
|
| Si usas un navegador diferente al Internet Explorer® de Microsoft, las imágenes de esta web pueden no aparecer. Si te ocurre esto debes desactivar el bloqueo de pop-ups en tu navegador. |
| Debido a la gran cantidad de imágenes que contiene esta web, en función de la velocidad de tu conexión, la página puede tardar bastante en cargarse totalmente. |
BENCENO Y DERIVADOS.
2. La estructura del benceno. 3. Los orbitales moleculares del benceno. 4. Calores de Hidrogenación. 5. Comparación entre la reactividad química del benceno y la de los alquenos. 6. Regla de Hückel. 7. Sistemas de anillos condensados: Naftaleno, Antraceno y Fenantreno.
9. El mecanismo de la Sustitución Electrófila Aromática (SEAr): halogenación, nitración, sulfonación. 10. Efecto orientador de los sustituyentes en las reacciones SEAr. 11. Reacción de alquilación de Friedel - Crafts. 12. Reacción de acilación de Friedel - Crafts. 13. Reacción de Sustitución Nucleofílica Aromática. 14. Hidrogenación de anillos aromáticos. 15. Reacción de Birch. 16. Reacciones de la cadena lateral en los derivados del benceno. |
1. Introducción. El benceno se aisló
por primera vez en 1825 por Michael Faraday por condensación de
una mezcla aceitosa obtenida del gas del alumbrado. La fórmula
empírica del nuevo compuesto era CH. La síntesis de este
compuesto se consiguió en 1834 por Eilhard Mistscherlich al calentar
ácido benzoico, aislado de la goma de benjuí, en presencia
de cal. En inglés benjuí es benzoin y como el nuevo
compuesto derivaba de la goma de benjuí, que es una resina balsámica
obtenida de varias especies de Styrax, una especie vegetal que
se encuentra en la India, al nuevo compuesto se le denominó en
inglés benzene. En la última
parte del siglo XIX se descubrieron muchos otros compuestos que parecían
estar relacionados con el benceno pues tenían bajas relaciones
hidrógeno/carbono y aromas agradables y por eso a este grupo de
compuestos se les denominó hidrocarburos aromáticos. El
término aromático se aplica en la actualidad a una serie
de compuestos cuya estabilidad y reactividad es semejante a la del
|
2. La estructura del benceno. La pimera estructura para el benceno fue propuesta por el químico alemán Friedrich August Kekulé von Stradonitz (1829-1896) en 1865 y consistía en una mezcla en equilibrio de dos ciclohexatrienos, formados con enlaces sencillos y dobles alternados. En la estructura de Kekulé los enlaces sencillos serían más largos (1.47 Å) que los enlaces dobles (1.33 Å). Cuando se desarrollaron los métodos físicos de determinación estructural y se pudo medir la distancia de enlace C-C del benceno se encontró que todas las distancias eran iguales y median 1.39 Å, que es un promedio entre la distancia de un enlace doble (1.33 Å) y un enlace simple (1.47 Å).
|
 |
Aparte de las características
físicas del benceno, que no resultan explicadas por las estructuras
de 1,3,5-ciclohexatrieno, existen una serie de propiedades químicas
del benceno que tampoco resultan explicadas por las estructuras de enlaces
dobles alternados que propuso Kekulé. Por ejemplo, el benceno no
reacciona con halógenos (X2, X=Cl, Br, I) o con haluros de hidrógeno
(HX, X=Cl, Br, I) como lo haría un compuesto poliénico.
La hidrogenación del benceno es mucho más lenta que la de
los alquenos y requiere condiciones muy drásticas: alta presión
de hidrógeno y empleo de catalizadores muy activos. El benceno es, en
comparación con los alquenos y los polienos, un compuesto más
estable y la estructura del 1,3,5-ciclohexatrieno no puede explicar esta
estabilidad adicional. La remarcable estabilidad
del benceno se puede explicar si se admite la deslocalización de
la densidad electrónica asociada a los orbitales p. Las estructuras
resonantes se diferencian en la distribución de la densidad electrónica
pero no en la posición relativa de los átomos que las integran.
