomiting, headaches, and disturbances of equilibrium, all of which may be similar to the symptoms that initially triggered the use of the drug. Withdrawal symptoms, of course, are most likely to occur in patients who have been on the medication for many days. Abrupt with- drawal of the benzodiazepines can lead to gen- eralized seizures. Addiction-prone individuals should be under careful surveillance while receiving benzodiazepines because of the pre- disposition of such patients to habituation and dependence.
Because of the multiple effects of each of these drugs, possible drug interactions should always be considered before use. The drugs with anticholinergic properties and the benzo- diazepines are contraindicated in patients with glaucoma unless they are receiving appropriate therapy. The antihistamines should not be used in patients with asthma, emphysema, chronic pulmonary disease, or difficulty in urinating because of an enlarged prostate. The benzodi- azepines are generally not recommended for children younger than 6 months. Transdermal scopolamine should not be used in children because it is unknown whether the amount of scopolamine released will produce serious adverse effects in children. Extra caution should be taken when treating elderly patients because of a higher risk of drug-related prob- lems or toxic effects.20 Medicines with no or fewer anticholinergic properties (e.g., prochlo- rperazine) are favored over medicines with more anticholinergic properties (e.g, promet- hazine). Short-acting sedating medicines (e.g., lorazepam) at low doses are favored over long- acting sedating medicines (e.g., diazepam). Because of its potential for producing memory loss, hallucinations, and alterations in mental status, scopolamine should be used with extreme caution in older people.
All of these drugs should be used with cau- tion in patients with renal or hepatic impair- ment because of their multiple actions, particularly their anticholinergic properties. Meclizine and dimenhydrinate are considered
safe to use during pregnancy.21 An increased risk of congenital malformations associated with diazepam during the first trimester of pregnancy has been suggested in several stud- ies. Small doses of benzodiazepines may be safe later in pregnancy.21 Diazepam is excreted in breast milk and, because neonates metabo- lize diazepam more slowly than adults, accu- mulation of the drug and its metabolites to toxic levels is possible.
What to Tell the Patient
These medications are used to suppress vertigo and the commonly associated nausea and vom- iting. They treat the symptoms but do not address the underlying cause of the vertigo. It is not a good idea to use vestibular suppressants on a chronic basis because they can interfere with the brain’s ability to compensate for the underlying problem. These medications typi- cally are sedating, so at least initially they should not be used when performing activities that require a high level of alertness, such as driving, operating machinery, or performing athletic activities. Patients should not take other drugs without discussing it with the doctor. Avoid excessive alcohol consumption since the effects could be additive.
The antiemetic drugs are used to suppress nau- sea and vomiting regardless of the cause. They can be used with vestibular suppressants when treating vertigo, nausea, and vomiting due to vestibular lesions or they can be used in isola- tion when treating nausea and vomiting due to other causes. These drugs are directed at the areas of the nervous system controlling vomiting (Fig. 19–1). Dopamine, histamine, acetylcholine, and serotonin are all neurotrans- mitters believed to act on these sites to produce vomiting.22 The emetic center is not a discrete localized area in the brain. The final process involved in emesis is coordinated in the reticu- lar formation of the brain stem, but input is received from numerous areas of the brain stem and cortex.23 The chemoreceptor trigger zone, located in the area postrema, is a major relay in triggering emesis.
Many antiemetics act by suppressing activity in the reticular formation or area postrema. Others suppress major centers feeding into the chemoreceptor trigger zone or the emesis cen- ter. Still others suppress activity in visceral afferents running from the gastrointestinal to the emetic center. Each of the antiemetic drugs probably have multiple sites of action within the CNS (Fig. 19–1). The cannabinoids and benzodiazepines are thought to work at the level of the cerebral cortex, controlling input to the brainstem vomiting center.24 The antidop- aminergics block input to the emetic center from the chemoreceptor trigger zone located in the area postrema.25 Serotonin antagonists, particularly antagonists to the 5-HT3 receptors,
block the activity of visceral afferents that run
from the gut to the emetic center and the area postrema.26 Newer 5-HT3 receptor antagonists
and Neurkinin-1 (NK-1) antagonists work
centrally although the exact mechanism of action is unclear. Finally, antihistamines and anticholinergics appear to work directly on the emetic center.
How to Use Them
The indications for using any of the common antiemetic drugs are summarized in Table 19–3. The antihistamines, anticholinergics, and benzodiazepines are most effective when treat- ing nausea and vomiting associated with vestib- ular lesions; the cannabinoids, antidopaminer- gics, and antiserotonergics are most useful in treating nausea and vomiting associated with surgery, chemotherapy, and radiation treat- ment. Through its antidopaminergic and antise- rotonergic actions, metoclopramide suppresses the central chemoreceptor trigger zone and diminishes the activities of visceral afférents in the gut;28 it is particularly effective for treating postoperative nausea and vomiting and in com- bination with other agents for treatment of postirradiation and chemotherapy-induced nausea and vomiting.29 Ondansetron is a well-tolerated selective blocker of 5-HT3 that has been effec-
tive for treating chemotherapy-induced nausea
and vomiting.30 Palonosetron is a newer 5HT
drugs in the NK-1 receptor agonist class.27 The benzodiazepines, as a single agent, are of limited use for treating nausea and vomiting, but they can be used in combination with other antiemetic drugs because of their action on the central emetic centers and their ability to reduce akathisia and anxiety.31 Of the vestibular suppressants, promethazine and droperidol are the most effective antiemetic drugs.32
Domperidone is chemically unrelated to the butrophenones, phenothiazines, or metoclopr- amide; however, it shares pharmacologic prop- erties with these agents. In the medulla, the drug produces a direct blocking effect of dopamine receptors in the chemoreceptor trig- ger zone. Like metoclopramide and haloperi- dol, domperidone is a peripheral dopamine antagonist; however, it does not cross the blood–brain barrier and produce CNS effects.
Precautions
Extrapyramidal symptoms and signs occur with use of phenothiazines and butyrophenones, and they may be confused with CNS signs of undiagnosed primary disease responsible for the vomiting.33 Tardive dyskinesia, a syndrome consisting of potentially irreversible involun- tary dyskinetic movements, also commonly occurs with using these drugs. The risk of developing the syndrome and the likelihood that it will become irreversible are believed to increase as duration of treatment and total cumulative dose administered to the patient increase. Potentially fatal symptoms often referred to as neuroleptic malignant syndrome have been associated with phenothiazines and butyrophenones.34 Alcohol should be avoided with all of these drugs because of possible addi- tive side effects. Because of the multiple effects of each of these drugs, possible drug interac- tions should always be considered before use.
The antidopaminergic antiemetic drugs should be used with great caution in children because there is a high incidence of extrapyra- midal side effects in children. Although the incidence of extrapyramidal side effects is less than 1%–2% in adults, it can be as high as 25% in children.35,36 These reactions frequently
receptor antagonist with a higher
3
binding
develop during the first few days of treatment.
affinity and longer half-life than ondansetron.27 Aprepitant and casopitant are new antinausea
Sometimes the drugs can be restarted at a lower dose once the acute reaction subsides.
![]()
412 ClinicalNeurophysiologyoftheVestibularSystem
![]()
![]()
+
+
+
+
Domperidone
+
+
+
+
Dephenidol
+
+
+
+
Dronabinol
+
+
+
+
+
+
Ondansetron
+
+
+
+
+
+
Droperidol
+
+
+
+
+
+
+
+
Prochlorperazine
+
+
+
+
Perphenazine
IndicationsforUseofCommonAntiemeticDrugs
IndicationsforUseofCommonAntiemeticDrugs
+
+
+
+
Chlorpromazine
+
+
+
+
+
+
Metoclopramide
+
+
Benzoquinamide
+
+
Trimethobenzamide
+
+
+
+
Promethazine
Table19–3
Table19–3
TypeofNausea andVomiting
TypeofNausea andVomiting
Vestibular induced Postoperative
Radiation induced Chemotherapy induced
Vestibular induced Postoperative
Radiation induced Chemotherapy induced
+
+
+
+
Scopolamine
Chlorpromazine should be administered cautiously to persons with hepatic disease because patients with a history of hepatic encephalopathy from cirrhosis may have an increased sensitivity to the CNS effects of chlorpromazine. Most of the antidopaminergic drugs are excreted at least in part by the kidneys and therefore should be used with cau- tion in patients with renal failure. Most of these drugs have not been assessed adequately regarding their safety during pregnancy, and therefore use during pregnancy must be weighed against possible hazards to mother and child.
These drugs are used to suppress nausea and vomiting but do not treat the underlying cause. Patients should understand the symptoms of extrapyramidal side effects and with tardive dyskinesia so that they can report them to the physician immediately if any become evident. If a child shows any type of unusual behavior or excessive excitement, the drug should be with- held and the symptoms should be immediately reported to the physician. As with all of these medications, adults should not drive or operate machinery until the full effects of drug are real- ized. Sedation may range from mild drowsiness to severe sleepiness.
Scopolamine (Transderm Sco¯ p)
Scopolamine transdermal system is an anticho- linergic agent classified as a belladonna alka- loid that is most useful for prevention of nausea and vomiting associated with motion sickness. Because of its short half-life in plasma and dose-dependent side affects, oral or parenteral administration is of limited use. The transder- mal system of scopolamine is programmed to deliver 0.5 mg at a constant rate (5 µg/hour) to the systemic circulation over a 3-day lifetime of the system.37 To be most effective in
preventing motion sickness, the system should be applied about 8 to 12 hr before exposure to motion. The scopolamine transdermal system should be used with great caution in older people because they are particularly susceptible to the CNS side effects, such as memory loss, delusions, and hallucinations. Drug withdrawal symptoms, including dizziness, nausea, vomiting, headaches, and disturbances of equilibrium, can occur after discontinuation of the transdermal sys- tem, particularly if it has been used for several weeks.19 Sometimes these withdrawal symp- toms are confused with the symptoms that ini- tiated the use of the transdermal scopolamine system.
Buclizine Hydrochloride (Bucladin-S)
Buclizine hydrochloride is a vestibular sup- pressant and antiemetic agent that is useful for treating vertigo due to vestibular lesions and for preventing motion sickness. It is a piperazine-type antihistamine with anticho- linergic properties. Like the other antihista- mine anticholinergic drugs, sedation and dry- ness of the mucous membranes are the most common side effects. The tablets can be taken without swallowing water by placing the tablet under the tongue and allowing it to dissolve. This route of administration can be useful when the patient is having severe nausea and vomiting.
Diphenhydramine Hydrochloride (Benadryl)
Diphenhydramine hydrochloride is a vestibu- lar suppressant and antiemetic agent with mod- erate sedating properties. It is an ethanolamine antihistamine that is also an anticholinergic agent. It is most useful for treating mild to moderate episodes of vertigo and for preven- tion of motion sickness. The most common side effects are dryness of the nose, throat, and mouth and moderate sedation. Diphen- hydramine is often effective for treating the extrapyramidal side effects that occur with the antidopaminergic drugs.38
Meclizine is a vestibular suppressant and anti- emetic agent that is less sedating than most of the other vestibular suppressant drugs.39 It is a piperazine-type antihistamine that also has anticholinergic properties. Peak effect is not reached until 7 to 9 hr after ingestion. It is par- ticularly useful for treating mild to moderate episodes of vertigo and for suppressing motion sickness. It should be taken several hours before exposure to motion to be effective in preventing motion sickness. Because it has a very low risk of teratogenicity, it can be useful for treating nausea and vomiting during pregnancy. As with the other antihistaminic anticholinergic agents, the main side effects are dryness of the mucous membranes and mild sedation.
Dimenhydrinate (Dramamine)
Dimenhydrinate is a vestibular suppressant and antiemetic agent that is primarily used for
particularly useful for treating severe vertigo with recurrent nausea and vomiting. It is an antidopaminergic agent and also has antihista- minic and anticholinergic properties. As an antihistamine, it acts by competitive antago- nism but does not block release of histamine; it antagonizes in varying degrees most pharmaco- logic effects of histamine.41 Although infre- quent, extrapyramidal symptoms can occur secondary to promethazine, and occasionally such symptoms can be confused for CNS signs of an undiagnosed primary disease. Pro- methazine can be very sedating, particularly in older people, and it should not be used in patients with a history of sleep apnea. It can be more effective with less sedation by combining it with 25 mg of ephedrine.42 Because of its excellent antiemetic action, it can be combined with other vestibular suppressants that have fewer antiemetic properties, such as scopol- amine and the benzodiazepines.
Betahistine (Serc)
Betahistine is an H receptor antagonist that
preventing motion sickness. Like other drugs
in this class, it has antihistaminic and anticho-
has long been used larly vertigo
3
for
treating vertigo particu- with Meniere’s syn-
linergic activity and the main side effects are
dryness of mucous membranes and mild to
associated
drome.43–45 Betahistine was briefly approved by
the Food (FDA) for
moderate sedation. The sedation associated
and Drug Administration
use in the
ago, but
with dimenhydrinate and other H blockers
United States about 40 years
can be reduced or avoided by release formulations.40 Patients
1
using
slow-
65
subsequently approval was withdrawn due to lack of evidence for efficacy. It is still avail-
over age
may have a stronger reaction and require
smaller doses. Dimenhydrinate
able in Europe and Canada and can be
obtained through compounding pharmacies
should be used
with caution in patients with
in the United States. Curiously, by blocking
cardiovascular or respiratory disease and in patients with convul-
presynaptic H3
increased
receptors, betahistine causes an of histamine and activation of
sive disorders. For prevention of motion sick-
ness, the first dose should be given 30 min
release
H1 receptors (the opposite effect of antihista-
before exposure to motion. Since dimenhydri- nate may cause drowsiness or impair judgment and coordination, patients should be warned not to operate any machinery or drive a vehicle because of the sedative effect. They should avoid alcohol and drink plenty of fluids for dry mouth and to prevent constipation.
Promethazine Hydrochloride (Phenergan)
Promethazine hydrochloride is a potent ves- tibular suppressant and antiemetic drug that is
minic vestibular suppressants). However, by blocking H3 receptors in the vestibular periph-
ery, betahistine may decrease spontaneous
afferent activity.10,46
Metoclopramide (Reglan)
Metoclopramide is an antiemetic medication that appears to work both centrally on the chemoreceptor trigger zone and peripherally on the gastrointestinal tract.47 It has antidop- aminergic and anticholinergic properties and thus has the potential side effects of extrapyramidal symptoms and dryness of the
mucous membranes. Its gastrointestinal effects are mediated through blockage of serotonin receptors; blocking activity originates in vis- ceral afferents running from the gastrointesti- nal tract to the emetic center. Metoclopramide is an antiemetic agent useful for treating many causes of nausea and vomiting. It also can be used in combination with vestibular suppres- sants and other antiemetic drugs.19 It can be administered orally or by injection with onset of action within the first hour (the half-life is 5 to 8 hr).
Benzquinamide Hydrochloride (Emete-con)
Benzquinamide hydrochloride is a short-acting non–amine-depleting benzamide derivative, chemically unrelated to the phenothiazines and other antiemetic drugs. It has antihistaminic and anticholinergic properties, but the mecha- nism of action of its antiemetic properties is unknown. It is most useful for treating mild, persistent nausea but has sedating properties like all of the other antiemetic drugs. A sudden increase in blood pressure and transient arrhythmia has been reported following intra- venous administration of benzquinamide.
Trimethobenzamide Hydrochloride (Tigan)
Trimethobenzamide hydrochloride is a benz- amide derivative that has antihistaminic and anticholinergic properties. This drug is an anti- emetic agent that is suitable for mild to moder- ate nausea and vomiting. It can produce drowsiness and may be additive with other sedating medications. Extrapyramidal side effects can also occur.
Diazepam (Valium)
Diazepam is a benzodiazepine agent that is a vestibular suppressant and has antianxiety and sedative effects.48 It may also help control nau- sea associated with vertigo and motion sick- ness.49 Diazepam is a GABA agonist that decreases transmission of signals through the vestibular nucleus. It can be given intravenously
to rapidly suppress acute severe vertigo, but the intravenous route must be used with extreme caution particularly in patients with limited pul- monary reserve, because of the possibility of producing apnea or cardiac arrest. Abrupt with- drawal of diazepam may be associated with prominent withdrawal symptoms, including sei- zures. Patients should be warned about the simultaneous use of alcohol or other CNS depressant drugs because of the additive effect.
Droperidol (Inapsine)
Droperidol is a butyrophenone derivative that has prominent antiemetic effects. The onset of action is within 30 min after injection; thera- peutic levels persist 2 to 4 hr and vestibular suppression can be demonstrated for as long as 24 hr.50 Like the phenothiazines, its mode of action is blockage of dopaminergic transmis- sion in the chemoreceptor trigger zone.35 It can produce marked tranquilization and sedation and therefore should be used with great cau- tion in older people. The drug also produces a mild -adrenergic blockade. Hypotension, pos- sibly associated with hypovolemia, and extrapy- ramidal effects should be watched for. The FDA placed a black box warning on the use of droperidol because of the possibility of induc- ing fatal cardiac arrhythmias due to prolonga- tion of the QT interval.32
Diphenidol (Vontrol)
Diphenidol is a piperidinebutanol compound that has antiemetic properties but is chemically unrelated to the other antiemetic drugs. Diphenidol does have weak peripheral antich- olinergic effects and therefore can have the usual side effects associated with these agents. Because of its potential for producing halluci- nations, confusion, and disorientation, it should be used with caution in older patients, particu- larly those who are hospitalized. The mode of action of its antiemetic effect is unknown.
Prochlorperazine (Compazine)
Prochlorperazine is an antidopaminergic agent that is a piperazine-type phenothiazine. Like the other phenothiazines, it works directly on
the chemoreceptor trigger zone by blocking dopaminergic transmission. Antihistaminic effects have been shown during chronic treat- ment, and it is an -adrenergic blocker.36 Antiemetic effects are maximal 2 to 4 hr after the peak blood level is reached, and the half- life exceeds 8 hr. It has a high incidence of extrapyramidal effects, moderate sedative effects, and a low incidence of anticholinergic effects and orthostatic hypotension. In some patients, the sedation may impair driving performance.51
Dronabinol (Marinol)
Dronabinol is an antiemetic cannabinoid extracted from Cannabis sativa (marijuana) that, like other cannabinoids, has complex effects on the CNS; dronabinol is classified as an antinau- seant with antianorectic activities. It seems to work by diminishing activity from the cortex to the chemoreceptor trigger zone. It has been used mostly for treating nausea and vomiting associated with chemotherapy and radiation.
REFERENCES
Zajonc TP, Roland PS. Vertigo and motion sickness Part II: Pharmacologic treatment. Ear Nose Throat J. 2006;85(1):25.