En realidad el benceno es un híbrido de resonancia cuyos enlaces
pi están deslocalizados, con un orden de enlace de
|
 |
Por tanto, el benceno
consiste en un anillo formado por seis átomos de carbono con hibridación
sp2, enlazados entre sí mediante enlaces sigma Csp2-Csp2. Cada
uno de los átomos de carbono se enlaza además a un átomo
de hidrógeno mediante un enlace sigma Csp2-H1s. Todos los enlaces
C-C tienen la misma longitud y todos los ángulos de enlace son
de 120º. Como los átomos de carbono presentan hibridación
sp2, cada átomo de carbono tiene un orbital p perpendicular al
plano del anillo que se
|
 |
3. Los orbitales moleculares del benceno.
|
 |
La ocupación de los orbitales enlazantes en el benceno es óptima puesto que no se sitúa ningún electrón en los destructivos orbitales antienlazantes. Se puede afirmar, a la vista del diagrama anterior, que la estabilidad del benceno no se debe a la existencia de estructuras resonantes sino a un grupo de orbitales moleculares de baja energía que son capaces de acomodar de forma altamente eficiente toda la densidad electrónica asociada a los electrones pi.
|
4. Calores de Hidrogenación.
En la siguiente gráfica se representan los calores de hidrogenación determinados experimentalmente en la hidrogenación del ciclohexeno, del 1,4-ciclohexadieno, del 1,3-ciclohexadieno y del benceno. También se representa el calor de hidrogenación teórico del hipotético 1,3,5-ciclohexatrieno, un compuesto que no existe.
|
 |
Cuando el ciclohexeno
se hidrogena a ciclohexano se desprenden 28.6 kcal/mol. El 1,4-ciclohexadieno,
un dieno con conjugado, libera en la hidrogenación 57.4 kcal/mol,
aproximadamente el doble del calor de hidrogenación del ciclohexeno. La hidrogenación del 1,3-ciclohexadieno, un dieno conjugado libera 55.4 kcal/mol, 1.8 kcal/mol menos que el doble del valor del ciclohexeno. Una energía de resonancia de 1.8 kcal/mol es típica para un dieno conjugado. Para el hipotético 1,3,5-ciclohexatrieno se puede calcular un calor de hidrogenación de:
|
Al contrario que
los anteriores alquenos, que se hidrogenan a presión atmosférica,
la hidrogenación del benceno necesita de elevadas presiones de
hidrógeno y de catalizadores muy activos. Cuando se produce la
hidrogenación sólo se liberan 49.8 kcal/mol, 32.9 kcal/mol
menos que el hipotético calor de hidrogenación del 1,3,5-ciclohexatrieno.
A esta diferencia de energía se le conoce como energia de
|
5. Comparación entre la reactividad química del benceno y la de los alquenos.
|
 |
La mayor parte de los alquenos decoloran las disoluciones de bromo molecular en tetracloruro de carbono porque el bromo se agrega al doble enlace formando compuestos trans-dibromados incoloros. Cuando se agrega bromo al benceno no tiene lugar ninguna reacción y el color rojo del bromo permanece.
|
 |
Para que el bromo reaccione con el benceno es necesaria la adición de una cantidad catalítica de un ácido de Lewis, como el bromuro férrico (FeBr3). Sin embargo, el producto de la reacción no es el producto de adición de los dos átomos de bromo, sino un compuesto en el que se ha sustituido un átomo de hidrógeno por un átomo de bromo, formándose además HBr como subproducto.
|
 |
Más adelante se explicará el mecanismo de la reacción anterior que se denomina reacción de sustitución electrófila aromática (SEAr).
|
6. Generalización de la aromaticidad: Regla de Hückel. Durante muchos años se supuso que la gran energía de resonancia del benceno sería común en otros polienos cíclicos con enlaces dobles conjugados. De modo genérico se denominan anulenos todos los polienos cíclicos con enlaces simples y dobles alternados. Por ejemplo, el benceno es el anuleno de seis miembros y por tanto se le puede llamar [6]anuleno. El ciclobutadieno es el [4]anuleno, el ciclooctatetraeno es el [8]anuleno y el ciclopentaeno es el el [10]anuleno.
|
 |
Sin embargo, no todos
los polienos conjugados cíclicos gozan de la excepcional estabilidad
termodinámica asociada al benceno. Por ejemplo, el ciclobutadieno
([4]anuleno) nunca se ha aislado o purificado porque experimenta una rápida
dimerización de Diels-Alder. Para evitar esta reacción el
ciclobutadieno se debe preparar en bajas concentraciones y en fase gaseosa
como moléculas individuales, que se atrapan en argón a muy
baja temperatura. Este no es el comportamiento que El ciclooctatetraeno ([8]anuleno) se sintetizó en 1911 por Richard Willstätter y se pudo demostrar que no presentaba la química típica de los compuestos aromáticos. Por ejemplo, el bromo se decolora fácilmente en contacto con este compuesto y el permanganato oxida sus dobles enlaces. De hecho, y al contrario que el benceno, el cicloocatetraeno no es plano. Su conformación más estable es de bote lo que provoca un deficiente solapamiento entre los orbitales 2p adyacentes.
|
 |
Condiciones para la aromaticidad.
1) Su estructura
debe ser cíclica y debe contener enlaces dobles conjugados. 2) Cada átomo
de carbono del anillo debe presentar hidridación sp2, u ocasionalmente
sp, con al menos un orbital p no hidridizado. 3) Los orbitales
p deben solaparse para formar un anillo continuo de orbitales paralelos.
La estructura debe ser plana o casi plana para que el solapamiento de
los orbitales p sea efectivo. 4) Además
debe cumplir la regla de Hückel cuyo enunciado es
el siguiente: Para que
un compuesto sea aromático el número de electrones pi en
el sistema Si el número
de electrones pi en el sistema cíclico es 4n, siendo n un número Al emplear la regla
de Hückel se debe estar seguro que el compuesto bajo consideración
cumple con los criterios de un sistema aromático o antiaromático,
es decir debe tener un anillo continuo de orbitales p que se solapan en
una conformación plana. El benceno es un [6]anuleno con un anillo continuo de orbitales p que se solapan. En el benceno hay seis electrones pi de modo que es un sistema 4n+2, con n=1. La regla de Hückel predice que el benceno será aromático. El ciclobutadieno
es un [4]anuleno con un anillo continuo de orbitales p que se solapan
pero tiene cuatro electrones pi. Como es un sistema 4n, con n=1, la regla
de Hückel predice que el ciclobutadieno será antiaromático. El ciclooctatetraeno es un [8]anuleno con ocho electrones pi. Al aplicar la regla de Hückel al ciclooctatetraeno se predice que este compuesto debe ser antiaromático: 4n electrones pi con n=2. Sin embargo, este compuesto no muestra la alta inestabilidad asociada a los compuestos antiaromáticos, como el ciclobutadieno. Por otra parte, su reactividad tampoco es la de un compuesto aromático puesto que su comportamiento químico es el de un alqueno. El ciclooctatetraeno, al contrario que el benceno no es plano y por lo tanto no puede presentar un solapamiento continuo de orbitales p, que es una característica fundamental de los compuestos aromáticos. La regla de Hückel no se puede aplicar a este anuleno. El compuesto es no aromático. El [14]anuleno y
el [18]anuleno son sistemas con 4n+2 electrones pi (siendo n=3 y n= 4
respectivamente) y son compuestos aromáticos. El [16]anuleno, con 4n electrones pi (n=4) debería ser un compuesto antiaromático. Sin embargo, la falta de planaridad del compuesto hace que se comporte como un polieno parcialmente conjugado. El compuesto por tanto es no aromático.
|
 |
El [10]anuleno es
un compuesto con 4n+2 electrones pi y por tanto debería ser aromático.
Sin embargo, los dos átomos de hidrógeno centrales del anillo
impiden que el sistema adquiera la planaridad y en consecuencia se interrumpe
el solapamiento continuo de orbitales p, por tanto el compuesto es no
aromático. Los conceptos de aromaticidad y antiaromaticidad también permiten predecir el comportamiento químico y la estabilidad de compuestos con carga. Por ejemplo, el ciclopentadieno es más ácido de lo esperado porque la pérdida de un protón convierte al dieno conjugado, que es un compuesto no aromático, en el anión ciclopentadienilo aromático.
|
 |
El ciclopentadieno
contiene un átomo de carbono sp3 (C5) de modo que no puede haber
un sistema continuo de orbitales p que se solapen. La pérdida de
un protón del C5 deja un orbital ocupado con un par de electrones.