Baloh RW. Antiemetic and antivertigo drugs. In: Rowland LF, ed. Current Neurologic Drugs. 2nd ed. Philadelphia: Williams & Wilkins; 1998: 184.
Brizee KR: Mechanics of vomiting: a minireview. Can J Physiol Pharmacol. 1990;68:221.
MacGregor EA. Anti-emetics. Curr Med Res Opin. 2001;17(suppl 1):s22.
Doi K, Tsumoto T, Matsunaga T. Actions of excit- atory amino acid antagonists on synaptic inputs to the rat medial vestibular nucleus: an electrophysiological study in vitro. Exp Brain Res. 1990;82:254.
Sasa M, Takeshita S, Amano T, Kurisu K. Primary neurotransmitters and regulatory substances onto vestibular nucleus neurons. Biol Sci Space. 2001;15 (4):371.
Phelan KD, Gallagher J P. Direct muscarinic and nicotinic receptor–modulated excitation of rat medial vestibular nucleus neurons in vitro. Synapse. 1992;10:349.
Steinbusch HWM. Distribution of histaminergic neu- rons and fibers in rat brain. Acta Otolaryngol Suppl (Stockh). 1991;479:12.
Yabe T, de Waele C, Serafin M, et al. Medial vestibular nucleus in the guinea-pig: histaminergic receptors. II. An in vivo study. Exp Brain Res. 1993;93:249.
Chávez H, Vega R, Soto E. Histamine (H3) receptors modulate the excitatory amino acid receptor response
of the vestibular afferents. Brain Res. 2005;1064 (1-2):1.
Zhang J, Han XH, Li HZ, Zhu JN, Wang JJ. Histamine excites rat lateral vestibular nuclear neurons through activation of post-synaptic H2 receptors. Neurosci Lett. 2008;448(1):15.
Botta L, Tritto S, Perin P, et al. Histamine H1 recep- tors are expressed in mouse and frog semicircular canal sensory epithelia. Neuroreport. 2008;19(4):425.
Gliddon CM, Darlington CL, Smith PF. GABAergic systems in the vestibular nucleus and their contri- bution to vestibular compensation. Prog Neurobiol. 2005;75(1):53.
Bergquist F, Ruthven A, Ludwig M, Dutia MB. Histaminergic and glycinergic modulation of GABA release in the vestibular nuclei of normal and laby- rinthectomised rats. J Physiol. 2006;577(pt 3):857.
Gallagher JP, Phelan KD, Shinnick-Gallagher P. Modulation of excitatory transmission at the rat medial vestibular nucleus synapse. Ann NY Acad Sci. 1992;656:630.
Halberstadt AL, Balaban CD. Selective anterograde tracing of the individual serotonergic and nonsero- tonergic components of the dorsal raphe nucleus projection to the vestibular nuclei. Neuroscience. 2007;147(1):207.
Peppard SB. Effect of drug therapy on compensation from vestibular injury. Laryngoscope. 1986;96:878.
Rascol O, Hain TC, Brefel C, et al. Antivertigo medi- cations and drug-induced vertigo. A pharmacological review. Drugs. 1995;50:777.
Bennett DR. Drugs used for vertigo and vomiting. In: Bennett DR, ed. AMA Drug Evaluations Annual 1993. Chicago: American Medical Association; 1992: 423.
Fick DM, Cooper JW, Wade WE, et al. Updating the Beers criteria for potentially inappropriate medication use in older adults: results of a US consensus panel of experts. Arch Intern Med. 2003;163:2716.
Vlastarakos PV, Nikolopoulos TP, Manolopoulos L, Ferekidis E, Kreatsas G. Treating common ear problems in pregnancy: what is safe? Eur Arch Otorhinolaryngol. 2008;265(2):139.
Takeda N, Morita M, Hasegawa S, et al. Neuropharmacology of motion sickness and emesis. Acta Otolaryngol Suppl (Stockh). 1993;501:10.
Takeda N, Matsunaga T. Neurochemical basis of motion sickness and its treatment and prevention. In: Baloh RW, Halmagyi GM, eds. Disorders of the Vestibular System. New York: Oxford University Press; 1996: 529.
Torotrice PV, O’Connell MB. Management of chemotherapy-induced nausea and vomiting. Pharmacotherapy. 1990;10:129.
Borison HL. Area postrema: chemoreceptor circum- ventricular organ of the medulla oblongata. Prog Neurobiol. 1989,32:351.
Hesketh PJ, Gandara DR. Serotonin antagonists: a new class of antiemetic agents. J Natl Cancer Inst. 1991;86:613.
Navari RM. Antiemetic control: toward a new stan- dard of care for emetogenic chemotherapy. Expert Opin Pharmacother. 2009;10(4):629.
Andrews PLR, Davis CJ, Bingham S, et al. The abdominal visceral innervation and the emetic reflex: pathways, pharmacology, and plasticity. Can J Physiol Pharmacol. 1990;68:325.
Kris M, Gralla RJ, Tyson LB, et al. Controlling delayed vomiting: double-blind, randomized trial comparing placebo, dexamethasone alone, and metoclopramide plus dexamethasone in patients receiving cisplatin. J Clin Oncol. 1989;7:108.
Beck TM, Hesketh PJ, Madujewicz S, et al. A stratified, randomized, double-blind comparison of intravenous ondansetron administered as a multiple dose regimen versus two single dose regimens, in the prevention of cisplatin-induced nausea and vomiting. J Clin Oncol. 1992;10:1969.
Kearsley JH, Williams AM, Fiumara B. Antiemetic superiority of lorazepam over oxazepam and meth- ylprednisone as premedications for patients receiv- ing cisplatin-containing chemotherapy. Cancer. 1989;64:1595.
Richards JR, Schneir AB. Droperidol in the emer- gency department: is it safe? J Emerg Med. 2003;24(4): 441.
Rodgers C. Extrapyramidal effects of antiemetics pre- senting as psychiatric illness. Gen Hosp Psychiatry. 1992;14:192.
Leopold NA. Prolonged metoclopramide-induced dyskinetic reaction. Neurology. 1984;34:238.
Ferrando SJ, Eisendrath SJ. Adverse neuropsychi- atric effects of dopamine antagonist medications. Psychosomatics. 1991;32:426.
Isah AO, Rawlins MD, Bateman DN. Clinical phar- macology of prochlorperazine in healthy young males. Br J Clin Pharmacol. 1991;32:677.
Renner UD, Oertel R, Kirch W. Pharmacokinetics and pharmacodynamics in clinical use of scopolamine. Ther Drug Monit. 2005;27(5):655.
Baker FM, Cook P. Compazine complications: a review. JAMA. 1981;73:409.
Manning C, Scandale L, Manning EJ, Gengo FM. Central nervous system effects of meclizine and dimenhydrinate: evidence of acute tolerance to anti- histamines. J Clin Pharmacol. 1992;32:996.
Seibel K, Schaffler K, Reitmeir P, Golly I. A ran- domised, placebo-controlled study comparing two formulations of dimenhydrinate with respect to efficacy
inmotionsickness and sedation. Arzneimittelforschung. 2002;52(7):529.
Kubo N, Shirakawa O, Kuno T, Tanaka K. Antimuscarinic effects of antihistamines: quantitative evaluation of receptor-binding assay. Jpn J Pharmacol. 1987;43:277.
Wood CD, Stewart JJ, Wood MJ, et al. Therapeutic effects of antimotion sickness medications on the secondary symptoms of motion sickness. Aviat Space Environ Med. 1990;61:157.
Lacour M, Sterkers O. Histamine and betahistine in the treatment of vertigo: elucidation of mechanisms of action. CNS Drugs. 2001;15(11):853.
Tighilet B, Trottier S, Mourre C, Chotard C, Lacour
M. Betahistine dihydrochloride interaction with the histaminergic system in the cat: neurochemical and molecular mechanisms. Eur J Pharmacol. 2002;446 (1-3):63.
Brandt T, Zwergal A, Strupp M. Medical treatment of vestibular disorders.Expert Opin Pharmacother. 2009;10(10):1537.
Tritto S, Botta L, Zampini V, Zucca G, Valli P, Masetto
Calyx and dimorphic neurons of mouse Scarpa’s ganglion express histamine H3 receptors. BMC Neurosci. 2009;10:70.
Talley NJ. 5-Hydroxytryptamine agonists and antago- nists in the modulation of gastrointestinal motility and sensation: clinical implications. Aliment Pharmacol Ther. 1992;6:273.
Takeno K, Shimogori H, Takemoto T, et al. The systemic application of diazepam facilitates the reac- quisition of a well-balanced vestibular function in a unilateral vestibular re-input model with intracochlear tetrodotoxin infusion using an osmotic pump. Brain Res. 2006;1096(1):113.
McClure JA, Lycett P, Baskerville JC. Diazepam as an antimotion sickness drug. J Otolaryngol. 1982;11:253.
Baldwin RL. Droperidol in the treatment of vertigo.
South Med J. 1983;76:1271.
Shupak A, Gordon CR. Motion sickness: advances in pathogenesis, prediction, prevention, and treatment. Aviat Space Environ Med. 2006;77(12):1213.
This page intentionally left blank
![]()
ADAPTIVE CONTROL OF NORMAL VESTIBULAR REFLEXES
MECHANISMS FOR COMPENSATION AFTER VESTIBULAR LOSS
SPECIAL CIRCUMSTANCES
Vestibular Loss in Children Vestibular Loss in the Elderly Failure of Compensation
CONTROLLED TRIALS OF VESTIBULAR EXERCISES
STRATEGY FOR DESIGNING VESTIBULAR EXERCISES
Unilateral Vestibular Lesions Bilateral Vestibular Lesions Central Vestibular Lesions VESTIBULAR EXERCISES FUTURE DIRECTIONS APPENDIX 20–1
SAMPLE HOME EXERCISE PROGRAM
Head-Turning Practice Walking Practice Other Exercises Dizziness Exercises
When the vestibular system has been perma- nently damaged, the initial state of imbalance at the level of the brainstem nuclei results in acute vertigo (see Pathophysiology of vestibu- lar symptoms in Chapter 1). Gradually, the patient adapts to this imbalance through a pro- cess of compensation that requires intact vision and depth perception, normal proprioception in the neck and limbs, and intact sensation in the lower extremities. Central pathways are also integral to compensation, and damage to these areas results in less effective recovery.
Clinicians have long been aware that vestib- ular compensation occurs more rapidly and is more complete if the patient begins exercising as soon as possible after a vestibular lesion.1,2 Controlled studies in primates have supported this general clinical observation. Baboons whose hind limbs were restrained by plaster casts after a unilateral vestibular lesion showed markedly delayed recovery of balance com- pared with lesioned animals that had been allowed normal motor exploration.3 Visual experience is also necessary; lesioned animals
kept in the light compensated faster than those kept in darkness.4,5 Compensation was acceler- ated in cats by stimulant drugs (amphetamine) and slowed by sedating drugs (diazepam, dimenhydrinate), with the authors postulating that the effect of the stimulating drugs was due to increased physical activity levels.6 For these reasons, vestibular exercise programs probably should be instituted as soon as possible after an injury to the vestibular system has been identi- fied, and the use of sedating drugs should be limited to the acute stage.
In addition to producing the subjective sen- sation of vertigo, vestibular lesions interfere with reflexes controlling eye movements dur- ing active head motion and with postural right- ing reflexes.7,8 This interference can result in oscillopsia caused by head movements and a tendency to veer or fall when walking. These symptoms and the associated dizziness can be improved by active exercises designed to speed compensation. Many of the exercises will result in dizziness. This sensation is a necessary stimulus for compensation; antivertiginous
419
medications should be avoided during this period to allow the compensation process to occur.
Although there is a consensus that vestibular rehabilitation is helpful for patients with ves- tibular lesions,9,10 few physical therapists receive training in vestibular rehabilitation during their formal education.11 Most learn about vestibular rehabilitation at short continu- ing education courses or from colleagues. Standards need to be established and thera- pists need to be educated about the vestibular system.
ADAPTIVE CONTROL OF NORMAL VESTIBULAR REFLEXES
Adaptive control of vestibular reflexes is con- stantly occurring in all of us. A common example is the adjustment in the gain of the vestibulo-ocular reflex (VOR) that occurs in normal people when they wear magnifying lenses in glasses. In order to maintain clear vision during head movements, the gain of the VOR must be rapidly adjusted up and down as they take the glasses on and off. In the labora- tory, subjects show even more dramatic changes in the VOR gain when they wear more powerful magnifying or minifying lenses.12,13 An extreme example is the reversal in VOR direction that occurs in animals and humans who wear revers- ing prisms for several days.14,15 These gain adjustments occur within central VOR path- ways since there is no change in peripheral sen- sory input. In subjects taking their glasses on and off, the adjustment in gain occurs immedi- ately before there is any sensory conflict as though the brain has two separate gain settings that can be turned on and off like a switch. An example of this presetting of gain within the vestibulospinal reflex occurs when a normal subject briefly loses balance when stepping onto an escalator that is unexpectedly out of service. The brain has preprogrammed a change in posture to compensate for the impending movement of the escalator, so that a correction for movement is automatically made even though no actual motion is experienced when the subject steps onto the stationary surface. This ability of central pathways to preprogram motor responses plays an important role in compensation after loss of vestibular reflexes.
Adaptive changes in vestibular reflexes are typically context specific.12,13 Experimental studies in animals and humans have shown that adaptive changes in the angular VOR induced by head rotation can be gated in and out depending upon the static orientation of the head, even though the pattern of semicircular canal activation is the same. Therefore, physi- cal therapy programs that are attempting to promote adaptation—so-called adaptation exercises—should be used in a natural setting where the compensated vestibular reflexes will be used. Cognitive factors also play an impor- tant role in vestibular adaptation, even to the point that mental exercises may be able to sub- stitute for actual movements in the adaptation process. For example, imagined fixed or mov- ing visual targets can have the same effect in modifying VOR gain as real targets. By going through their routines in their minds, such as a professional golfer analyzing his or her swing, professional athletes can adapt a motor behavior without actually performing the motor act. Thus, psychological factors such as motiva- tion, attention, effort, and interest play a part in any therapy program aimed at vestibular rehabilitation.
Vestibular adaptation typically has a charac- teristic time course both for acquisition and loss (see Fig. 3–22 in Chapter 3).16 In order to maintain adaptation, one requires continued exposure to sensory inputs for the continued elaboration of the adapted response. Central pathways are key in maintaining adaptation, so obviously lesions within these pathways can lead to loss of adaptation. Adaptive capabilities seem to decline with normal aging, which may in part explain why vestibular compensation tends to be slower in older people.7,17 In addi- tion, older patients who lose vestibular func- tion may also have an impaired somatosensory or visual system and thus be less able to com- pensate for the loss of vestibular function.
MECHANISMS FOR COMPENSATION AFTER VESTIBULAR LOSS
Common mechanisms used for recovery after loss of labyrinthine function are summarized in Table 20–1. In patients with partial peripheral vestibular lesions, the gain of the vestibulo-ocular
Table 20–1 Mechanisms for Compensation after Loss of Vestibular Function
![]()
Adjust gain centrally
Use other sensory inputs Use other motor responses
Anticipate intended motor behavior Change movement strategy
![]()
Adjust central perception
and vestibulospinal reflexes can be adjusted to help compensate for the peripheral loss. For example, after a unilateral peripheral lesion, the gain asymmetry is gradually adjusted and the dominant time constant is shortened to help maintain the gain in the high-frequency range.18 The input from one normally functioning remain- ing labyrinth is enough to drive most of the vestib- ulo-ocular and vestibulospinal responses over a wide functional range. There are limitations to these central adjustments in gain, however, and there must be enough residual peripheral func- tion to allow the adaptive mechanisms to act. Also, these gain adjustments only work within a certain range of movements, so that compensation is not possible for high acceleration head thrusts (the basis of the bedside head-thrust test).19
Substitution with other sensory inputs and alternative motor responses is commonly used in the compensation process after vestibular lesions. Substitution exercises are used to enhance these other inputs. For example, input from cervical muscle proprioceptors can be used to help generate compensatory eye move- ments in patients with labyrinthine damage. Studies in patients with bilateral labyrinthine disease show that the cervical–ocular and opto- kinetic reflexes have increased gains compared to normal subjects.20,21 Subjects also can substi- tute alternative motor responses in lieu of the vestibular compensatory responses. For exam- ple, small anticompensatory saccades can be substituted for vestibular slow phases to help stabilize gaze during head rotation.
Patients with labyrinthine lesions develop strategies that use prediction or anticipation of intended motor behavior to substitute for ves- tibular reflexes.22 For example, patients who have lost labyrinthine function learn to prevent gaze overshoot during combined eye and head movements by preprogramming compensatory
slow phases and decreasing saccade size in anticipation of head movement.
Patients with vestibular lesions will com- monly alter their behavior to avoid excessive demands on the compromised vestibular reflexes.23 They may walk slower, widen their base, and avoid head movements to decrease symptoms of dizziness and imbalance. Although these changes in movement strategies may initially decrease symptoms, they may be counterproductive in the long run since they prevent the brain from receiving sensory sig- nals needed in the compensation process.
The brain has the ability to adjust central perception of movement that occurs in patients after vestibular lesions. For example, the oscil- lopsia that occurs after bilateral vestibular loss becomes less and less many years after the lesion. Patients usually have no problem driv- ing or carrying on other routine activities.24 Patients with central vestibular lesions and long-standing spontaneous nystagmus also report decreasing oscillopsia over time.
In summary, the vestibular compensation process after a vestibular lesion is a fragile and dynamic process. It requires complex senso- rimotor integrations within the brain, involving many different pathways. Intercurrent illness, injury, and excessive medication or alcohol use may lead to temporary reemergence of long- compensated symptoms from a prior vestibular lesion. The compensation process is generally slower and less complete in older patients, par- ticularly if they have comorbidities resulting in additional defects in sensory inputs from other modalities as occurs with peripheral neuropathy or impaired vision.
Vestibular Loss in Children
As a general rule, the vestibular compensation process is more robust in children than in adults. However, this is not to say that children do not show significant deficits from early-onset vestibular loss. In children who are born with vestibular loss or who lose vestibular function within the first year, motor milestones are seri- ously delayed during the first 2 or 3 years of life.25 However, during the preschool age, most children achieve standard landmarks of motor
development such as head control, independent walking, and running. By the time they enter elementary school (about 6 years of age), most have standard motor skills that would not be outside of the normal range. Many can swim even under water and maintain good balance even with eyes closed. These remarkable motor skills in the context of severe vestibular loss pre- sumably depend on compensatory input from visual, somatosensory, and proprioceptive senses and the maturation of motor systems in the cer- ebellum, basal ganglia, and motor cortex.