El carbono C5 pasa a tener una hibridación sp2 en el anión
ciclopentadienilo situando un par de electrones en un orbital p. De esta
forma el anión ciclopentadienilo consigue la planaridad y la
|
 |
Cuando el cicloheptatrienol
se trata con H2SO4 acuoso diluido se forma fácilmente el catión
cicloheptatrienilo, denominado catión tropilio. Algunas sales de
tropilio se puede aislar y almacenar durante meses sin que se descompongan.
El carbono sp3 (C7) que está unido al grupo hidroxilo cambia su
hidridación a sp2 cuando se ioniza. El orbital p vacío permite
el solapamiento continuo del sistema de orbitales p del catión
tropilio. El número de electrones pi deslocalizados cumple la regla
de Hückel
|
 |
En contraste con el catión tropilio, el anión tropilio es difícil de preparar. De hecho el cicloheptatrieno tiene una acidez similar a la del propeno (pKa = 43). El anión tropilio es muy reactivo lo que concuerda con la predicción de Hückel que indica que el anión tropilio es antiaromático con un total de 8 electrones pi (4n, n=2).
|
 |
7. Sistemas de anillos condensados. Los anillos fusionados
son aquellos que comparten dos átomos de carbono y un enlace entre
ellos. Los compuestos formados por dos o más anillos de benceno
fusionados se denominan hidrocarburos aromáticos polinucleares. El naftaleno es el hidrocarburo aromático polinuclear más simple. Consta de dos anillos de benceno fusionados. El naftaleno se representa empleando una de las tres estructuras resonantes de Kekulé o bien mediante la notación de círculo para los anillos aromáticos.
|
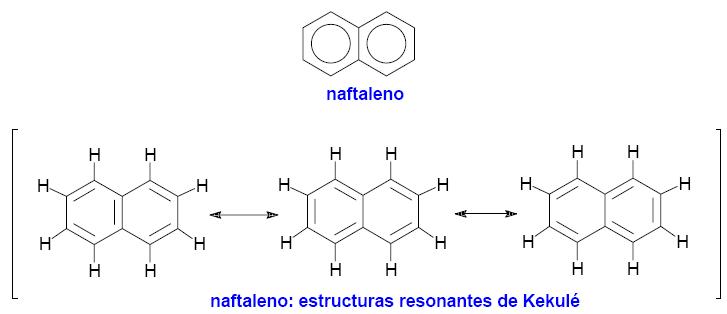 |
En el naftaleno hay
dos anillos aromáticos que contienen un total de 10 electrones
pi. El benceno contienen 6 electrones pi, y dos anillos bencénicos
aislados contendrán un total de 12 electrones pi, por lo que el
antraceno tiene un déficit de dos electrones pi. La energía
de resonancia del naftaleno es de 60 kcal/mol, lo que equivale a 30 kcal/mol
por cada anillo aromático. Este valor es menor que la energía El antraceno es un hidrocarburo polinuclear tricíclico. A medida que aumenta el número de anillos aromáticos fusionados, continúa decreciendo la energía de resonancia por anillo y los compuestos se hacen más reactivos.
|
 |
El antraceno tiene
una energía de resonancia de 84 kcal/mol, o sea 28 kcal/mol por
anillo aromático. El antraceno tiene 14 electrones p, en comparación
con 18 electrones pi que tendrían tres anillos aromáticos
aislados. El antraceno, al tener menos energía de resonancia que
el benceno, participa con frecuencia en reacciones de adición,
que son características de los compuestos poliénicos no
aromáticos.
|
 |
El fenantreno es también un hidrocarburo polinuclear tricíclico. Su energía de resonancia es de 91 kcal/mol, 30.3 kcal/mol por anillo aromático, ligeramente mayor que la del naftaleno.
|
 |
El fenatreno experimenta una adición 1,2 en las posiciones 9 y 10 para dar un producto con dos anillos completamente aromáticos.
|
 |
8. Compuestos heterocíclicos aromáticos. Piridina. En Química Orgánica todos los átomos distintos al carbono y al hidrógeno reciben la denominación genérica de heteroátomos. Cuando se sustituye una unidad CH del benceno por un átomo de nitrógeno se obtiene un compuesto aromático heterocíclico denominado piridina. El par no enlazante de electrones del nitrógeno se encuentra en un orbital híbrido sp2 situado en el plano del anillo aromático. La densidad electrónica asociada a este orbital no puede entrar en resonancia con el sistema de orbitales p deslocalizados porque se encuentra situado perpendicular a éstos.
|
 |
Las reacciones de la piridina son las propias de los compuestos aromáticos. Tiene una energía de resonancia de 26 kcal/mol y participa en reacciones de sustitución y no de adición. La piridina es básica y en disolución acuosa ácida el protón se une al par electrónico no enlazante del átomo de nitrógeno generando un catión piridinio, que continúa conservando la aromaticidad porque contiene 6 electrones pi (4n+2, n=1).
|
 |
Pirrol. El pirrol es un heterociclo
aromático de cinco eslabones formado por cuatro átomos de
carbono y uno de nitrógeno. El nitrógeno presenta una hibridación
sp2, pero, al contrario de lo que ocurre en la piridina, el par electrónico
libre del nitrógeno ocupa un orbital p y participa en el solapamiento
con los orbitales p de los átomos de carbono adyacentes formando
un anillo continuo de densidad electrónica. Los dos
|
 |
El pirrol (pKb = 13.6) es una base mucho más débil que la piridina (pKb = 8.8). Para enlazar el protón el pirrol necesita usar uno de los pares electrónicos del sexteto aromático. Cuando se protona el pirrol el átomo de nitrógeno cambia su hidridación de sp2 a sp3 y se interrumpe el anillo continuo de densidad electrónica. El pirrol pierde su aromaticidad al protonarse y por lo tanto la estabilidad asociada a los compuestos aromáticos, lo que explica la baja constante de equilibrio del proceso.
|
 |
Furano. El furano es un heterociclo
aromático de cinco eslabones, como el pirrol, pero con el oxígeno
como heteroátomo en lugar del nitrógeno. El átomo
de oxígeno presenta una hibridación sp2 de manera que uno
de los pares de electrones solitarios ocupa uno de los orbitales sp2.
Los otros dos orbitales sp2 se emplean en la formación de los enlaces
s con los átomos de carbono adyacentes. El otro par de electrones
no compartidos ocupa el orbital p no hidridizado que se solapa con los
orbitales p de los
|
 |
Tiofeno. El tiofeno es un hidrocarburo heterocíclico aromático semejante al furano pero con un átomo de azufre en lugar de uno de oxígeno. El átomo de azufre del tiofeno presenta hibridación sp2. Los tres orbitales híbridos sp2 se forman con la combinación de los orbitales s y p del tercer nivel cuántico. Dos de los orbitales sp2 se emplean en la formación de enlaces sigma Csp2-Ssp2. En el tercer híbrido sp2 se coloca un par de electrones no enlazantes. El segundo par de electrones no compartidos se sitúa en el orbital no hibridizado 3p y se solapa con los orbitales 2p de los átomos de carbono contiguos para formar el sexteto de electrones aromáticos. La energía de resonancia del tiofeno no es tan grande como la del furano debido a la diferencia de tamaño entre los orbitales 2p y 3p que ocasiona un solapamiento menos efectivo.
|
 |
Los hidrocarburos aromáticos, el tabaco y el cáncer.
|
 |
En la siguiente figura se representa esquemáticamente un fragmento de ADN que está constituido por dos cadenas complementarias que se mantienen unidas por los puentes de hidrógeno que se establecen entre las bases adenina - timina y guanina - citosina.
|
 |
Cuando se produce
la división celular las dos cadenas complementarias que constituyen
el ADN se separan y cada una de ellas da lugar a una nueva molécula
de ADN idéntica a la original porque la base adenina se empareja
con la timina (o viceversa) y la guanina con la citosina (o viceversa). Las bases que forman parte del ADN y del ARN contienen átomos nucleofílicos que pueden participar en procesos SN2. A continuación se indica un proceso de este tipo en el que el oxígeno carbonílico de la guanina, ayudado nucleofílicamente por el grupo NH contiguo, ataca mediante un mecanismo SN2 a un sustrato RX que contiene un grupo saliente.