Vestibular Loss in the Elderly
Vestibular compensation is more difficult and less complete in older patients than in young adults.26,27 All of the compensatory mechanisms outlined in Table 20–1 may be diminished in older people. Other sensory inputs such as somatosensory and visual sense are typically diminished so they are less able to compensate for the loss of vestibular sense. Their ability to substitute other motor responses for vestibular responses is also limited because of slowing of all motor responses due to aging; both periph- eral and central mechanisms are affected. The severity of age-related white matter abnormali- ties correlates with the severity of gait and motor abnormalities.28,29 Overall cognitive decline that occurs with normal aging may interfere with their ability to anticipate intended motor behavior and adjust central perception. Changing movement strategy may be one of the few remaining options, so they may adopt a sedentary lifestyle that may further interfere with the compensation process.
Failure of Compensation
A small percentage of patients continue to complain of persistent dizziness and imbalance months to years after an acute peripheral ves- tibular lesion. The cause of this lack of com- pensation and whether it could be prevented by a vestibular compensation exercise program are largely unknown. In one study 31% of 210 patients continued to complain of disequilib- rium more than 3 months after the surgical removal of an acoustic neuroma.26 Age >55.5 years, female gender, constant preoperative disequilibrium present for >3.5 months, and
central findings on electronystagmography were all significantly associated with this poor outcome. In another study 29 of 142 patients (20%) continued to have significant dizziness after they underwent surgical procedures to section the vestibular nerve for a variety of ves- tibular disorders.30 Possible reasons for the persistent dizziness were incomplete vestibular nerve section, poor central nervous system (CNS) compensation, new vestibular disease in the opposite ear, and the presence of other CNS diseases. A prospective study of 60 patients with vestibular neuritis found that only 34 (57%) reported complete relief from sub- jective symptoms at long-term follow-up.31 A common theme in these and other studies is that older patients with evidence of CNS dis- ease have the highest likelihood for persistent dizziness (failure of compensation) after an acute vestibular lesion.
CONTROLLED TRIALS OF VESTIBULAR EXERCISES
Most early studies of vestibular exercises in the treatment of patients with acute and chronic vestibular lesions were not controlled and focused on improvement in vestibular symp- toms, so it is difficult to separate the benefit of the vestibular exercises from other nonspecific effects. Several recent randomized controlled studies found that vestibular rehabilitation exercises improve the functional outcome after unilateral vestibular loss.32–36 A systematic review of the literature by The Cochrane Collaboration concluded that there is moder- ate to strong evidence from high-quality ran- domized trials supporting safety and efficacy of vestibular rehabilitation for unilateral periph- eral vestibular dysfunction.9,10
Importantly most of the vestibular rehabilita- tion trials were conducted in the population of patients with surgical lesions or groups of mixed or vaguely stated etiologies. Very few studies have been performed in the population of patients with vestibular neuritis. This difference in the etiology of the peripheral deficit is impor- tant because many patients with vestibular neu- ritis will have spontaneous return of vestibular function as the viral-inflammatory process resolves. On the other hand, patients with a fixed and complete surgical lesion will not have
One study in vestibular neuritis patients
Table 20–2 Unilateral Peripheral Vestibular Lesions
![]()
Goal Force compensation, rebalance, adaptation
found that the sway as measured by posturoga-
phy was significantly improved in patients treated with an intense vestibular therapy pro- gram compared to controls.32 However, the
Sample
exercises
Fixate targets, track targets; rapid head movements when sitting, then walking; walk in dark, on uneven surface
randomization procedure was violated in this study because 43 of the original 82 randomized patients were excluded because they showed a partial or complete caloric response recovery on day 30. Other outcomes including ocular torsion and subjective visual vertical were not significantly different between the groups. The intervention in this study involved physical therapists treating the study group for 5–7 days in the hospital, which is likely to be more intense therapy than is practical in routine care.
A second trial in a vestibular neuritis popula- tion did not use a placebo group.37 Instead this trial compared a program of home training (e.g., oral and written vestibular therapy instruction) to the same program of home training but also with additional physical ther- apy sessions (i.e., three 40-minute supervised sessions during the first week and then one ses- sion per week for 9 weeks). No differences were seen in any of the subjective outcomes (i.e., rating of vertigo or imbalance) or objec- tive outcomes (i.e., caloric testing, Romberg test, one-leg stance, and tandem walking) at any of the follow-up intervals (1 week, 10 weeks, or 6 months).
Customized vestibular rehabilitation pro- grams appear to be superior to general instruc- tions that simply emphasize the need for exposure to movement in patients after surgery for an acoustic neuroma.35 For patients who are to undergo unilateral ablative surgery, begin- ning vestibular exercises a few weeks before the surgery and continuing after surgery may speed up recovery.38
STRATEGY FOR DESIGNING VESTIBULAR EXERCISES
Unilateral Vestibular Lesions
Persons with unilateral vestibular lesions demon- strate a series of deficits requiring compensation
Expect Complete recovery, minimal subtle
deficits
![]()
(Table 20–2). A near-complete recovery of nor- mal abilities can be expected, although specific deficits will remain.
GAZE STABILITY
Because of the unilateral loss of the VOR in the acute patient, there is spontaneous nystagmus that results in a complete loss of gaze stability. This can be complicated by diplopia as part of the ocular tilt reaction. These acute severe visual symptoms resolve gradually over the first several days to a week as the remaining vestib- ular labyrinth restores VOR function or if the function of the lesioned side recovers. As this occurs, gaze stability improves but may not return to normal. Quick movements toward the side of the lesion can result in a sense of oscillopsia and dizziness. In some patients, these symptoms will remain permanent. Treatment involves maintaining full neck mobility to allow increased input from neck proprioceptors during head turns and ocular fixation practice while at rest and at low and high head accelerations in both the vertical and horizontal planes.
BALANCE
Before compensation occurs, otolith input is asymmetric. This results in a perceptual illu- sion that the environment is tilted. There is a tendency to veer toward the side of the lesion when walking. Because of an increased depen- dence on visual input, falls can occur if lighting is poor or if the eyes are closed. This visual dependence is complicated in the acute situa- tion by the presence of spontaneous nystagmus and skew diplopia. After the acute nystagmus and skew deviation disappear within a few days, balance improves but there are still deficits,
Bilateral Vestibular Lesions
Very few randomized studies have tested ves- tibular therapy interventions in patients with a bilateral vestibulopathy. Persons with bilateral vestibular loss demonstrate a series of perma- nent deficits that require compensation (Table 20–3). Some compensation involves strength- ening of existing reflexes such as the cervical- ocular reflex and smooth pursuit, while the remainder requires trained behaviors such as substituting centrally programmed eye move- ments for the lost VOR.39
GAZE STABILITY
Because of the absence of the VOR, head movements that are of sufficient velocity to exceed the smooth pursuit system (>1 Hz) result in retinal slip, which leads to symptom- atic oscillopsia. Treatment strategies center on attempting to substitute other sensory input systems for the vestibular loss. Exercises are used to maintain full neck mobility to allow the
Table 20–3 Bilateral Peripheral Vestibular Lesions
![]()
Goal Substitute other sensory information to replace vestibular
cervical-ocular reflex to provide help with com- pensatory eye movements. Slow head oscilla- tions can be used to strengthen pursuit abilities during head movements, and saccade exercises (looking back and forth between targets) may help optimize the voluntary use of fast eye movements. Ultimately, some patients learn that to obtain maximum visual acuity, they must hold the head still.
One small randomized trial of vestibular exercises assessed the efficacy of the exercises on improvement of dynamic visual acuity (e.g., visual acuity under movement) and subjective intensity of oscillopsia in patients with bilateral vestibulopathy.39 The exercises used as the intervention in this study were “adaptation” exercises (i.e., focusing on a target during head movements while the target is either stationary or moving) and balance exercises. The investi- gators chose to remove two out of seven con- trol subjects after randomization because one subject was moving her head during the pla- cebo exercises and the other had better- than-expected improvement in dynamic visual acuity. The study found that the intervention group did improve dynamic visual acuity per- formance, while the placebo group did not. Although one would think improvement in dynamic visual acuity would lead to less symptomatic oscillopsia, this study found that the change in dynamic visual acuity did not correlate with change in subjective ratings of oscillopsia.
BALANCE
In the absence of peripheral vestibular input, postural control relies upon the ankle and step- ping strategies. Loss of the vestibulospinal reflex leads to an increased dependence on ankle proprioception and cutaneous sensation from the feet and ankles to provide balance. These other senses are used to substitute for the vestibular loss. Such patients are also visu- ally dependent, but there are limitations to their compensation because of the associated
Sample
exercises
Track targets with and
without head movement Learn to hold head still to
read
Walk on foam and uneven surfaces with vision
oscillopsia. Deficits become apparent when they are exposed to poor support surfaces (soft or shifting surfaces, narrow support base), par- ticularly if visual inputs are misleading. Treatment strategies for balance include main- taining or improving ankle strength and mobil-
Expect Mild to moderate persistent
limitations
![]()
ity, increasing cutaneous input in the lower extremities (e.g., use of supportive high-top
shoes, shoes with firm soles), gait and balance exercises, and stressing the importance of solid footing and good lighting at all times.
Central Vestibular Lesions
Recovery from central vestibular lesions is typ- ically much slower than recovery from periph- eral vestibular lesions (Table 20–4). No doubt this can be traced to the fact that structures involved in the recovery process are themselves damaged. The cerebellum is a key structure for compensation and patients with cerebellar lesions improve the least with vestibular rehabilitation.40
GAZE STABILITY
Patients with central vestibular lesions often will have spontaneous nystagmus that persists for months to years. Although the oscillopsia associated with this spontaneous nystagmus may decrease over years, it rarely completely disappears. Often there is a null region where the nystagmus is less, or maybe even absent, so patients can learn to hold their eyes near the null region when best visual acuity is critical (for example, when reading). If a null point is present, then the use of an eyeglass prism or ocular surgery may be beneficial. There are no data to suggest that either head or eye exercises will decrease the magnitude of the eye move- ment or the subjective oscillopsia. In some cases, such exercises seem to aggravate the problem by causing nausea and even vomiting.
BALANCE
Central balance disorders tend to be much more severe than those associated with peripheral
Table 20–4 Central Vestibular Lesions
![]()
Goal Suppress nausea, dizziness, diminish oscillopsia, diplopia, help compensation
vestibular lesions; thus, patients are at a much higher risk of falling. Physical therapy should be aimed at maintaining strength, particularly in the lower extremities, and instructing the patient on using proper support such as a cane or a walker. Regular walking (with support) is encouraged because it maintains strength and mobility, but there are no adequate randomized studies to indicate the benefit of a vestibular rehabilitation program in central disorders.
Vestibular exercises should begin as soon as the acute stage of nausea and vomiting has ended and the underlying disease process is subsid- ing. Many of the exercises will result in dizzi- ness. This sensation is a necessary stimulus for compensation; antivertiginous medications should be avoided as much as possible during this period to maximize the beneficial effect. Exercises should be done at least twice daily for several minutes but may be done as often as the patient can tolerate (see Appendix 20–1 for sample exercises that can be given to the patient).
While nystagmus is present, adaptation exer- cises should begin with the patient attempting to focus the eyes and to move and hold them in the direction that provokes the most dizziness. Once the nystagmus diminishes to the point that a target can be “held” visually in all direc- tions, the patient should begin eye and head coordination exercises. A useful exercise involves staring at a visual target while oscillat- ing the head from side to side or up and down. The speed of the head movements can be grad- ually increased, as long as the target can be kept in good focus. Target changes using com- bined eye and head movements to jump quickly back and forth between widely separated visual targets are also useful. Blinking during these fast head turns can help reduce symptoms of dizziness or blurring of vision.
Gait and balance exercises should begin by having the patient try to stand and walk while
Sample
exercises
Expect
Fixate near and far, different gaze positions
Walk touching wall, up and down slopes
Moderate to severe limitations, particularly in older people
nystagmus is still present. It may be necessary to walk in contact with a wall or to use an assistant in the early stages. Slow, supported turns should be made initially. As improvement occurs, head movements should be added while standing and walking—at first slow
![]()
side-to-side or up-and-down movements, then fast head turns in all directions. Learning to combine fast head turns with brief eye closure or blinks during walking turns can increase sta- bility and decrease dizziness.
Compensation can require as long as 2 to 6 months. Dizziness that persists beyond this time indicates either the presence of an ongo- ing, recurrent vestibular illness or poor com- pensation. The patient’s history should be reviewed, and any vestibular suppressants should be discontinued. Evidence of central involvement or impairment of vision, proprio- ception, or sensation should be evaluated. If all areas are normal, no evidence of active disease is present, and no medications are in use, a pro- gram of habituation to dizziness is generally the next step. All movements that provoke dizziness should be identified, and they should then be repeated as often as possible to maximize the symptoms. This type of therapy can gradually result in habituation to the provoking stimulus.
With the recent rapid advances in virtual tech- nology, virtual environments could theoreti- cally be useful for training patients to adapt to complex, multimodel environments.41 Individ- ualized programs could be developed and ther- apists could more easily assess the patient’s progress and need for further therapy. Improved understanding of the cellular and molecular mechanisms involved in hair cell and nerve fiber regeneration after injury may make it possible to replace damaged hair cells and nerve fibers by injecting growth factors or other factors that enhance the regeneration process.42 As described in Chapter 2, BDNF induces hair cell regeneration after ototoxic damage in chin-
acceleration in the planes of the semicircular canals and provides a pulse-frequency modula- tion of the ampullary nerve from each canal. When such a devise was implanted in chinchil- las whose vestibular system was ablated with gentimicin, partly compensatory vestibulo- ocular responses were recorded in multiple planes.44 However, current spread beyond the electrode’s targeted nerve branch was a major problem. No doubt progress will be made in the design of the acceleration sensor and elec- trodes and in surgical techniques for implant- ing the devises.
REFERENCES
Cawthorne T. The physiological basis for head exer- cises. J Chart Soc Physiother. 1944;30:106.
Cooksey FS. Rehabilitation in vestibular injuries. Proc Soc Med. 1945;39:273.
Lacour M, Roll JP, Appaix M. Modifications an devel- opment of spinal reflexes in the alert baboon (papio papio) following unilateral vestibular neurectomy. Brain Res. 1976;113:255.
Igarashi M, Levy JK, O-Uchi T, et al. Further study of physical exercise and locomotor balance after uni- lateral labyrinthectomy in squirrel monkeys. Acta Otolaryngol (Stockh). 1981;92:101.
Igarashi M, Ishigawa K, Ishii M, et al. Physical exercise ands balance compensation after total ablation of ves- tibular organs. Prog Brain Res. 1988;76:395.
Peppard SB. Effect of drug therapy on compensation from vestibular injury. Laryngoscope. 1986;96:878.
Horak FB. Postural orientation and equilibrium: what do we need to know about neural control of balance to prevent falls? Age Ageing. 2006;35(suppl 2):ii7.
Schubert MC, Hall CD, Das V, Tusa RJ, Herdman SJ. Oculomotor strategies and their effect on reduc- ing gaze position error. Otol Neurotol. ePubh ahead of print Oct 31, 2009.
Burton MJ, Monsell EM, Rosenfeld RM. Extracts from The Cochrane Library: vestibular rehabilitation for unilateral peripheral vestibular dysfunction (review). Otolaryngol Head Neck Surg. 2008;138(4):415.
Hillier SL, Hollohan V. Vestibular rehabiliatation for
chillas, but so far the regenerated hair cells do not form functional synapses with the afferent nerves. Finally, development of a vestibular prosthesis is a reasonable goal considering the dramatic success with cochlear prosthetic devises. Vestibular prosthetic devises have been developed and implanted in mammals, including primates, but they are still in the early stages of development.43,44 The prototypi-
unilateral peripheral vestibular dysfunction. Cochrane
Database Sys Rev. 2007;17:CD005397.
Cohen HS, Gottshall KR, Graziano M, Malmstrom EM, Sharpe MH. International survey of vestibular rehabilitation therapists by the Barany Society Ad Hoc Committee on Vestibular Rehabilitation Therapy. J Vestib Res. 2009;19(1-2):15.
Melvill Jones G. How and why does the vestibulo- ocular reflex adapt? In: Baloh RW, Halmagyi GM, eds. Disorders of the Vestibular System. New York: Oxford University Press; 1996: 85.
Miles A, Lisberger SG. Plasticity in the vestibulo-
cal devise is a head-mounted implantable angular accelerometer that measures angular
ocular reflex: a new hypothesis. Annu Rev Neurosci. 1981;4:273.
Melvill Jones G, Davies P. Adaptation of cat vestibulo- ocular reflex to 200 days of optically reversed vision. Brain Res. 1976;103:551.
Melvill Jones G, Guitton D, Berthoz A. Changing pat- terns of eye-head coordination during 6 h of optically reversed vision. Exp Brain Res. 1988;69:531.
Miles FA, Eighmy BB. Long-term adaptive changes in primate vestibuloocular reflex. I. Behavioral observa- tions. J Neurophysiol. 1980;43:1406.
Paige GD. Senescence of human visual–vestibular interactions. J Vestib Res. 1992;2:133.
Baloh RW, Honrubia V, Yee RD, Hess K. Changes in the human vestibulo-ocular reflex after loss of periph- eral sensitivity. Ann Neurol. 1984;16:222.
Halmagyi GM, Curthoys IS. A clinical sign of canal paresis. Arch Neurol. 1988;45:737.
Kasai T, Zee DS. Eye–head coordination in lab- yrinthine-defective human beings. Brain Res. 1978;144:123.
Bronstein AM, Hood JD. The cervico-ocular reflex in normal subjects and patents with absent vestibular function. Brain Res. 1986;373:399.
Curthoys IS, Halmagyi GM. Vestibular compensation: a review of the oculomotor, neural and clinical con- sequences of unilateral vestibular loss. J Vestib Res. 1995;5:67,22.
Herdman SJ. Vestibular rehabilitation. In: Baloh RW, Halmagyi GM, eds. Disorders of the Vestibular System. New York: Oxford University Press; 1996.
MacDougall HG, Moore ST, Black RA, Jolly N, Curthoys IS. On-road assessment of driving perfor- mance in bilateral vestibular-deficient patients. Ann NY Acad Sci. 2009;1164:413.
Kaga K. Vestibular compensation in infants and chil- dren with congenital and acquired vestibular loss in both ears. Int J Pediatr Otorhinolarynol. 1999;49: 215.
Driscoll CL, Lynn SG, Harner SG, Beatty CW, Atkinson EJ. Preoperative identification of patients at risk of developing persistent dysequilibrium after acoustic neuroma. Am J Otol. 1998;19:491.
Hall CD, Cox LC. The role of vestibular rehabilita- tion in the balance disorder patient. Otolaryngol Clin North Am. 2009;42(1):161, xi.
Baezner H, Blahak C, Poggesi A, et al. Association of gait and balance disorders with age-related white matter changes: the LADIS study. Neurology. 2008;70(12):935.
Kerber KA, Enrietto JA, Jacobson KM, Baloh RW. Disequilibrium in older people: a prospective study. Neurology. 1998;51:574.
Thedinger BS, Thedinger BA. Analysis of patients with persistent dizziness after vestibular nerve section. Ear Nose Throat. 1998;77:290,295.
Okinaka Y, Sekitani T, Okazakai H, Miura M, Tahara
Progress of caloric response if vestibular neuronitis.
Acta Otolaryngol (Supp). 1993;503:18.
Strupp M, Arbusow V, Maag KP, Gall C, Brandt T. Vestibular exercises improve central vestibulospinal condensation after vestibular neuritis. Neurology. 1998;51:838.
Yardley L, Donovan-Hall M, Smith HE, Walsh BM, Mullee M, Bronstein AM. Effectiveness of primary care-based vestibular rehabilitation for chronic dizzi- ness. Ann Intern Med. 2004;141(8):598.
Venosa AR, Bittar RS. Vestibular rehabilitation exer- cises in acute vertigo. Laryngoscope. 2007;117(8): 1482.
Vereeck L, Wuyts FL, Truijen S, De Valck C, Van de Heyning PH. The effect of early customized vestibu- lar rehabilitation on balance after acoustic neuroma resection. Clin Rehabil. 2008;22(8):698.
Giray M, Kirazli Y, Karapolat H, Celebisoy N, Bilgen C, Kirazli T. Short-term effects of vestibular reha- bilitation in patients with chronic unilateral vestibular dysfunction: a randomized controlled study. Arch Phys Med Rehabil. 2009;90(8):1325.
Kammerlind AS, Ledin TE, Odkvist LM, Skargren E. Effects of home training and additional physical ther- apy on recovery after acute unilateral vestibular loss: a randomized study. Clin Rehabil. 2005;19:54.
Magnusson M, Kahlon B, Karlberg M, Lindberg S, Siesjö P, Tjernström F. Vestibular “PREHAB”. Ann NY Acad Sci. 2009;1164:257.
39 Herdman SJ, Hall CD, Schubert MC, Das VE, Tusa RJ. Recovery of dynamic visual acuity in bilateral ves- tibular hypofunction. Arch Otolaryngol Head Neck Surg. 2007;133(4):383.
Brown KE, Whitney SL, Marchetti GF, Wrisley DM, Furman JM. Physical therapy for central vestibular dysfunction. Arch Phys Med Rehabil. 2006;87(1): 76.
Kenyon RV, Leigh J, Keshner EA. Considerations for the future development of virtual technology as a reha- bilitation tool. J Neuroeng Rehabil. 2004;1(1):13.
Matsui JI, Ryals BM. Hair cell regeneration: an excit- ing phenomenon…but will restoring hearing and bal- ance be possible? Rehabil Res Dev. 2005;42(4 suppl 2): 187.
Merfeld DM, Haburcakova C, Gong W, Lewis RF. Chronic vestibulo-ocular reflexes evoked by a ves- tibular prosthesis. IEEE Trans Biomed Eng. 2007;54 (6 pt 1):1005.
Della Santina CC, Migliaccio AA, Patel AH. A multichannel semicircular canal neural prosthesis using electrical stimulation to restore 3-d vestibular sensation. IEEE Trans Biomed Eng. 2007;54(6 pt 1): 1016.
APPENDIX 20–1. SAMPLE HOME EXERCISE PROGRAM
Head-Turning Practice
These exercises can be performed while sitting in a chair. They should be repeated several times at each session, and they can be practiced as many times as you wish during the day. Sessions should be done at least twice daily, as a minimum. Gradually increase how long and how often you perform these exercises.
FIXATION PRACTICE
Select a target directly in front of you. While looking at the target, slowly turn your head from side to side. Try to keep the target from moving or jiggling as you turn. Repeat this, using up-and-down head movements. If you can keep the target perfectly still while you move, practice doing the head turns a bit faster.
Extend your hand out in front of you and use your thumb as a target. Move your arm back and forth, and follow your thumb with your eyes. Next, combine head and target move- ments. For example, while moving your arm to the left, move your head to the right, all the time keeping your eyes on your thumb. Go back and forth.
TARGET CHANGES
Select two targets, one off to your left, the other off to your right, and far enough apart so that you have to turn your head to look from one to the other. Look at one target; then, as fast as you can, close or blink your eyes and turn your head to look at the other. Go back and forth between the two targets as quickly as possible.
Walking Practice
You should try to spend at least 30 min a day practicing your walking. The more time you spend, the quicker you will adjust.
GUIDED WALKING
Stand next to a wall in a long hallway or a length of wall that you can walk along for at least
15 feet. Touch the wall with your hand and keep it in contact with the wall as you walk along it. When you reach the other end, turn around and go back, using the other hand. Once you are able to walk in both directions along the wall without bumping into it or need- ing to use your hand for support, start walking close to the wall with your hands at your side. Your goal is to walk in a straight line down the middle of the hall or room without needing to use your hands or the wall for balance.
WALKING TURNS
Pick a wall several feet in front of you and place a piece of tape or other target in the middle of it at eye level. Walk toward the target. When you reach the wall, keep your head and eyes locked in place on the target as you start to turn with your shoulders and body. When you can no longer turn without moving your head, close your eyes and rapidly turn your head, opening your eyes as soon as the turn is completed. Walk in a straight line away from the wall. Repeat, turning in the other direction.
OUTDOOR WALKS
Go for walks daily, beginning with a 5-min walk and increasing by at least 5 min every day until you are walking for 30 min. Try to walk at a normal pace. Turn your head from side to side to look at the scenery, closing your eyes or blinking to reduce dizziness.
Other Exercises
Spend as much time as you wish doing these exercises. Aim for at least 5 min each day.
READING PRACTICE
Read anything that you like. If needed, you can balance your head on your hand to keep the words steady as you read. Try to keep the letters in focus while you read.
ROCKING CHAIR EXERCISES
Slowly rock in a rocking chair while you watch TV, or pick a target and try to keep focused on it as you rock. As you improve, rock faster. Turn your head to one side and pick something to
focus on as you rock; then practice rocking with your head turned to the other side.
RIDING IN CARS
Whenever you have the opportunity, ride as a passenger in a car. While the car is in motion, slowly turn your head from one side to the other with eyes closed, then with eyes open. Have the driver accelerate or slow down periodically. It is normal to feel unusual sensa- tions of motion at first, but these diminish with practice.
Dizziness Exercises
These exercises can be done as often as you wish, to keep dizziness under control.
HEAD MOVEMENT DIZZINESS
If dizziness occurs only when your head is turned, check to see if it goes away when you turn your head with your eyes closed. If it does, try to blink or close your eyes while you turn your head, and turn your head faster
than normal. Use the target practice exercises listed earlier. At least twice a day, do the fixa- tion practice exercise, but turn your head quickly to make yourself feel as dizzy as possi- ble. The more you repeat this, the quicker your dizziness will go away. If dizziness occurs even with the eyes closed, you will need to practice turning your head with eyes closed—slowly at first, and gradually increasing the speed until you become used to the dizziness.
EYE MOVEMENT DIZZINESS
Use this exercise if dizziness occurs when you move your eyes, even when your head is held perfectly still. Lie down with your eyes closed. Slowly rotate them around as far to the sides, up, and down as you can. If you feel increased dizziness in one particular direction, try to keep your eyes turned in that direction as you count to 10. Then open your eyes and try to focus on something. As soon as the dizziness decreases (a few seconds), close your eyes and again turn them in the most unpleasant direction. Keep repeating this exercise until the dizziness is decreased.
This page intentionally left blank
![]()
When the first edition of this book was written in the late 1970s, there was great excitement regarding the potential for engineering models to describe vestibular function in normal sub- jects and patients. Steinhausen’s pendulum model of semicircular canal function was shown to reliably predict the flow of action potentials in isolated ampullary nerves in several mam- mals, including primates. A simple first-order approximation of this linear model remarkably predicted vestibulo-ocular responses in normal subjects and provided a framework for inter- preting abnormal responses in patients. These models consisted of “black boxes” represented by transfer functions that described the rela- tionship between the input and output. Little was known about what actually went on in these black boxes. David A. Robinson, one of the pioneers in oculomotor modeling, warned that “Block diagrams of oculomotor organiza- tion serve as a compact description of system behavior but seldom have much bearing on the way in which the real system, composed of nerve and muscle, actually operates. The models thus do not contribute much to the neurophys- iology (or neurology) of eye movements and incur the danger of suggesting that there actu- ally are segregated portions of the nervous system which perform differentiation, integra- tion and other operations indicated in the boxes of the diagrams.”1 But progress in understand- ing basic cellular and molecular mechanisms awaited technical advances in cell and molecular biology. As technical advances have occurred, we can now focus on these black boxes. How do the mechanosensory channels and the ribbon synapses in hair cells work? What transmitters are released and what recep- tors are expressed at peripheral and central
vestibular synapses? What ion channels are expressed in the peripheral and central vestib- ular pathways, and how do these ion channels explain the observed physiology? What changes occur at the molecular level after damage to peripheral and central vestibular structures? In this fourth edition, answers to these questions are beginning to be addressed, but we are still in the early stages of understanding. Future scientific discoveries will no doubt improve our understanding of the vestibular system in health and disease. A better understanding of this intricate system will hopefully lead to the future ability to modulate it or even to substi- tute for it—advances that could enhance the care of patients with vestibular disorders.
Meaningful scientific progress requires translating basic science discoveries into inter- ventions that improve our well-being—a step that hinges on rigorous clinical studies. A poster child for scientific progress in the field of clini- cal neurophysiology of the vestibular system is benign positional vertigo (BPV). BPV is now a well-defined entity, and we have a solid under- standing of the underlying pathophysiology of the disorder. The diagnostic tests for it are reli- able and valid. A hypothesis introduced in the 1960s led to the development and refinement of a treatment, and an early case series in the 1980s suggested its usefulness. Numerous ran- domized clinical trials followed over the past 20 years, and the efficacy of the treatment was solidly established by several independent systematic reviews of the clinical trials. As a result, clinical practice guidelines have been published within the past year.
As noted previously, some future advances will require new scientific discovery, but others will require determining ways to optimize the
431
432 Clinical Neurophysiology of the Vestibular System
use of services already available. Which of our vestibular patients will meaningfully benefit from the interventions we direct them toward andwhichwillbeharmedbythem? Randomized controlled trials are the gold standard method to study the efficacy of any intervention, yet other than a few examples adequate trials are lacking in many areas important for the man- agement and treatment of our patients. Beyond randomized trials, we also need research that defines the effectiveness of our interventions in the real world and that helps us better identify the patients most likely to benefit from what we have to offer.
Our clinical tests need later phase diagnostic test research to better define their value. Many sophisticated tests have been developed to make laboratory measurements of the vestibu- lar system. Most of these tests have been refined for optimal performance. Others are still in the early development phases. These tools play an essential role in research studies. Clinically, some of these tests have proven to be very good discriminators of vestibular pathology, but others have not, particularly when used in certain populations such as the elderly or patients with mild or early-stage dis- orders. Most have not undergone rigorous assessment of the reliability (reproducibility) of the test or the test interpretation outside of expert centers. Furthermore, later phase diag- nostic test research has not been performed with most of these tests. The validity of these tests for important endpoints (imaging find- ings, clinician consensus diagnosis, future diag- nosis or outcome) has not been assessed in populations of patients having important levels of clinical uncertainty. Finally, none of the tests has been subjected to the gold standard deter- minant of the clinical value of a diagnostic test. The gold standard is to assess whether the patients who receive the test have better future meaningful outcomes than patients who do not receive the test. In these designs, the test is
considered the intervention and patients are randomized to have it. When considered at the aggregate level of utilization, the routine use of tests prior to these later phase assessments may result in more unintended consequences (e.g., inconvenience to the patient, adverse events, adverse emotional effects of false-positive results, and excess costs) than meaningful benefit to the patient.
More research to define the clinical value of bedside information (i.e., medical history and examination findings) may help in deciding which patients are likely to benefit from tests or therapies and which are not. For example, the head-thrust test seems to be an exception- ally effective way to assess the likelihood of stroke in patients with acute vestibular syn- drome presentations, but what is the reliability of the test in these circumstances (particularly among frontline clinicians who do not regularly use it) and would it perform better than a validated scale of stroke risk factors?
Finally, we need to understand why one of the most efficacious and simple interventions in all of clinical medicine—particle reposition- ing for BPV—is substantially underutilized. In fact, patients with BPV are more likely to receive a brain imaging study than to be cured with repositioning. Many disorders are desper- ately in need of the discovery of a cure, but BPV already has one. What good are efforts to discover new treatments if they ultimately go unused or misused? Is the underutilization a matter of “clinical inertia” or could underuti- lization stem from other factors, such as policy disincentives?
REFERENCE
1. Robinson DA. Models of oculomotor neural organiza- tion. In: Bach Y, Rita P, Collins C, Hyde J, eds. The Control of Eye Movements. New York: Academic Press;1971:519.
Video 6-1. The past pointing test
With eyes closed, the patient is instructed to lift his arm off the target and then bring the finger back down in the same spot. The patient in the video consistently misses the target to the right side, which is the affected side. Note that the patient in this video is the same patient in Video 6-5.
Video 6-2. The head thrust test
The patient’s eye stays fixed on the target after quick, small amplitude movements of the patient’s head to the left, which is consistent with an intact vestibular ocular reflex on the left side (i.e., negative head thrust test). However, after head movements to the patient’s right side, the patient’s eyes move off the target and then a re-fixation (corrective) saccade is required to bring the eyes back to the target, which is consis- tent with a positive head thrust test on the right side indicating de-afferentation or a lesion of the right vestibular system. Note that the patient in this video is the same patient in Video 6-5.
Video 6-3. The head thrust test
Note that after quick, small amplitude move- ments of the patient’s head to the right side, the patient needs to make a corrective saccade to bring the eyes back to the target (i.e., a posi- tive head thrust test). But, the eyes stay on tar- get after the head thrust test to left side (i.e., a negative head thrust test). This video is partic- ularly helpful for demonstration purposes because the voluntary corrective saccade to the right side is delayed on the first test.
Video 6-4. Spontaneous nystagmus
Patient with high velocity spontaneous left beating nystagmus which increases velocity when the patient looks to the left side and then stops when the patient looks to the right side.
This recording was made on the first day of a vestibular neuritis presentation.
Video 6-5. Spontaneous nystagmus
The patient has spontaneous left-beating nystag- mus. The patient was videotaped on the second day of a vestibular neuritis, and thus the velocity of the nystagmus is less than seen in Video 6-4.
Video 6-6. Second degree nystagmus
The nystagmus velocity increased when the patient looks to the left. The nystagmus stops when the patient looks to the on right gaze. Note that the patient in this video is the same patient in Video 6-5.
Video 6-7. Spontaneous down-beating nystagmus
In primary position, the patient has persistent small amplitude down-beating nystagmus.
Video 6-8. Spontaneous up- beating nystagmus
In primary position, the patient has persistent small amplitude up-beating nystagmus.
Video 6-9. Gaze-evoked nystagmus (multi-directional)
In a patient presenting with acute vertigo and imbalance Note the prominent left-beating nystagmus in left gaze. Next, note that the patient clearly also develops right- and down- beating nystagmus on right gaze. Because the nystagmus changes direction, this is a central pattern even though the velocity is greater in one direction.
433
434 Video Legends
Video 6-10. Gaze-evoked nystagmus and impaired smooth pursuit
The patient develops right beating nystagmus when the patient looks to the right side and left-beating nystagmus when looking to the left side. Also note the impairment of smooth pursuit (i.e., saccadic pursuit) as the patient follows a target back and forth.
Video 6-11. Gaze-evoked downbeating nystagmus
On gaze to either side, the patient develops down-beating nystagmus.
Video 6-12. Benign positional vertigo, posterior canal
The patient was just placed in the right head- hanging position (i.e., right Dix-Hallpike posi- tion). After a brief delay, a burst of upbeat and torsional nystagmus is seen. The duration of the nystagmus is about 12 seconds.
Video 6-13. Benign positional vertigo, posterior canal, testing using goggles
Video goggles were used in this video. The patient is placed in the left Dix-Hallpike posi- tion and a burst of upbeat torsional nystagmus is seen.
Video 6-14. Convergence retraction nystagmus
The patient has spontaneous convergence nys- tagmus. Also note that the patient has impaired upgaze.
Video 6-15. Ocular flutter
Spontaneous bursts of back and forth horizon- tal saccades consistent with ocular flutter. Note
that there is no pause between the individual saccades.
Video 7-1. Saccade dysmetria
The patient is being instructed to look back and forth from one target to another. With each saccade eye movement, the patient over- shoots the target then has to make another sac- cade back to the target.
Video 10-1. Epley Maneuver for right posterior canal benign positional vertigo
First, the patient’s head is turned to the right side. Next, the patient is rapidly brought down to the right head hanging position (i.e., the right Dix-Hallpike test). The clinician observes for the typical burst of upbeat and torsional nystag- mus (see Video 6-7). Next the patient’s head is turned toward the left and the patient rolls over onto the left side, making certain not to allow the patient to lift the head up. At the end of this position, the patient is lying on the left side with the face turned so the patient is looking at the ground. This position is maintained for about 30 seconds. Then, the patient is rapidly brought back up to the sitting position.
Video 10-2. The Gufoni maneuver for patients with left horizontal canal benign positional vertigo
From the seated position, the patient is quickly placed in the right decubitus position. This position is maintained for approximately 10-20 seconds. Next, the patient’s head is turned so that the patient is looking into the table. This position is maintained for approximately 20-30 seconds, then the patient is quickly brought back up to sitting position.