|
 |
Si un proceso como el anterior ocurre se provoca un cambio en el tamaño y la forma de la base nitrogenada. Este cambio puede interferir en el proceso de unión por puentes de hidrógeno entre adenina - timina y guanina - citosina y puede llevar a errores en el proceso de transcripción genética que pueden desembocar en mutaciones celulares. Algunas de estas mutaciones pueden llevar a un proceso de división celular incontrolado que resulte finalmente en un tumor cancerígeno. El benzopireno es un hidrocarburo aromático constituyente, entre otros centenares de compuestos, del humo del tabaco. El benzopireno mismo no es cancerígeno pero en el proceso de inhalación del humo del tabaco el benzopireno penetra en el organismo y resulta oxidado por determinadas enzimas, como la P-450-mono-oxigenasa, que lo convierte en el epóxido A. Este compuesto es hidrolizado por el enzima epóxido-hidratasa al diol B. Una segunda epoxidación enzimática del compuesto B por la P-450-mono-oxigenasa lleva al epoxidiol C.
|
 |
Si el compuesto C reacciona con la guanina del ADN se forma el compuesto E. Cuando se produce la división celular esta parte de guanina que se ha convertido en el compuesto E no puede emparejarse con la citosina lo que lleva a una transcripción errónea del mensaje genético que puede desembocar en última instancia en una división celular incontrolada.
|
 |
9. El mecanismo de la sustitución electrófila aromática: halogenación, nitración, sulfonación. Los alquenos reaccionan como especies nucleofílicas atacando a los electrófilos para dar productos de adición. Los electrones pi de los alquenos están enlazados con menor firmeza a los núcleos que los electrones sigma, y atacan a los electrófilos para generar un carbocatión que a continuación es atacado por un nucleófilo para dar finalmente el producto que resulta de la adición al doble enlace.
|
 |
El benceno, al igual
que un alqueno, tiene nubes electrónicas pi por arriba y por debajo
del plano molecular que forman los enlaces sigma. Aunque los electrones
pi del benceno forman parte de un sistema aromático estable pueden
atacar a un electrófilo fuerte y generar un carbocatión
que está relativamente estabilizado por deslocalización
de la carga positiva. Este carbocatión ciclopentadienilo se denomina
complejo sigma, porque el electrófilo está
unido al anillo bencénico por un nuevo enlace sigma. El complejo
sigma no es aromático porque el átomo de carbono unido al
electrófilo presenta una hibridación sp3 e interrumpe el
anillo de orbitales p. Esta pérdida de aromaticidad del complejo
sigma contribuye a que el ataque del electrófilo sea muy endotérmico.
El complejo sigma puede recuperar la aromaticidad por inversión
del primer paso, volviendo a los reactivos, o por la pérdida de
un protón en el átomo de carbono sp3 ocasionado por el ataque
de una base al complejo sigma. El producto final es formalmente el producto de sustitución de uno de los hidrógenos del anillo aromático por un electrófilo y por tanto a este mecanismo se le denomina Sustitución Electrófilica Aromática (SEAr).
|
 |
 |
Halogenación.
|
 |
Al contrario que el ciclohexeno, el benceno no reacciona con bromo para formar un producto dibromado. Se ha calculado que la adición de bromo al benceno sería una reacción endotérmica en 2 kcal/mol debido a la pérdida de aromaticidad que supondría este proceso.
|
 |
Para que el benceno reaccione con el bromo el proceso se debe efectuar en presencia de un ácido de Lewis, como el tribromuro de hierro (FeBr3), y el producto de la reacción es un producto de sustitución: el bromobenceno. La reacción es exotérmica y desprende 10.8 kcal/mol.
|
 |
La reacción
de bromación sigue el mecanismo general de Sustitución Electrofílica
Aromática. Como el bromo no es suficientemente electrofílico
para ser atacado por el benceno la reacción se lleva a cabo en
presencia de cantidades catalíticas de FeBr3. Uno de los átomos
de bromo de la molécula Br2 interacciona con el átomo de
hierro del FeBr3 de forma que uno de pos pares electrónicos libres
del
|
 |
Una vez formado el intermedio Br2·FeBr3, altamente electrofílico, se produce el ataque nucleofílico del benceno lo que lleva a la formación del complejo sigma. Este intermedio es atacado por el ión bromuro para regenerar la aromaticidad y formar el bromobenceno y HBr.