* Videos can be found at the following website: www.oup.com/us/cns/vestibular
Abducens nerve, 77, 78, 88, 238
Aberrant torsional eye movements, 199, 201f Ablative procedures, for Meniere’s syndrome, 283 Abscess
epidural, 239 Acetazolamide (Diamox)
for episodic ataxia type 2, 132, 397 for familial periodic ataxia, 132
for ion channel disorder, 293 for Meniere’s syndrome, 282 for migraine, 294, 298t, 299 for spinocerebellar ataxia, 398
Acoustic compliance, 223 Acoustic impedance, 223, 224f Acoustic reflex, 224–25
Acquired disorders, 384 Acquired hearing loss, 384
Acquired syphilitic labyrinthitis, 248 Actin, vestibular hair cell
electromotility and, 40, 40f Actinomycosis, 250
Active head rotation, 192–93, 198f Acyclovir
for sudden sensorineural hearing loss, 247 for viral infection, 247
Adenoid cystic carcinoma, 339, 340 Aditus ad antrum, 25
Adrenocorticotropic hormone (ACTH), for multiple sclerosis, 313
Adults
benign recurrent vertigo of, 291, 291t brain stem gliomas in, 347
Chiari type I malformation in, 391 dizziness in, 226
inner ear viral infections in, 242 malignant tumors in, 340 metastatic cerebellar tumors, 348
maternally inherited mitochondrial disorders in, 388 neurologic disorders in, 274
otoconia mineralization and turnover, 48 sudden deafness in, 245
syringobulbia in, 392 toxic/metabolic disorders in, 377 vestibular neuritis in, 244
Afferent nerve activity, hair cell influence on, 42–43 Agoraphobia, 135
acute toxic effects of, 368–70, 369f and thiamine deficiency, 368–70
acute alcohol intoxication, 368 cerebellar degeneration, 370, 371f Wernicke’s encephalopathy, 370
Alpha2-adrenergic receptor gene, 143
Alpha-tocopherol transfer protein (-TTP), 395 Alport’s syndrome, 368, 385–86, 385t Alprazolam, for postconcussion syndrome, 364 American Academy of Ophthalmology, 290 Amikacin, 374t
Aminoglycosides, 374–75 dizziness caused by, 137
vestibular and auditory ototoxicities of, 374t Amitriptyline, for migraine, 298t
Amoxicillin, for acute otitis media, 235 Amphetamine
for Meniere’s syndrome, 281t vestibular rehabilitation, 419
Amphotericin, mycotic mastoiditis, 250
Ampulla
Ampullofugal endolymph flow, 8, 44, 54, 69, 70, 76, 77,
77f, 80, 105, 178, 179, 192, 196, 197, 199, 260, 260f,
Ampullopetal endolymph flow, 8, 44, 54, 69, 70, 76, 77,
77f, 80, 105, 178–80, 178f, 192, 196, 197, 199, 266, 268,
Angiography, for vertebrobasilar ischemia, 328–29 Angular velocity, 193
sinusoidal changes in, 193–95 step changes in, 193
Anterior canal benign positional vertigo, 265, 268 Anterior inferior cerebellar artery (AICA), 35, 36f, 320,
anatomy of, 324, 324f, 326f internal auditory artery, 324f and lateral medullaty infarction
symptoms and signs in, 323t recurrent penetrating arteries, 324f terminal cerebellar branches, 324f
Anterior vestibular artery, 35, 36f Anterior vestibular vein, 35, 36f Antiamphiphysin antibody, 310t Antibiotics
for acute otitis media, 235–36 for bacterial labyrinthitis, 237
Antibodies, associated with cerebrellar ataxia, 315
Anticoagulants, for transient ischemic attacks, 330 Anti-CV2 antibody, 310t
Antidopaminergic drugs, for nausea and vomiting associated with vertigo, 406t, 411
Antiemetic drugs, 410–13 instructions to patients, 413
for Meniere’s syndrome, 280, 281t precautions, 411, 413
435
Antigen-specific laboratory testing, in autoimmune inner ear disease, 308
Anti-glutamic acid decarboxylase (anti-GAD), 315 Anti-Hu antibody, 310t, 311
Antihypertensive drugs, dizziness caused by, 137 Anti-inflammatory drugs, ototoxicity of, 375–76 Anti-Ma antibody, 310t
Antiplatelet agents, for stroke prevention, 330 Anti-Ri antibody, 310t, 311
Antiserotonergic drugs, for nausea and vomiting associated with vertigo, 406t, 411
Anti-Ta antibody, 310t Anti-Tr antibody, 310t
Antivertigo drugs, 406t, 413–16 for Meniere’s syndrome, 281t
Antiviral agents, for sudden sensorineural hearing loss, 247 Anti-Yo antibody, 310t, 311
Antoni type A schwannoma, 342 Antoni type B schwannoma, 342 Antrum, 235
Apogeotropic nystagmus, 267 Apoplectiform cerebral congestion, 273 Apraxia
Aprepitant, for nausea and vomiting, 411
Arabinase, for sudden sensorineural hearing loss, 247 Arnold-Chiari malformation, 160
Arnold nerve, glomus tumors in, 340 Arousal, 81
Ascending tract of Deiters (ATD), 15 Aspirin
for hearing loss, 377 for migraine, 298t
for transient ischemic attacks, 330 Assimilation of the atlas, 391 Astrocytoma, 348
Asymmetric gaze-evoked nystagmus, 162 Ataxia
autosomal dominant spinocerebellar, 393–94, 395t autosomal recessive spinocerebellar, 394–96 cerebellar, and multiple sclerosis, 314–15
episodic, and vertigo syndromes, 396 Friedreich’s, 394–95
inherited spinocerebellar, 393–98
spinocerebellar, 183, 393, 398
telangiectasia, 185 Atenolol, for migraine, 298t Atherosclerosis, 368
ATP1A2 gene, and familial hemiplegic migraine, 293 Audiogram, 221–22, 222f
for cerebellopontine angle tumors, 344 Audiometry, 221–23
audiogram, 221–22, 221f, 222f, 222t speech recognition tests, 223 Stenger test, 223
for brain trauma, 363 impedance, 223, 224f
for Meniere’s syndrome, 278–80, 279f Audiovestibular loss. See also Hearing loss genetic syndromes with, 384–86, 385t
dominantly inherited hearing loss, 386t, 387 inherited vestibular loss with normal hearing, 388 maternally inherited mitochondrial disorders, 387–88 recessively inherited hearing loss, 386–87, 386t
Audio vestibular system infectious diseases of, 233
Audiovestibular testing, for autoimmune inner ear disease, 307–8
Auditory-evoked responses, 225–26, 226f brainstem auditory-evoked response, 226 electrocochleography, 226
Auditory nerve, 11f, 37 Aura
migraine without, 288, 296, 296t
Autoimmune injury, and Meniere’s syndrome, 278 Autoimmune inner ear disease, 303–9
systemic immune-mediated diseases, 306–7 diagnosis of
audiovestibular testing, 307–8
Autosomal dominant spinocerebellar ataxia syn- dromes, 393–94
horizontal eye movement abnormalities with, 394, 395t
Autosomal recessive spinocerebellar ataxia syn- dromes, 394–96
Baclofen and periodic alternating nystagmus, 161 Bacterial labyrinthitis, 236–37, 246
Barbeque” maneuver, for geotropic nystagmus, 267, 267f Barbiturates
dizziness caused by, 137t Basal cell carcinoma, 339
Basal ganglia, lesions of, 138, 139f, 184t Basilar impression, 390
diagnosis of, 296–97 symptoms of, 289, 289t
Bechterew’s nucleus. See Superior vestibular nucleus Bedside examination
of hearing, 131–32, 131f, 220–21 of vestibular system, 149–67
Behavioral audiometry, 221–23, 221f, 222f, 222t Behavioral therapy, for phobiac dizziness, 137
Benign paroxysmal positional nystagmus, 178f Benign paroxysmal positional vertigo. See Benign
positional vertigo (BPV)
Benign positional vertigo (BPV), 123–24, 255–69, 256f, 354 anterior canal benign positional vertigo, 268 background, 255–57
of childhood, 290 diagnosis of, 259t
horizontal canal benign positional vertigo, 265–68 mimics of, 268–69
posterior canal variant of, 258–65 pathophysiology, 260–61
Benign positional vertigo, 364t
Benign recurrent vertigo of adulthood, 291, 291t Benzathine penicillin, for syphilitic inections, 249 Benzodiazepines, 137, 405, 407, 412t
for nausea and vomiting associated with vertigo, 408, 410, 411
Benzquinamide hydrochloride (Emete-con), 406t for mild, persistent nausea, 415
for nausea and vomiting associated with vertigo, 412t Beta blockers
for ion channel disorder, 293 for migraine, 299
for vertigo associated with Meniere’s syndrome, 414 Bilateral peripheral lesions, 200–202
Bilateral sequential vestibular neuritis, 244 Bilateral vestibular lesions, 424–25, 424t
Bilateral vestibular schwannomas, 342–43 Binaural diplacusis, 220
Biopsy, for brainstem and cerebellar tumors, 349 Bird, labyrinth of, 32f
Bithermal caloric test, 177–83, 180f, 181f, 182t in central lesions, 182–83
mechanism of stimulation, 177–79, 178f methodology for, 179–80
in peripheral lesions, 181–82 Bleeding diathesis, 332
Blood supply, in inner ear, 35–36
Bone morphogenetic protein 13 (BMP13), 391
Brain-derived neurotrophic factor (BDNF), 72, 426 Brain imaging, for vertebrobasilar ischemia, 326–28 Brain stem
Brainstem auditory-evoked response (BAER), 226, 227f, 228f, 389
in acoustic neuroma, 344 in brain trauma, 363
in multiple sclerosis, 313 in whiplash injuries, 363
Brain stem hemorrhage, vertigo and, 332 Brain trauma
dizziness due to brainstem trauma, 362 management of, 364
postconcussion syndrome, 362–63 temporal bone fractures, intracranial
diagnosis and management of, 348–49 Bright’s disease, 385
Buclizine hydrochloride (Bucladin-s), 406t, 409t for preventing motion sickness, 413
for vertigo due to vestibular lesions, 413 Burst tonic (BT) neurons, 78
Butyrophenones, extrapyramidal side effects of, 411
and familial hemiplegic migraine, 293
Calcium channel blockers, for ion channel disorder, 293 Caloric fixation suppression index, 181
bithermal, 177–83, 180f, 181f, 182t in central lesions, 182–83 mechanism of
in peripheral lesions, 181–82 cold, 155–56
Canalithiasis theory, 163, 257, 260
Canal-ocular reflex, 193, 195f Canal segment, of facial nerve, 28
Cannabinoids, for nausea and vomiting associated with vertigo, 406t, 411
Carbamazepine
for vestibular paroxysmia, 335 Carboplatin, ototoxicity of, 376 Carcinoma
Cardiac output, impaired, near-faint dizziness, 133 Casopitant, for nausea and vomiting, 411 CCA1/KRIT1, 348
Center of pressure (COP), 208, 209f Central auditory speech tests, 228–29
Central compensation for vestibular lesions, 21–22 Central hearing disorders, 220
Central positional nystagmus, 164
vs peripheral positional nystagmus, 164t Central processor, in orientation, 22
Central spontaneous nystagmus, 159–60
vs peripheral spontaneous nystagmus, 159t Central vestibular
Central vestibular lesions, 202–3, 425, 425t
Central vestibular pathways, vestibular nuclei, 11–12 Central vestibular system, 63
cervico-ocular reflexes, 87–89 subjective vestibular
vestibulo-ocular reflexes, 72–86
vestibulospinal reflexes, 97–102
visual–vestibular interaction, 89–97 Cerebellar arteries, branches of, 325, 326f
Cerebellar ataxia, and multiple sclerosis, 314–15 Cerebellar–vestibular interaction, 100–101, 101f Cerebellopontine angle
lesions of, 184t vertigo in, 130t
surgical approaches to, 347 tumors of, 341–47
brainstem auditory-evoked responses in, 344 diagnostic algorithm of, 345f
management of, 346–47 Cerebellopontine angle cistern, 36
Cerebellopontine angle tumors, diagnosis of, 345f Cerebellum, 64f
degeneration of
in alcoholism, 370, 371f paraneoplastic, 309–10
hemorrhage into, 332–34, 333f infarction of, 324–25
lesions of
caloric testing in, 182t, 183 disequilibrium in, 138, 139f rotational testing in, 156, 202
smooth pursuit and, 187 Cerebellum, lesions of, 183
Cerebral cavernous malformations (CCM), 348 Cerebral edema, after temporal bone fracture, 362 Cerebrospinal fluid (CSF), 33, 311, 354, 357
Cerumen, impacted, 149, 156, 179, 219, 224
Cervical synostosis, congenital, 391 Cervical VEMPs (cVEMPs), 211–13 Cervicodorsal roots, stimulation of, 87, 88
Cervicomedullary compression, 392 Cervico-ocular reflexes¸421
anatomic and physiologic basis, 87–88
neck-induced eye movements, characteristics of, 88–89 and vestibulo-ocular, synergistic interaction of, 88, 89f
management of, 392–93, 393f Children
acute otitis media in, 234 astrocytoma in, 348
autosomal recessive spinocerebellar ataxia syndromes in, 396
bacterial meningitis in, 237 basilar migraine in, 289
benign paroxysmal vertigo of, 290 brain stem gliomas in, 347 cerebellar tumors in, 348
congenital hearing loss in, 384, 385 with congenital hypothyroidism, 368 dizziness in, 226
medulloblastomas in, 348 Meniere’s syndrome in, 274 migraine without aura in, 296 opsoclonus in, 310
rhabdomyosarcoma in, 340 sensorineural hearing loss in, 220 and suppurative labyrinthitis, 237 toxic/metabolic disorders in, 377 vestibular loss in, 421–22
for nausea and vomiting associated with vertigo, 412t Cholesteatoma, 235–36, 236f
diagnosis of, 235–36, 236f management of, 236
Choline acetyltransferase (ChAT), 52, 56
Chromosome 12p, and Meniere’s syndrome, 277 Chronic anxiety, 136
Chronic subjective dizziness. See Psychophysiologic dizziness
Cilia, of hair cell, 5, 5f, 6. 42f Cisplatin, dizziness caused by, 137 Cis-platinum, ototoxicity of, 376
Clavulante, for acute otitis media, 235 Clonazepam, for opsoclonus, 312 Coccidioidomycosis, 250
COCH gene mutation, hearing loss associated with, 387 Cochlea, 11f
Cochlear aqueduct, 33–35, 33f, 36f, 237, 357, 359f
Cochlear hearing loss, 225, 227, 229
Cochleosaccular dysgenesis, 389 Cochrane Collaboration, The, 422
Cogan’s syndrome, with inner ear involvement, 305 Cogwheel pursuit, 187
Cold caloric test, 155–56 Comatose patient
reflex eye movements in, 153 Common cochlear artery, 35, 36f
Compensatory eye movements, 77–78, 77f Computed tomography angiography (CTA)
for vertebrobasilar ischemia, 329 Computerized tomography (CT), 206
for atlantoaxial dislocation, 392
for brain stem and cerebellum hemorrhage, 333, 333f for brain stem gliomas, 349
for cerebellopontine angle tumors, 344
for inner ear abnormalities with Klippel-Feil syndrome, 391
for inner ear genetic disorders, 390
for internal auditory canal and cerebellopontine angle tumors, 345
for middle ear and temporal bone tumors, 340 for otosclerosis, 373
for semicircular canal dehiscence syndrome, 361f for temporal bone trauma, 355, 355f
of temporal bones
with Mondini malformation, 389, 389f with cholesteatoma, 236f
for vertebrobasilar ischemia, 327–28
for whiplash injuries, 363 Concussion
labyrinthine, 353–54, 355t symptoms after, 363t
Conductive hearing loss, 219 Congenital cervical synostosis, 391
Congenital cytomegalovirus (CMV), and acquired hearing loss, 384
Congenital spontaneous nystagmus, 160–61 Congenital syphilitic labyrinthitis, 248 Conventional contrast angiography, 329
for recurrent vertigo attacks, 329–30f Conventional rotational chair, 190t Convergence retraction nystagmus, 165 Corneal–retinal potential, definition of, 171 Cortical spreading depression, 294 Corticosteroids
for autoimmune inner ear disease, 308 for bacterial labyrinthitis, 237
for sudden sensorineural hearing loss, 247 Counterrolling, definition of, 207
Cows (cold opposite, warm state), 178 Cranial nerve
Cranial vertebral junction disorders, 390–93 atlantoaxial dislocation, 391
properties of afferent nerve fibers in, 56f Crocodile, labyrinth of, 32
displacement of, 45–47 Cupula
anatomy of, 7, 9f, 17f Cupulolithiasis, 256, 257
Cupulometry test, 105 Current of silence,” 41 Cuticular plate
anatomy of, 5, 5f Cyclizine, 406t, 409t
Cyclophosphamide, for immune-mediated inner ear disease, 308
Cyst, epidermoid, of cerebellopontine angle, 343 Cytosine, for sudden sensorineural hearing loss, 247 Cytosine arabinase, for viral infection, 247
Davis mechanoelectric theory, of hair cell function, 41 Deafness. See Audiovestibular loss; Hearing loss
Deep tendon reflex, 19, 36, 152, 395
Deiters’ nucleus. See Lateral vestibular nucleus Delayed endolymphatic hyrdrops, 275 Demonstrated positional nystagmus, 348 Demyelination, in multiple sclerosis, 312–13
Dephenidol, for nausea and vomiting associated with vertigo
Descending (inferior) vestibular nucleus, 67 Destructive procedures, 282–83
Developmental disorders, 383–98
presyncopal light-headedness and, 368 vascular changes with, 367
Diazepam (Valium), 161, 406t, 409t, 410 for Meniere’s syndrome, 280, 281t
for nausea associated with vertigo and motion sickness, 415
for postconcussion syndrome, 364 vestibular rehabilitation, 419
Dibekacin, ototoxicity of, 374t
Diffuse cerebellarIntrinsic brain stem, lesions of, 184t Dihydroxyergotamine, for migraine, 298t Dimenhydrinate (Dramamine), 406t
for Meniere’s syndrome, 281t for nausea and vomiting, 414
during pregnancy, 410 side effects of, 414
vestibular rehabilitation, 419 Dimorphic afferents, 54f Dimorphic unit, 52, 55
Diphenhydramine hydrochloride (Benadryl), 406t, 409t for mild to moderate episodes of vertigo, 413
for motion sickness, 413 side effects of, 413
Diphenidol (Vontrol), 406t, 415
for nausea and vomiting associated with vertigo, 412t Direct fluorescent antigen assay (DFA), for herpes zoster
Directional preponderance
with caloric stimulation, 182t formula for, 181
Disequilibrium
common causes, 138, 139f diagnosis and management of, 140 falls in older people, 139–40
gait disorders in older people, 138–39 Dissociated/disconjugate gaze-evoked nystagmus, 162 Dissociated-spontaneous nystagmus, 165
Dix-Hallpike positioning test, 158, 163, 177, 262, 268 for benign positional vertigo, 355
Dizziness, 127. See also Disequilibrium, Vertigo after whiplash injuries, 363–64
due to brainstem trauma, 362, 364t epidemiology of, 121–25
burden on patients, 124 health-care utilization, 125
population prevalence of, 122f specific disorders, 123–24
falls in older people due to, 139–40, 139t head trauma induced, 257–58, 259t hypoglycemia, 138
Dizziness (Cont.) mechanisms of, 128t and migraine, 287
near-faint, 132–34 causes of, 132t
with hyperventilation, 134 with impaired cardiac
output, 133 with orthostatic
with vasovagal attacks, 133–34 ocular, 140–42, 141f
psychophysiologic, 134–37, 135t
and systemic metabolic disorders, 367–71 alcohol and thiamine deficiency, 368–70 diabetes mellitus, 367–68
vestibular and nonvestibular types, 144–45, 144t DNA mutations, mitochondrial, 388 Dolichoectasia, definition of, 334
Dominantly inherited hearing loss, 386t, 387 Domperidone, 406t, 411, 412t
indications for use, 412t
for nausea and vomiting associated with vertigo, 408, 411, 412t
Dorsolateral pontine nucleus, in visual-vestibular interaction, 94–95, 94f
causes of, 159–60 Dronabinol (Marinol), 406t
extracted from Cannabis sativa, 416
for nausea and vomiting associated with vertigo, 412t Droperidol (Inapsine), 406t, 409t, 411, 415
for nausea and vomiting associated with vertigo, 408, 412t Drug-induced dizziness, 137–38, 137t
Drug intoxication syndrome, 137 Drug ototoxicity, risk factors for, 377t Dynamic ocular counterrolling, 190t
Dynamic theory of endolymph volume, 34, 276 Dynamic visual acuity test, 155
Dysrhythmia
with caloric stimulation, 182t Dysrhythmic nystagmus, 183, 184t, 202
Ear. See also Inner ear; Middle ear cross section of, 26f
embryological development of, 38f examination of, 149–50, 150f
Ear drum. See Tympanic membrane Ear infection, 233–50
intracranial extension of, 238–41 diagnosis of, 240–41, 240f management of, 241
routes of spread in, 238–39 mycotic, 250
syphilitic, 248–49, 249f Eccentric head rotation, 207
Ecchymosis in temporal bone fracture, 353 Echocardiography, for vertebrobasilar ischemia, 328 EDN, 385
Edrophorium test, in pseudo medial longitudinal fascicle nystagmus, 162
Efferent vestibular neurons, 56–57 Eighth nerve lesions, 182
Electrocochleography (ECoG), 226, 226f, 279 for Meniere’s syndrome, 279
Electrode systems for electroculography, 171–72, 172f
Electronystagmography (ENG), 162, 171, 172f, 174f for brain trauma, 363
for perilymph fistula, 357 test battery, 175t
for whiplash injuries, 363 Electrooculography, 85, 171
for ocular counterrolling, 207 Encephalomyelitis
in paraneoplastic immune disorders, 310–11 Endolymph, 34
volume, regulation of, 34 Endolymphatic duct and sac, 37
Endolymphatic hydrops. See Meniere’s syndrome Endolymphatic sac, 26f, 33, 33f, 34, 277, 282, 303, 359f
autoimmune injury to, 278 tumors, 332
ENG. See Electronystagmography EOG. See Electrooculography
Ephedrine, for Meniere’s syndrome, 281t Epidermoid cysts of cerebellopontine angle, 343, 345 Epidural abscess, secondary to ear infection, 239 Episodic ataxia and vertigo syndromes, 396
Episodic ataxia type 1 (EA-1), 396 Episodic ataxia type 2 (EA-2), 396, 397f Episodic ataxia type 5 (EA-5), 396
Episodic ataxia type 6 (EA-6), 396
Episodic vertigo, and transient ischemic attacks (TIAs), 320–21
Epitympanic recess, 25 Epley maneuver, 257, 263f
for posterior canal variant of benign positional vertigo, 262f, 263, 264
Equilibrium
loss of. See Disequilibrium
maintenance of, vestibular reflexes in, 13
Etanercept, for immune-mediated inner ear disease, 309 Ethacrynic acid, ototoxicity of, 375
Eustachian tube
abnormalities of, in otitis media, 234
Evoked potentials, vestibular, laboratory evaluation of, 209–10, 210f
Evoked responses, auditory. See Auditory evoked responses; Brain stem auditory evoked responsees
Exercise, after vestibular loss, 422–26. See also Vestibular exercises
Experimental lesions, effect of, 80 External otitis
Eye-head (EH) neurons, 78 Eye motion, pattern of, 77f, 80 Eye movements
characteristics of, 82f, 84–85, 85f compensatory, 77–78, 77f, 82f
neck-induced, characteristics of, 88–89 off-center axis rotation, 86
off-vertical axis rotation, 86 recording methods, 171–74
electrooculography, 171–72, 172 infrared video recording, 172 magnetic search coils, 172
saccadic, 184, 184f, 186f by sinusoidal angular
acceleration, 14f, 16f vestibular-induced, 92
visually guided tracking, 91–92
Eye velocity versus head velocity, 197f
Facial nerve, 11f, 26f, 28–29 schwannomas, 342
Falls, in older people, 139–40, 139t Familial bilateral
Familial hemiplegic migraine (FHM), 292–93 genetic factors of, 293
Familial periodic ataxia and vertigo, 132, 396 Fast component generation, 79–80
Fastigial nucleus, 101–2, 101f Fatigue
saccadic abnormalities in, 185
smooth pursuit abnormalities in, 187
Fetal alcohol syndrome, inner ear disorders in, 384, 390
Fibrous dysplasia of temporal bone, 373 First-degree nystagmus, 157
Fish
vestibular labyrinth of, 30, 30f vestibular nuclei of, 65
Fistula, Perilymph. See Perilymph fistula Fistula test, 150
Fixation, caloric testing with, 179–80 Fixation exercise, 423, 429
Fixation suppression index, 181 Flat tympanogram, 224
Floccular target neurons (FTNs), 97 Florical, for otosclerosis, 374
Fluid-attenuated inversion recovery (FLAIR), 313, 326,
for Wernicke’s encephalopathy, 370 Flunarizine, for migraine, 298t
Fluorescent treponemal antibody absorption (FTA-ABS) test, 248
Fluoxetine, for migraine, 298t Forced prolonged position”
for geotropic nystagmus, 267–68
Fourth ventricle lesions of, 183
Fracture of temporal bone, 353
intracranial complications associated with, 362 Frataxin gene mutation, 395, 397
Free-floating otoconia, and benign positional vertigo, 258 Frenzel glasses, 157f
Friedreich’s ataxia (FA), 394–95
Frontal lobe, lesions of, 138, 139f Frontoparietal cortex, lesions
Functional brain imaging, in normal human sub- jects, 104–5
Functional magnetic resonance imaging (fMRI), in normal human subjects, 104
Gabapentin
for vestibular paroxysmia, 335 Gait
and balance exercises, 425 disorders, in older people, 138–39
Gamma-amino butyric acid (GABA), 43, 161
Gastropod, orientation in, 11, 13
Gaze-evoked nystagmus, 161–62, 176–77
asymmetric, 162 causes, 161t dissociated, 162
symmetric, 161 Gaze stability
bilateral vestibular lesions, 424 central vestibular lesions, 423
during horizontal head rotations, 200f unilateral vestibular lesions, 423
Gegenhalten, 139 Gelatin layer, 8f Gene chips, 390, 397
with audiovestibular loss, 384–86, 385t Genetics
and Meniere’s syndrome, 277–78 and migraine, 292–93
Genetic testing, for congenital hearing loss, 390 Genetic testing
in congenital inner ear disorders, 390 in spinocerebellar ataxia, 393
for Meniere’s syndrome, 283 Geotropic nystagmus, 265–67
GJB2 gene mutation, hearing loss associated with, 386, 387, 390
Glatiramer acetate, for multiple sclerosis, 314 Glioma
of brain stem, 347–48 of cerebellum, 348
Glomus body tumors (paragangliomas), 340
Glossopharyngeal nerve, glomus tumors in, 340 Glutamate, 43, 52
Glycerol test, in Meniere’s syndrome, 279–80 Glycine, 52
Greater superficial petrosal nerve, 28, 29f Group A Streptococcus, 234
Gufoni maneuver, for geotropic nystagmus, 267 Guild theory, of endolymph volume, 34
Habituation, nystagmus and 81 Hair cell, 4–6, 39–43
activation
and direction of force, relationship between, 41 mechanism of, 41–42, 41f
afferent nerve activity, influence on, 42–43 cilia, 5, 6
function, Davis mechanoelectric theory of, 41
hair cell/afferent nerve junction, signal processing at, 43
hyperpolarization, 6, 7f in mammals, 39, 39f
morphologic characteristics, 39–40
transduction, model for, 40f types, 5–6, 5f
Haloperidol, for nausea and vomiting associated with vertigo, 408, 411
Head acceleration
angular, cupula displacement during, 9f, 10, 45–47, 46f linear
eye movements produced by, 77, 77f ololith displacement during, 6
Headache
migraine, after caloric testing, 179 migraine aura without, 297
in basilar migraine, 296–97 Head injury, 131
benign positional vertigo, 354 fracture, 353
labyrinthine concussion, 353–54 Head movement
dizziness associated with, exercise for, 140 force associated with, 3
oscillopsia with, 21t, 141, 141f in space, 143
Head rotarion. See also Rotational testing active (autorotation), 192–93
compensatory eye movements associated with, 15, 16f, 77, 77f
eccentric (off-center), 190f, 190t, 207 high-acceleration small-amplitude, 192 off-vertical, 190f, 190t, 207–8
Head-shaking nystagmus, 164 Head-thrust test, 153, 154f
Head tilt, compensatory eye movement associated with, 82, 82t
Head trauma
brain stem injury in, 362 dizziness after, 362, 364t
loss of consciousness after, 361 positional vertigo after, 257
postconcussion syndrome after, 362–63, 363t Head-turning exercises, 428
Hearing, clinical evaluation of auditory-evoked responses, 225
brainstem auditory-evoked response, 226 electrocochleography, 226
speech recognition tests, 223 Stenger test, 223
central auditory speech tests, 228–29 impedance audiometry, 223, 224f potentials, generating, 226–27
results in patients, 227–28 test methodology, 227
Hearing disorders, types of, 219–20 central hearing disorders, 220 conductive, 219
sensorineural, 219–20 Hearing level (HL), 221
Hearing loss. See also Audiovestibular loss acoustic reflex testing in, 224–25 acquired, 384
auditory-evoked responses in, 225–26, 226f auditory tests in, summary of, 229, 229t bedside testing for, 220
behavioral audiometry in, 221–23
brainstem auditory-evoked responses in, 225–26 central, 220
central auditory speech tests for, 228–29 conductive, 219
hereditary, 384–88, 385t, 386t impedance audiometry in, 223, 224f sensorineural, 219–20
in autoimmune inner ear disease, 304–7, 306f in Meniere’s syndrome, 357
patterns of, 222f sudden, 245–46
speech recognition tests for, 223 Stranger test for, 223 tympanometry in, 224, 225f viruses associated with, 242t
Heat-shock protein 70 (HSP-70), 305
Hemophilus influenza, 234, 239 Hemorrhage
into brain stem and cerebellum, 332–34 diagnosis and management of, 333–34
Hereditary disorders, 384–88 inherited syndromes, 384–86, 385t
nonsyndromic audiovestibular loss, 386–88, 386t
Hereditary nephritis. See Bright’s disease Heredity, and Meniere’s syndrome, 277–78 Herpes simplex virus type 1 (HSV-1), 244, 267 Herpes zoster oticus, 243–44
High-acceleration test, advantage of, 192 Hindbrain, 38f
congenital malformation of, 391, 392 efferent vestibular neurons in, 56
Horizontal canal benign positional vertigo, 265–68, 267f apogeotropic nystagmus, 267
geotropic nystagmus, 265–67 treatment of, 267–68, 267f
Horizontal canal-ocular reflex, 13–15, 14–15f Horizontal eye movement abnormalities
with autosomal dominant spinocerebellar ataxia syndromes, 394, 395t
type and degree of, 395t Huntington’s disease, 185
Hydrocephalus, otitic, 240, 241 Hyperactive responses
with caloric stimulation, 182t Hyperbilirubinemia, 384
Hypermobile tympanogram, 224 Hyperventilation
near-faint dizziness with, 134 nystagmus due to, 164
Hypoglycemia
dizziness due to, 138 functional, 138
Hyponatremia, 368 Hypotension
Ibuprofen¸for migraine, 298t Imbalance. See Disequilibrium
Immune assays, in autoimmune inner ear disease, 305 Immune-based therapy, for multiple sclerosis, 315 Immune disorders, paraneoplastic, 309–12
antineuronal antibodies in, 309, 310f clinical features of, 309–11 diagnosis of, 311
Immune-mediated disorders, 303–15, 314–15 Immunohistochemical staining, of vestibular nuclei, 65f Immunosuppression, for paraneoplastic immune
Impaired fixation suppression with caloric stimulation, 182t
Impaired saccade accuracy, 185 Impedance audiometry, 223, 224f Incudomalleal articulation, 27
Incus, 27, 27f, 355 Infarction
lateral medullary, 322–23, 322f, 323t lateral pontomedullary, 323–24, 324f treatment for, 331–32
Inferior vestibular nucleus. See Descending (inferior) vestibular nucleus
Infrared video recording, of eye movements, 172, 173, 173f
Inheritance. See Heredity
Inherited bilateral vestibulopathy, 388
Inherited spinocerebellar ataxia syndromes, 393–98 autosomal dominant, 393–94
episodic ataxia and vertigo syndromes, 396 episodic ataxia type 1, 396
episodic ataxia types 3–7, 396
diagnosis of, 396–97 gene location, 394t management of, 397–98
Inherited vestibular loss with normal hearing, 388 patients with, 388
Inner ear, 359f, 383–90 acquired disorders, 384
blood supply, 35–36 cross-section of, 33f diagnosis of, 389–90
hereditary disorders, 384–88 inherited syndromes, 384–86, 385t
nonsyndromic audiovestibular loss, 386–88, 386t innervation, 11f, 36–37
mycotic infections of, 249–50 pathology, 388–89
receptor organs, stimulus specificity of, 10 structure, 32–33, 33f
systemic immune-mediated diseases of, 304t, 306–7 viral infections of, 242–47, 242t
versus other causes of peripheral cochleovestibular loss, 246–47
Inner ear vestibular receptors, 43–49 otolith organs
physiology of, 49 semicircular canals
physiology of, 44–47 Innervation
of inner ear, 36–37 of macules, 52
Instantaneous firing rate (IFR), 74f Interferon-beta, for multiple sclerosis, 314
Internal auditory artery, occlusion of, 35, 321, 324f Internal auditory canal, 11f, 25, 36–37
lesions of, 29, 29f, 130, 130t
diagnosis of, 344–46, 345t, 346f management of, 346–47
Internal auditory canal tumors, 341–47
Internuclear ophthalmoplegia, 162
Interstitial keratitis, and syphilic infections, 248 Interstitial nucleus, of vestibular nerve, 67–68 Intracranial complications of ear infections, 238–41
diagnosis of, 240–41 algorithm for, 240f
lateral sinus thrombophlebitis, 239 meningitis, 239
otitic hydrocephalus, 240 routes of spread, 238–39 management of, 241
Intralabyrinthine conductive hearing loss, 225 Intralabyrinthine hemorrhage, 332
diagnosis and management of, 332 Intrinsic brain stem, lesions of, 184t
Intrinsic membrane properties, of secondary vestibular neurons, 70–71
Inverse ocular bobbing, 166 Ion channel
differential expression of, 53 disorders, 293, 293t
expression pattern in rodent ganglion somata, 53t Iron chelators, for prevention of aminoglycoside
Jacobson’s nerve, glomus tumors in, 340 Jellyfish
Jervell and Lange-Nielsen syndrome, 385, 385t Jugular foramen syndrome, 340
K+ secretion, 34
Kanamycin, ototoxicity of, 374t KCNA1, 396
inner ear abnormalities with, 391 Korsakoff’s syndrome, 371
Laboratory examination, of vestibular system, 171–213 nystagmography, 171–89
vestibular-evoked potentials, 209–13
vestibulo-ocular reflexes, 189–203
vestibulospinal testing, 208–9
visual–vestibular interaction, 204–7 Labyrinth
bony, 4, 32–33, 129, 150, 151, 235, 245, 340, 356, 357,
embryonic development of, 37–38, 38f hemorrhage into, 332
fluid chemistry of, 34–35 fluid dynamics of, 33–34 innervation of, 36–37
phylogeny, 29–32, 30–32f structure of, 32–33, 33f
for Meniere’s syndrome, 283 spontaneous nystagmus after, 16, 17f
Labyrinthine artery, 35, 36f Labyrinthine circulation, 36f Labyrinthine concussion, 332, 353–54
diagnosis of, 354–55 Labyrinthine fluid dynamics, 34 Labyrinthine infarction, 321–22
Labyrinthine ischemia, 36, 246–47
Labyrinthine lesions, Symptoms and signs after, 20–21, 21t
Labyrinthine trauma, 131 Labyrinthitis
hyperventilation-induced nystagmus associated with, 164
Lamina cribrosa, 36 Lamprey
labyrinth of, 32 vestibular nuclei of, 65
Latency associated transcript, 244
Lateral medullary infarction, 167, 322–23, 322f Lateral pontomedullary infarction, 323–24 Lateral sinus thrombophlebitis, 239
Lateral vestibular nucleus, 66–67
Lateral vestibulospinal tract, 98–99, 100f Lateropulsion, 323
Leukemia, intralabyrinthine hemorrhage in, 332 Light-headedness, 132, 133
presyncopal, diabetes mellitus and, 368 Linear acceleration, 208
Linear track, 190t Locus cerruleus, 136
Longitudinal fracture of temporal bone, 353, 355f Longitudinal theory, 34
Longitudinal theory, of endolymph volume, 34 Long process of the incus, 27
Long process of the malleus, 27
Long Q-T syndrome. See Jervell and Lange-Nielsen syndrome
Loop diuretics, 375 ototoxic effects of, 375
Loose otoconia
and benign positional vertigo, 258 Lorazepam, 406t, 409t
Low-frequency sinusoidal test, in patients with central lesions, 205f
Low-salt diet, for Meniere’s syndrome, 281
Macrosaccadic oscillations, 166 Macrosquare wave jerks, 165–66 Macules
baseline firing rate of, nerve endings and, 49
mechanism of stimulation of, 49 nerve fiber diameter in, 50
Magnetic resonance angiography (MRA), 340
for middle ear and temporal bone tumors, 340 for vertebrobasilar ischemia, 328–29, 329f
Magnetic resonance imaging (MRI), 206
for brain stem and cerebellum hemorrhage, 333 for brain stem gliomas, 348–49, 349f
of cerebellar artery branches, 326
for cerebellopontine angle tumors, 344 for Chiari malformation, 392
for episodic ataxia type II, 397t
for internal auditory canal and cerebellopontine angle tumors, 344