|
 |
 |
En el esquema anterior
se pone de manifiesto que el FeBr3 no se consume en la reacción
y por tanto actúa de catalizador del proceso. La formación
del complejo sigma determina la velocidad, y el estado de transición
que conduce a su formación ocupa el punto máximo en el diagrama
de energía. Este primer paso del proceso es muy endotérmico
debido a que se forma un carbocatión no aromático. El segundo
paso, el ataque de la base al carbocatión, es exotérmico
porque se recupera la aromaticidad del sistema. La reacción general
es
|
 |
Cloración del benceno. La cloración del benceno se lleva a cabo de modo parecido a la bromación, empleando como ácido de Lewis el AlCl3.
|
 |
Yodación del benceno. La reacción de yodación se lleva a cabo con yodo en presencia de HNO3 como agente oxidante. El HNO3 se consume en la reacción y por tanto no es un catalizador. La misión del HNO3 es oxidar al yodo y generar el ion yodonio (I+) que es el electrófilo que resulta atacado por el benceno.
|
 |
Nitración del benceno.
|
 |
El ácido sulfúrico reacciona con el ácido nítrico generando el ion nitronio (+NO2), que es el electrófilo de la reacción de sustitución electrofílica aromática.
|
 |
El ion nitronio reacciona con el benceno formando el complejo sigma, que a continuación, pierde un protón para dar lugar al nitrobenceno.
|
 |
Sulfonación.
|
 |
El benceno ataca al SO3 formando el complejo sigma. La pérdida de un protón produce el ácido bencenosulfónico.
|
 |
La reacción de sulfonación es reversible y el grupo SO3H se puede eliminar calentando el ácido bencenosulfónico en ácido sulfúrico diluido.
|
 |
La desulfonación sigue el mecanismo general de las reacciones SEAr. El protón es el electrófilo del proceso y el grupo saliente es el SO3.
|
 |
10. Efecto orientador de los sustituyentes en las reacciones SEAr. Las reacciones de
Sustitución Electrofílica Aromática son 25 veces
más rápidas con el tolueno que con el benceno y por ello
se dice que el tolueno está activado, respecto del benceno, frente
a la reacción de sustitución electrofílica aromática. La nitración del tolueno da una mezcla de productos, en la que los mayoritarios son los que resultan de la sustitución en las posiciones orto y para. Se dice que el grupo metilo del tolueno es orto y para dirigente.
|
 |
Las relaciones de
los productos indican que la orientación de la sustitución
no es al azar. Si cada posición fuera igualmente reactiva habría
cantidades iguales de producto orto y meta y la mitad de producto para
debido a que hay dos posiciones orto, dos meta y una para. El paso que determina
la velocidad de la reacción es el primer paso, que corresponde
a la formación del complejo sigma. Este es también el paso
en el que el electrófilo se enlaza al anillo, determinando el tipo
de sustitución. La formación del complejo sigma es una reacción
endotérmica y por tanto la estructura del estado de transición
que conduce al complejo sigma se asemeja al producto de la reacción
(postulado de Hammond). Por tanto, se puede justificar el empleo de las
estabilidades de los complejos sigma como indicadores de las energías
de los estados de transición que conducen a su formación. Cuando el benceno reacciona con el catión nitronio, el complejo sigma tiene la carga positiva distribuida sobre tres átomos de carbono secundarios:
|
 |
No obstante, en el caso de la sustitución orto o para del tolueno, la carga positiva está repartida sobre dos átomos de carbono secundarios y uno terciario.