for inner ear genetic disorders, 390
for intralabyrinthine hemorrhage, 332 for lateral medullary infarction, 327f
for lateral pontomedullary infarction, 327f for Meniere’s syndrome, 280
for middle ear and temporal bone tumors, 340 for multiple sclerosis, 313, 314f
for vertebrobasilar ischemia, 326–28 for Wernicke’s
encephalopathy, 370 Magnetic search coils, 172 Main cochlear artery, 35, 36f
Mal de debarquement syndrome, 144 Malignant external otitis, 241–42
algorithm for, 242f definition of, 241
management of, 241–42 algorithm for, 242f
Mammal, labyrinth of, 32f Manubrium, 27, 28
Marijuana, 137 Mastoidectomy
for chronic mastoiditis, 236 for petrositis, 238
Mastoiditis
Mastoid portion of temporal bone, 25, 221 Mastoid portion of temporal bone, 25, 26f
Maternally inherited mitochondrial disorders, 387–88 Maximum slow-component velocity (MSCV), 181, 191 Mean sway velocity, 210f
Mechanoelectric theory, of hair cell function, 41 Mechanosensory ion channels, 40
Meclizine (antivert, bonine), 406t, 409t for benign positional vertigo, 264
for Meniere’s syndrome, 280, 281t
for mild to moderate episodes of vertigo, 414
for nausea and vomiting during pregnancy, 410, 414 side effects of, 414
for suppressing motion sickness, 408, 414
Medial longitudinal fasciculus (MLF), 15, 66, 67, 151,
Medial vestibulospinal tract, 99, 100f Medulloblastomas, 348
Meniere’s syndrome, 124, 130, 273–83, 368, 387, 391
clinical features of, 275 diagnosis of, 278–80
audiometric testing, 278–80, 279f imaging, 280
dilated membranous labyrinth in, 276f electrocochleography in, 279
infection/autoimmune injury, 278
hearing loss in, 275, 278–79 glycerol test in, 279-280 idiopathic, 274
medical management of, 280–82, 281t prophylaxis, 281–82
symptomatic treatment of acute spells, 280 and migraine, 278, 289–90
pathophysiology of, 275–77, 276f surgical management of, 282–83
destructive procedures, 282–83 pressure pulse treatment, 282 shunts, 282
and syphilic infections, 248 tinnitus in, 275
Meniett device, for Meniere’s syndrome, 282 Meningiomas, 342, 343
congenital inner ear disorders in, 248 labyrinthitis associated with, 248 secondary to ear infection, 239 syphilitic ear infection in, 248
and acquired hearing loss, 384 Meningogenic bacterial
Mercury intoxication, 378 Metabolic disorders367–78 systemic, 367–71
Metals, heavy, neurotoxicity of, 377–78 Metastatic cerebellar tumors, 348 Metastatic tumors, 344, 348 Methotrexate
for immune-mediated inner ear disease, 308–9 Methylprednisone, for SSNHL, 247 Methysergide, for migraine, 298t Metoclopramide (Reglan), 298, 406t, 414–15
indications for use, 412t for migraine, 298, 298t
for nausea and vomiting associated with vertigo, 411, 415
Microphonic potential, definition of, 42
Microsomal triglyceride transfer protein (MTP), 395 Midbrain
stimulation of, ocular tilt reaction and, 166–67, 167f Middle cerebellar peduncle, infarction of, 323
Middle ear, 27–28 cross-section of, 27f
inflammation of. See Otitis media tumors of, 339–41
Middle fossa approach
for cerebellopontine angle tumors, 347
for semicircular canal dehiscence syndrome, 360 Midline cerebellar hemorrhage, 333
Migraine, 122, 132, 287–99 auditory symptoms with, 289 with aura, 288
without aura, 288 diagnosis of, 296, 296t
aura without headache, 296 attack, phases of, 296 background, 287–88
and benign positional vertigo, 257 clinical features of, 288, 292
and meniere’s syndrome, 289–90 migraine equivalents, 290–92 migraine with aura, 288
migraine without aura, 288 migrainous vertigo, 288–89 common drugs for treating, 298t
drugs for treating, 298t diagnosis of, 295–97 incidence of, 291t management of, 297–99
symptomatic and abortive treatment, 298 prophylactic treatment, 298–99
and Meniere’s syndrome, 278 pathophysiology of, 292–95
spreading wave of depression, 294 vasomotor abnormalities, 294–95, 295f
symptoms, factors triggering, 297t Migraine aura without headache, 296 Migraine equivalent, 296
Migrainous positional vertigo vs benign positional vertigo, 289
Migrainous vertigo, 124, 125, 288–89, 297
Mild traumatic brain injury, 361
MITF gene, 385 Mitochondria, anatomy of, 5f
Mitochondrial DNA, mutations in, 388
Mitochondrial encephalomyelopathy with lactic acidosis and stroke-like episodes (MELAS), 388
Mollusk, mechanoreceptors of, 31 Monaural diplacusis, 220
Mondini malformation, 388, 389, 389f
Motion perception and orientation, 19–20
Motion sickness, 409t definition of, 142 genetic factors in, 143 symptoms of, 142
Motorists’s disorientation syndrome, 136 Moving-platform posturography, 208–9, 210f Mucoepidermoid carcinoma, 339
clinical features of, 313 diagnosis of, 313
Multisensory dizziness, management of, 142 Muscular tone, maintenance of, 18 Myasthenia gravis
gaze-evoked nystagmus in, 161 saccadic abnormalities in, 185
Mycobacterium tuberculosis, 250
Myelin basic protein (MBP), 313 Myokymia, in episodic ataxia type 1, 396 Myxine, labyrinth of, 32f
Na-K-Cl cotransporter, in endolymph production, 34 Naproxen
National Hospital Ambulatory Medical Care Survey (NHAMCS), 125
antidopaminergic drugs for, 406t, 411 antiemetic drugs for, 410–13, 412t vestibular suppressants for, 407–10
Near-faint dizziness, 132 causes of, 132t hyperventilation, 134
orthostatic hypotension, 132–33
postural tachycardia syndrome (POTS), 133 vasovagal attacks, 133–34
Neck, soft tissue injury to, 363, 364
Neck-induced eye movements, characteristics of, 88–89.
See also Eye movements Neck-vestibular interaction, 363
Neighboring rostral reticularis tegmenti pontes nucleus (RTPN), 94
Nephritis, hereditary, in Alport’s symdrom, 368 Nervous system, immune-based attack on, 309, 314 Netilmicin, totoxicity of, 374t
Neurofibromatosis type 1 (NF1), vertigo in, 132 Neurofibromatosis type 2 (NF2), vertigo in, 132, 342–43, 343t Neuroleptic malignant syndrome, 411
Neuronal mechanisms, 78–79, 79f Neuronal reflex arc, 13t Neurotoxins, 377–78
of vestibular nuclei, 68 NF2 gene, 342–43
Nimodipine, for migraine, 298t Nitric oxide synthase (NOS), 57
N-methyl -d-aspartate (NMDA), 43, 68, 375 antagonists, for prevention of aminoglycoside
Nonsteroidal anti-inflammatory drugs (NSAIDs), ototoxicity of, 375
Nonsyndromic audiovestibular loss, 386–88, 386t.
See also Audiovestibular loss
dominantly inherited hearing loss, 386t, 387 inherited vestibular loss with normal hearing, 388 maternally inherited mitochondrial
recessively inherited hearing loss, 386–87, 386t Notch signaling, 38
Nucleus of optic tract, 93, 93f Number needed to treat” (NNT), 263 Nylen, 162
bithermal caloric test, 177–83
eye movements recording methods, 171–74 interpreting the recording, 174–75 pathologic nystagmus, recording, 175–77 visual–ocular control, tests of, 183–89
central origin spontaneous, 159 central positional, 164
congenital spontaneous, 160–61
after-nystagmus, 90f, 91 paroxysmal positional, 163, 163f pathologic, 156–57, 158
periodic alternating nystagmus (PAN), 161 peripheral positional, 164t
rebound, 162 recording, 191f second-degree, 157
third-degree, 157–58 types of, 156t vibration-induced, 164
Occipitalization of the atlas. See Assimilation of the atlas Occipital lobe, lesions of, 130, 130t
Ocular and spinal vestibular reflexes, comparison of, 97–98
Ocular counterrolling, 85–86, 207
Ocular dipping, 166 Ocular dizziness
convergence retraction nystagmus, 165 dissociated spontaneous nystagmus, 165 ocular bobbing, 166
saccadic intrusions, 165–66 voluntary ocular oscillations, 165
Ocular-otholith-canal reflex interaction, model of, 82 Ocular tilt reaction
bedside examinations of, 166–67, 167f in lateral medullary infarction, 323
Ocular tilt reflex, 17–18 Oculocephalic reflex, 153 Oculogravic illusion, 106, 106f Oculomotor apraxia, 185, 396
Off-center axis rotation (OCAR), 86 Off-vertical axis rotation (OVAR), 86 Off-vertical head rotation, 207–8
disadvantages of, 208 Ondansetron, 406t, 412t
for chemotherapy-induced nausea and vomiting, 411 indications for use, 412t
Onside pitch rotation, 190t Ophthalmoscope, for nystagmus testing, 157 OPRM1 gene, 388
Opsoclonus-myoclonus syndrome, 166
in paraneoplastic immune disorders, 310 Optokinetic after-nystagmus (OKAN), 90f, 91
abnormalities of, in peripheral lesions, 188, 188f velocity of, 187, 187f
Optokinetic nystagmus (OKN), 89–92, 90f, 91f, 187–89, 188f, 189f
abnormalities of, in peripheral lesions, 188, 188f cortical (active), 91
high-frequency (stare), 90 interaction with smooth pursuit, 91 low-frequency (look), 90
methods of testing and results in normal subjects, 187–88
in parieto-occipital lesions, 206 slow-phase velocity of, 89, 90
subcortical (passive), 91 results in patients, 188–89
Organic solvents, neurotoxicity of, 378 Orientation, 19–20
vestibular sensations during, 104–5 Orthostatic hypotension, 132–33 Oscillopsia, 141f
Osteogenic sarcoma, of temporal bone, 340 Osteopenia, and benign positional vertigo, 257 Osteopetrosis, 373
and benign positional vertigo, 257 Otalgia, in otitis media, 234
Otholith-ocular canal reflex interaction, model of, 82
Otitic hydrocephalus, 240 Otitis externa
diagnosis and management of, 234–35 hearing loss, 234
labyrinthitis and, 236–37 progression patterns, 234f serous, 234
Otoacoustic emissions, in congenital inner ear disorders, 389
Otoconia
production and maintenance of, 47–48 Otocyst, 37, 38f
Otolaryngology Committee on Hearing and Equilibrium, 290
Otolithic membrane, 6, 10 Otolith–ocular reflxes
eye movements with, 83–84, 83f, 84t laboratory examination of, 207–8
Otolith organs, 106–7, 106f anatomy of, 47–48
production and maintenance of, 47–48 physiology of, 49
Otologic manifestations of syphilis, features of, 249t Otomastoiditis, 233–35
diagnosis and management of, 234–35 Otorrhea
in tuberculous mastoiditis, 250 Otosclerosis, 371–73
pathologic process of, 372, 372f surgery for, 374
anti-inflammatory drugs, 375–76
diagnosis of, 376 dizziness caused by, 137 hearing loss due to, 220 “loop” diuretics, 375
for Meniere’s syndrome, 283 platinum compounds, 376 risk factors for, 377t
rupture of, 357 Oxcarbazepine
Palato-ocular myoclonus, 166 Palonosetron
for chemotherapy-induced nausea and vomiting, 411 Panic attacks, 135
medications for, 137 symptoms of, 135t
eye movements produced by, 106
Paramedian pontine reticular formation (PPRF), 16–17, 79–80
Paraneoplastic immune disorders, 309–12 antineuronal antibodies, 309, 310t background, 309
clinical features of, 309–11 cerebellar degeneration, 309–10
Parieto-insular vestibular cortex (PIVC), 102, 104 Parietooccipital region, lesions of, 187
Parkinson’s disease diagnosis of, 140
saccadic abnormalities in, 187 Paroxetine, for panic attacks, 137 Paroxysmal positional
nystagmus, 163, 163f peripheral vs central, 164t
Particle repositioning maneuver
for posterior canal variant of benign positional vertigo, 264, 265f
for posttraumatic positional vertigo, 356
Passive whole-body yaw rotation, 191–92, 193–95
Pathologic nystagmus, 156–57, 158
hyperventilation-induced nystagmus, 164 methods of examination, 157–58 positional nystagmus, 162–64
spontaneous nystagmus, 175–76, 176f spontaneous nystagmus, 158–61
vibration-induced nystagmus, 164 Pathophysiology of vestibular symptoms, 20–21 PAX-3, 385
Pendred (enlarged vestibular aqueduct) syndrome, 385, 385t, 387
Pendrin gene mutation, hearing loss associated with, 385
Pendulum model, 44, 45–46, 46f
Penicillin
for syphilitic inections, 249 Perilymph, 33–34
Perilymph fistula, 247, 364t, 387
algorithm for diagnosis and management of, 356f diagnosis of, 357–58
Periodic alternating nystagmus (PAN), 161 Periodic ataxia and vertigo, 132
Peripheral positional nystagmus, 164
vs central positional nystagmus, 164t Peripheral spontaneous nystagmus, 159, 176f
vs central origin spontaneous nystagmus, 159t Peripheral vestibular receptors, 4–10
inner ear receptor organs, 10 macules, 6
Peripheral vestibular system, 25 efferent vestibular neurons, 56–57 hair cell, 39–43
and direction of force, relationship between, 41 mechanism of, 41–42
afferent nerve activity, influence on, 42–43
hair cell/afferent nerve junction, signal processing at, 43 morphologic characteristics, 39–40
inner ear
inner ear vestibular receptors, 43 otolith organs, 47–49
semicircular canals, 43–47 primary vestibular neurons, 49–56
physiology, 52–56 temporal bone, 25–29, 26f
tympanic membrane, 26–27 Perphenazine, 406t
for nausea and vomiting associated with vertigo, 412t Persistent dizziness
after brain trauma, differential diagnosis, 363, 364t reasons for, 422
Persistent positional nystagmus, 163–64 Perverted nystagmus
with caloric stimulation, 182t definition of, 183
Petromyzon, labyrinth of, 32f Petrositis, 237–38
Petrous bone
anatomy of, 25, 26f infection of, 237, 238
Phenergan, for Meniere’s syndrome, 280 Phenobarbital, 161
Phenothiazines, extrapyramidal side effects of, 411 Phenytoin, 161
dizziness caused by, 137 Phobic dizziness, 135
Physical rehabilitation, for brain injury, 364 Physiologic dizziness
mal de debarquement syndrome, 144 motion sickness, 142–43
space sickness, 143 Physiologic nystagmus, 17f, 156
Phytanoyl-CoA hydroxylase (PAHX), 396 Pill-rolling” tremor, 140
Plasma membrane calcium ATPase 2 (PMCA2), 48 Plasmapheresis, for paraneoplastic immune
Platinum compounds, ototoxicity of, 376 Platybasia. See Basilar impression Pneumatoscopy, 150
Polyarteritis nodosa, with inner ear involvement, 304, 306 Polychondritis, relapsing, 307
Polymerase chain reaction (PCR), for herpes zoster oticus, 245
Pons
Pontomedullary reticular formation, stimulation of, 100 Positional nystagmus, 162–64, 177
horizontal canal, 265–68 peripheral versus central, 164t
Positional vertigo, benign, 255–69
Position-vestibular pause (PVP) neurons, 78 Positron-emission tomography (PET), 136, 311
in normal human subjects, 104 Postconcussion syndrome, 362–63, 364t
Posterior canal variant of BPV (PC-BPV), 258–65 clinical features of, 258–59, 259t
management of, 261–65 pathophysiology of, 260–61, 260f
Posterior cerebellar artery (PCA), 329f
Posterior inferior cerebellar artery (PICA), 320, 324f, 326f, 327f, 328f, 329f
and lateral medullaty infarction symptoms and signs, 323t
Posterior spiral vein, 36f Posterior vestibular artery, 35, 36f Posterior vestibular artery, 35, 36f
Posttraumatic positional vertigo, 354 diagnosis of, 355
Postural tachycardia syndrome (POTS), 133 Postural vertigo, phobic, 135
Posture
Potassium, in pathogenesis of migraine, 293 Prednisione
for autoimmune inner ear disease, 308 for herpes zoster oticus, 247
for immune-mediated inner ear disease, 309 plus steroids, 249
Prestin, cochlear hair cell electromotility and, 42 Presyncope, 132
Pretectal region
lesions of, effects on eye movements of, 80 in visual-vestibular interaction, 93, 95
Primary afferent neurons, 17f, 52, 53 anatomical and physiological properties, 55 classification, 53t
Primary afferent vestibular nerve fibers, 66 distribution of, 50f, 64f
Primary cholesteatomas, 343 Primary vestibular neurons, 49–56
innervation of the cristae, 50–52 neurotransmitters, 52
distribution of, 64f physiology of, 52
afferents from cristae, 53–55 afferents to macules, 55–56 spontaneous firing rates, 52–53, 53t
Primidone, dizziness caused by, 137 Prochlorperazine (Compazine), 406t, 410, 415–16
for Meniere’s syndrome, 280, 281t for migraine, 298t
for nausea and vomiting associated with vertigo, 412t Promethazine hydrochloride (Phenergan), 406t, 409t,
for benign positional vertigo, 264 for Meniere’s syndrome, 281t for migraine, 298
for severe vertigo with recurrent nausea and vomiting, 408, 414
Propranolol, for migraine, 298t Proteus, 235
Pseudomonas aeruginosa, 235, 239
Psychological factors, in vestibular rehabilitation, 420 Psychophysical studies
of semicircular canals, 105 Psychophysiologic dizziness, 134
diagnosis and management of, 136–37 panic disorder, 135
phobic dizziness, 135 Pure-bouton afferents, 54f Pure-calyx afferents, 54f Pure tone audiogram, 221f
Pure tone average (PTA), 221, 222t Purkinje cells, 160
Pursuit system. See Smooth pursuit
Push-pull mechanism, 15 Quinine, ototoxicity of, 375
Radiating arterioles, 35 Radiation therapy
for brainstem and cerebellar tumors, 349
for middle ear and temporal bone tumors, 341 Ramsay Hunt syndrome. See Herpes zoster oticus Reading exercises, 428
Reafference principle, 73 Rebound nystagmus, 162, 177f
Recessively inherited hearing loss, 386–87, 386t Recurrent penetrating arteries (RPA), of AICA, 324f Refsum’s syndrome, 396
Relapsing polychondritis, 304f, 307 Renal dialysis, 371
Retrocochlear hearing loss, 226, 229
Reversible saccade slowing, 185 Rhabdomyosarcoma, of middle ear, 340
Rheumatoid arthritis, cervicomedullary compression, 391, 393
Ribbon synapse, 5f, 40, 54f Riding in car practice, 429 Rinne test, 220–21
Rituximab, for paraneoplastic immune disorders, 312 Rocking chair exercises, 428–29
Roll, head rotation in, 207 Romberg test, 152
Rostral reticularis tegmenti pontes nucleus (RTPN), 94 Rotational nystagmus, 156, 191
Rotational testing, of horizontal vestibulo-ocular reflex, 156, 190f
Rotational vertebral artery syndrome, 335 Rotational vestibulo-ocular reflexes, 73–81, 70f
compensatory eye movements, 77–78, 77f
experimental lesions effect, 80 eye motion, pattern of, 77f, 80
fast component generation, 79–80 level of arousal and habituation, 81 neuronal mechanisms, 78–79, 79f
semicircular canal–ocular connections, 75–77, 75t, 76f Round window
anatomy of, 11f, 27f, 28, 33f rupture of, 357
Rubella, congenital inner ear disorders in, 384
Saccadic eye movements, 183–86, 184f, 186f abnormalities of, lesion location and, 184t accuracy of, 184
in lateral medullary infarction, 323 methods of testing and results in normal
Saccular macules, 6, 8f, 47, 49, 84t anatomy of, 32, 32f
mechanism of stimulation of, 49 organization of, 84
Salicylates, ototoxicity of, 375–76
Salt restriction, for Meniere’s syndrome, 280, 281 San Francisco Syncope Rule, 134
Sarcoma, of temporal bone, 340 Scarpa’s ganglion, anatomy of, 11f, 37 Scheibe deformity, 389
Schwalbe nucleus. See Medial vestibular nucleus Schwannomas, 341–42
Antoni type A, 342 Antoni type B, 342
Scopolamine (Transderm Sco¯ p), 406t, 409t, 410 indications for use, 412t
for nausea and vomiting associated with motion sickness, 408, 413
Sea anemone, mechanoreceptors of, 30f, 31f Second-degree nystagmus, 157
Sedating drugs
dizziness caused by, 137 Seesaw nystagmus, 165
Selective serotonin reuptake inhibitors (SSRIs), for depression, 364
Semicircular canal(s), 4, 6, 105
nerve, cross section of, 51f orientation of, 9f physiology of, 44–47
pendulum model, 45–47 Semicircular canal dehiscence syndrome
audiogram with, 361f diagnosis of, 360, 361f management of, 360
pathophysiology of, 358–60 symptoms and signs, 358, 359f
Semicircular canal–ocular connections, 75–77 with eye muscles, 75t
excitatory and inhibitory pathways between, 76f Semicircular canal–otolith interaction, 86, 87f
Semont maneuver, for posterior canal variant of benign positional vertigo, 263, 263f, 264
Sensorineural hearing loss, 219–20, 222 in autoimmune ear disease, 304
in Meniere’s syndrome, 307 in systemic lupus
Serologic tests, in autoimmune ear disease, 405 Serotonin antagonists, 406t, 411
Serotonin reuptake inhibitors, for panic attacks, 137 Serous labyrinthitis, 236, 237, 246
Shunts, for Meniere’s syndrome, 282
Sick headache. See Migraine without aura Signal processing, at the hair cell/afferent nerve
junction, 43 Sinusoidal rotation
changes in angular velocity, 193–95 nystagmus responses to, 203f
Sisomicin, ototoxicity of, 374t SLC1A3, 396
Slow-component velocity (SCV), 191, 191f, 194f Smooth pursuit system, 90–91, 186–87, 187–88f
methods of testing and results in normal subjects, 186 results in patients, 187
Sodium fluoride, for otosclerosis, 374 Soft-tissue injuries, management of, 364 Sound pressure level (SPL), 221
Space, caloric testing in, 179 Space phobia, 136
Speech detection threshold (SDT), 223 Speech discrimination test, 223
Speech reception threshold (SRT), 223 Speech recognition tests, 223
Spinocerebellar ataxia (SCA) syndrome, 183, 393, 398
Spinocerebellar atrophy, vertigo in, 132 Spiral ganglion, anatomy of, 11f
Spiral modiolar vein, of patient with autoimmune inner ear disease, 306f
Spondyloepiphyseal dysplasia, 391 Spontaneous downbeat nystagmus, 159, 176f
Spontaneous nystagmus, 16, 17f, 158–59, 175–76, 176f
periodic alternating nystagmus, 161 peripheral, 159
Spontaneous vestibular nystagmus, 80
Squamous cell carcinoma, of middle ear, 339 Squamous portion of temporal bone, 25, 26f Square wave jerks, 165
Stapedius muscle, 27f, 28, 29f acoustic reflex testing of, 225
Staphylococcus aureus, 235 Static-force platform
Static ocular counterrolling, 190t Static posture test, 152
Static tilt experiments, 106 Statocyst, 4
of ctenophore comb, 30, 30f octopus, 31f
Stenger test, 223 Step stimulus
Stereotaxic radiosurgery, for vestibular schwannomas, 347 Steroids
for multiple sclerosis, 313 plus penicillin, 249
for syphilitic inections, 249 Streptococcus pneumoniae, 234, 239 Streptomycin, ototoxicity of, 374t
for Meniere’s syndrome, 283 Striola, 6, 8f
labyrinthine infarction, 321–22 lateral medullary infarction, 322–23
lateral pontomedullary infarction, 323–24 treatment for, 331–32
Subarachnoid hemorrhage, 362 Subclavian steal syndrome, 319 Subjective vestibular sensation
functional brain imaging in normal human subjects, 104–5
response properties of thalamic relay neurons, 104 response properties of vestibular cortex neurons, 104 vestibulocortical pathways in patients, lesions of, 105 vestibulothalamocortical connections, 102–4, 103f
Suboccipital surgery, for cerebellopontine angle tumors, 347
Sudden deafness atherosclerosis and, 321 in head trauma, 354
viral infections associated with, 245–46
Sudden sensorineural hearing loss (SSNHL), 242, 245 diagnosis of, 245
and labyrinthine concussion, 353–54 Sumatriptan, for migraine, 298t Superior cerebellar artery (SCA), 329f
Superior vestibular nucleus (Bechterew’s nucleus), 66 Suppurative labyrinthitis, 236, 246
Surgical resection, for brainstem and cerebellar tumors, 349
Symmetric gaze-evoked nystagmus, 161 Symptomatic treatment of acute spells, 280 Syncope
vasodepressor light headedness and, 134 Synostosis, congenital cervical, 391
Syphilis otologic manifestations of, 248–49, 249t diagnosis of, 248–49
Syphilitic labyrinthitis, 246, 248–49
Systemic lupus erythematosus (SLE), 306
Systemic metabolic disorders, and dizziness, 367–71
Tandem gait tests, 152 Tardive dyskinesia, 411
T cell surface protein defect, in autoimmune ear disease, 304
Temporal bone, 25–29 facial nerve, 28–29, 29f middle ear, 27–28 medial view of, 26f
metabolic disorders of, 371–74 diagnosis of, 373
Paget’s disease, 373 trauma to
intracranial complications associated with, 362 labyrinthine concussion, 353–54
management of, 355–56 posttraumatic positional vertigo, 354
tympanic membrane, 26–27 Temporal lobe
lesions of, vertigo in, 130t, 131 Tensor tympani muscle, 27
Terminal cerebellar branches, of AICA, 324f TGFB1, 372
Thalamic relay neurons, response properties of, 104 Thiamine deficiency, in alcoholism, 368–70
Third-degree nystagmus, 157–58
Three-dimensional (3D) FLAIR, 332 Thrombolysis, for basilar artery occlusion, 331 Thrombolytics, for acute ischemic stroke, 331 Thrombophlebitis, 238
Ticlopidine, for transient ischemic attacks, 330 Tilt-translation ambiguity, solving, 73, 74f
Tip-links, 5, 5f, 40, 40f Tobramycin, 374t Tone decay test, 229
Topiramate, for migraine, 298t, 299 Top shelf vertigo,” 258
Toxic labyrinthitis, 236 Tranquilizers
dizziness caused by, 137t saccadic abnormalities and, 185
smooth pursuit abnormalities and, 187
Transcranial Doppler (TCD) imaging, for vertebrobasilar ischemia, 328
Transesophageal echocardiography (TEE), for vertebrobasilar ischemia, 328
Transient ischemic attacks (TIAs), 320–21 treatment for, 329–31
Transient migrainous accompaniments,” 292 Translabyrinthine surgery, for Cerebellopontine angle
Translational vestibulo-ocular reflex, 17, 81–85
eye movements, characteristics of, 82f, 84–85, 85f otolith–ocular connections, 83–84, 83f
Transmastoid approach, for semicircular canal dehiscence syndrome, 360
Transverse fracture of temporal bone, 353, 355f management of, 356
benign positional vertigo after, 258
to brain, 360–64. See also Brain trauma to temporal bone, 353–56
Tricyclic amines
for depression, 364 dizziness caused by, 137
for ion channel disorder, 293 for migraine, 299
Trigeminal nerve, compression of, 341
Trigeminal vascular system, in pathogenesis of nerve, 294 Trimethobenzamide hydrochloride (Tigan), 406t, 415
for mild to moderate nausea and vomiting, 415 Trinucleide repeat syndromes, 395
Triptans, for migraine, 297, 298
Truncal ataxia, in cerebellar infarction, 325 Tuberculous mastoiditis, 249–50
of middle ear and temporal bone malignant tumors, 339–40
glomus body tumors (paragangliomas), 340 diagnosis of, 340
of internal auditory canal and cerebellopontine angle schwannomas, 341–42
metastatic tumors, 344 diagnosis of, 344–46, 345f management of, 346–47
brain
diagnosis and management of, 348–49
12S Ribosomal ribonucleic acid (rRNA), 375, 388 Tympanic cavity. See Middle ear
Tympanic membrane anatomy of, 26–27, 26f appearance of, 150f
Tympanic portion of temporal bone, 25, 26f Tympanometry, 224, 225f
Ultrasound, for vertebrobasilar ischemia, 328 Unilateral peripheral lesions, 196–200 Unilateral vestibular lesions, 423–24, 423t
Upright pitch rotation, 190t Uremia, 368
Usher 1B syndrome, 384–85, 385t Utricle
connection with eye muscles, 84t and semicircular canals, 37
Utricular macule, 6, 8f anatomy of, 48f, 49
stimulation of, ocular tilt reaction and, 167
Vagus nerve, glomus tumors in, 340 Valacycolvir, for herpes zoster oticus, 247
Valproate, for migraine, 298t Valproic acid, for migraine, 299
Vascular compression syndromes, 334–35 rotational vertebral artery syndrome, 335 vertebrobasilar dolichoectasia, 334
Vascular occlusion, and sudden deafness, 321 Vasospasm, and migraine, 294–95
Velocity storage effect, 73–75, 74f, 78
Venereal disease research laboratory (VDRL) test, 248 Ventricle, fourth
lesions of, perverted caloric nystagmus in, 183 tumor of, 348
Verapamil, for migraine, 299 Vermian cortex, 101, 101f
Vertebrobasilar dolichoectasia, 334, 334f Vertebrobasilar ischemia (VBI), 319–32
labyrinthine infarction, 321–22 lateral medullary infarction, 322–23
lateral pontomedullary infarction, 323–24 transient ischemic attacks, 320–21 treatment, 329–32
transient ischemic attacks, 329–31 vertigo in, 319
and brain stem hemorrhage, 332
central versus peripheral causes, 128–29, 129t compensation, 131
diagnosis and management, 132 common causes, duration of, 129, 129t family history, 131–32
lifetime prevalence of, 123f precipitating factors, 129–30
predisposing factors, 131 recurrent attacks, 329–30f symptoms of, 130–31, 130t time course, 129
and vertebrobasilar ischemia, 319–20 symptoms associated with, 321t
Vestibular afferent neurons, classification of, 54f Vestibular aqueduct
enlarged, 385, 387, 390 vein at, 35, 36f
Vestibular compensation, failure of, 422
Vestibular cortex neurons, response properties of, 104 Vestibular disorders, 131
Vestibular-evoked myogenic potentials (VEMPs), 210–13, 212f, 389–90
mechanism of stimulation, 210–11 for multiple sclerosis, 313 normative data, 211
results in patients, 211–13 test methodology, 211
Vestibular-evoked potentials, 209 brain stem and cortical, 209–10
vestibular evoked myogenic potentials (VEMPs), 210–13
Vestibular exercises, 420, 425–26 controlled trials of, 422–23
strategy for designing, 423–25, 423t, 424t, 425t Vestibular lesions
unilateral, 423–24, 423t Vestibular loss
mechanisms for compensation after, 420–21, 421t Vestibular Meniere’s syndrome, 290
Vestibular neurectomy
for Meniere’s syndrome, 283 Vestibular neuritis, 244–45, 423
diagnosis of, 245–46 pathologic findings, 243f
Vestibular neurons
compensation after labyrinthectomy, 71–72, 71f intrinsic membrane properties of, 70–71
in rodent medial vestibular nucleus, 70, 71t types of, 68–70, 70f
afferent and efferent connections of, 12f anatomy, 66–68
descending (inferior), 67 field potential in, 69f
immunohistochemical staining of, 65f interstitial, 67–68
Vestibular-only (VO) neurons, 102 Vestibular paresis
with caloric stimulation, 182t definition of, 181
Vestibular paroxysmia, 334–35 Vestibular physical therapy, 247 Vestibular reflexes, 12–19
adaptive control of, 420 basic elements, 13f
horizontal canal-ocular reflex, 13–15 nystagmus, 15–17
translational vestibulo-ocular reflexes, 17 vestibulo-autonomic reflexes, 19
vestibulospinal reflexes, 18–19
Vestibular rehabilitation, 419–26
Vestibular schwannoma (acoustic neuroma), 342, 343t, 346f
Vestibular sensation, subjective. See Subjective vestibular sensation
Vestibular suppressants, 407–10 instructions to patients, 410 precautions, 408, 410
Vestibular system, anatomy and physiology of, 3 central vestibular pathways, 11–12
lesions, central compensation for, 21–22 motion perception and orientation, 19–20 peripheral vestibular receptors, 4–10
inner ear receptor organs, 10 macules, 6
symptoms, pathophysiology of, 20–21 vestibular reflexes, 12–19
horizontal canal-ocular reflex, 13–15 nystagmus, 15–17
translational vestibulo-ocular reflexes, 17 vestibulo-autonomic reflexes, 19
vestibulospinal reflexes, 18–19 Vestibular system
bedside examination of, 149–67 central, 63–107
orientation role of, 3 peripheral, 25–57
Vestibular testing
for Meniere’s syndrome, 280 and suborgans, 190t
Vestibule-ocular reflexes and vestibulo-ocular reflexes, comparison of, 92, 92f
Vestibulo-autonomic reflexes, 19
Vestibulocerebellum, 11, 12f Vestibulocochlear vein, 35, 36f Vestibulocochlear vein, 35, 36f Vestibulo-collic reflexes, 101–2
Vestibulocortical pathways in patients, lesions of, 105 Vestibulo-ocular reflex (VOR), 72–86, 74f, 179, 368, 420,
and cervico-ocular, synergistic interaction of, 88, 89f gain and phase of, 199f
response to step rotation in cerebellar atrophy patients, 206f
rotational, 73–81, 75t, 76f, 77f, 79f semicircular canal–otolith interaction, 86, 87f tests of, 153
cold caloric test, 155–56 doll’s eye test, 153 dynamic visual acuity, 155 head-thrust test, 153–55
translational, 81–85, 82f, 83f, 84t, 85f
and vestibule-ocular reflexes, comparison of, 92, 92f with vision, adaptive modification of, 96, 96f
Vestibulo-ocular reflexes, rotational testing of, 189, 190f stimulus and response, relationship between, 191–93
high-acceleration small-amplitude rotation, 192 passive whole-body yaw rotation, 191–92
results in normal subjects, 193–96 active head rotation, 196
high-acceleration, low-amplitude rotation, 195–96, 196f
passive whole-body yaw rotation, 193–95 results in patients, 196–203
bilateral peripheral lesions, 200–202 central vestibular lesions, 202–3 unilateral peripheral lesions, 196–200
Vestibulospinal connections, 98–100, 99f lateral vestibulospinal tract, 98–99, 100f medial vestibulospinal tract, 99, 100f reticulospinal tract, 99–100
Vestibulospinal reflexes, 18–19, 18f, 421, 424
cerebellar–vestibular interaction, 100–101, 101f ocular and spinal vestibular reflexes, comparison
vestibulo-collic reflexes, 101–2
vestibulospinal connections, 98–100, 99f, 100f tests of, 151
Vestibulospinal testing, 208 bedside test, 151f
moving-platform posturography, 208–9 static-force platform posturography, 208
Vestibulospinal tract ipsilateral, 67
Vestibulothalamocortical projections, 102–4, 103f Vibration-induced nystagmus, 164 Videonystagmography (VNG), 172, 173f
Video recordings, of eye movements, 173–74 Visual acuity test, dynamic, 155
Visually guided tracking eye movements, organization of, 91–92
Visual-ocular control, laboratory tests of, 183, 184t Visual–ocular control, tests of, 183–89
methods of testing and results in normal subjects, 187–88
results in patients, 188–89 saccadic eye movements, 183–86
methods of testing and results in normal subjects, 183–85
results in patients, 185–86 smooth pursuit, 186–87
methods of testing and results in normal subjects, 186
Visual ocular control abnormalities, summary of, 184t Visual tracking eye movements, 89–91 Visual–vestibular conflict, in motion sickness, 143 Visual–vestibular interaction, 95, 95f, 204
cellular basis for, 96–97, 97f methodology, 204
results in normal subjects, 204–5, 204f results in patients, 205–7
vestibular -and visual-induced eye movements, comparison of, 92
vestibulo-ocular reflext with vision, adaptive modification of, 96, 96f
visually guided tracking eye movements, organization of, 91–92
visual tracking eye movements, 89–91 visual–vestibular interaction, 95
cellular basis for, 96–97, 97f
visuo-vestibulo-ocular connections, 92–95, 93f, 94f Visualvestibulo-ocular reflex (VisVOR), 204
Visuo-vestibulo-ocular connections, 92–95, 93f, 94f Vitamin E deficiency, in spinocerebellar ataxia, 395 Voluntary ocular oscillations (voluntary nystagmus), 165 Vomiting. See Nausea and vomiting
von Hippel-Lindau disease, 132, 332
Waardenburg type 1 syndrome, 385, 385t, 389 Wackenheim’s clivus–canal line, 392
Walking, delayed, in congenital vestibular loss, 383 Walking exercises, 425
Wallenberg’s syndrome. See Lateral medullary infarction Warfarin, for transient ischemic attacks, 330
Wernicke’s encephalopathy, 370
Western blot assay, in autoimmune inner ear disease, 305 Whiplash injuries