|
 |
Los complejos sigma
que se generan como consecuencia del ataque a las posiciones orto y para
del tolueno se describen adecuadamente mediante la contribución
de tres estructuras resonantes: dos son de tipo carbocatión secundario
y una de tipo carbocatión terciario. Esta última estructura
es la que explica la mayor estabilización del catión ciclohexadienilo,
en comparación con el catión ciclohexadienilo que se genera
en la nitración del benceno, que se describe mediante tres estructuras
resonantes de carbocatión secundario. Por otra parte, el
complejo sigma que resulta del ataque meta tiene la carga positiva repartida
sobre tres carbonos secundarios. Este intermedio tiene energía
semejante al intermedio que se genera en la reacción de nitración
del benceno. Por tanto, cuando el ataque del electrófilo se produce en las posiciones orto o para, el grupo metilo del tolueno estabiliza al complejo sigma, y en igual medida al estado de transición que lo genera. En consecuencia, el tolueno reacciona con más rapidez que el benceno y lo hace preferentemente en las posiciones orto y para. A continuación se comparan los perfiles de energía de las reacciones de nitración del tolueno y del benceno.
|
 |
Los resultados que se observan con el tolueno son generales para cualquier alquilbenceno que sufre una reacción de Sustitución Electrofílica Aromática. En el siguiente esquema se indican las proporciones de isómeros obtenidos en la reacción de bromación del etilbenceno.
|
 |
Se observa que los productos mayoritarios de la reacción SEAr sobre el etilbenceno son los isómeros orto y para. Además, la reacción de bromación es más rápida que la misma reacción sobre el benceno debido al efecto estabilizante que ejerce el grupo etilo sobre el catión ciclohexadienilo. En general, los grupos alquilo son activantes y orto y para dirigentes.
|
Orientación en compuestos aromáticos con sustituyentes donadores pi.
|
 |
Este resultado parecerá
extraño porque el oxígeno es un átomo muy electronegativo
y sin embargo, como se verá a continuación, es capaz de
ceder densidad electrónica para estabilizar el estado de transición
y por tanto el complejo sigma. Esto ocurre porque los electrones no enlazantes
del átomo de oxígeno adyacente a un carbocatión estabilizan
la carga positiva mediante resonancia. En la siguiente figura se indican las dos estructuras resonantes de un carbocatión con la carga positiva contigua a un átomo de oxígeno.
|
 |
La estructura resonante
más importante, la que se parece más al híbrido de
resonancia, es la II a pesar de que la carga positiva está situada
sobre el átomo de oxígeno. Esta estructura resonante tiene
más enlaces covalentes que la I y además los átomos
de carbono y de oxígeno tienen sus octetos completos. Se dice que
el átomo de oxígeno es un donador pi porque cede densidad
electrónica mediante un ¿Cuál
es el efecto del grupo metoxi en la reacción de nitración
del anisol? Cuando se produce el ataque en orto, el catión ciclohexadienilo
se puede describir mediante la intervención de cuatro estructuras
resonantes. En la cuarta estructura resonante el oxígeno comparte
la carga positiva. Esta estructura resonante también existe en
el ataque en para pero no en el ataque en meta. Por tanto, los estados
de
|
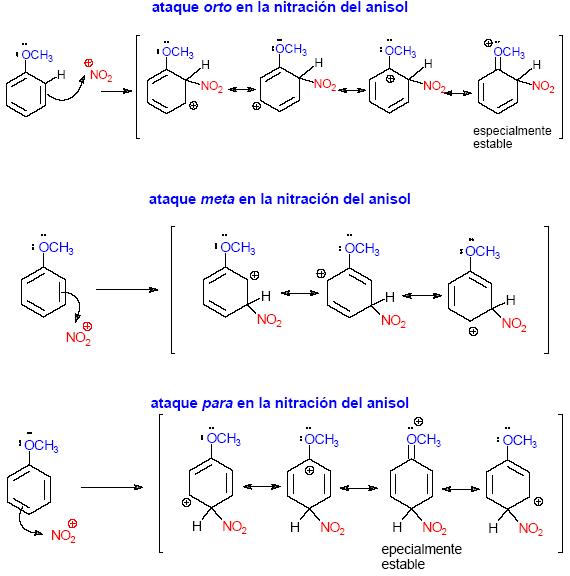 |
El grupo metoxi es un potente activante de las posiciones orto/para y la reacción del bromación del anisol se puede efectuar rápidamente en agua y en ausencia de catalizador. Si la reacción se efectúa en presencia de un exceso de bromo la reacción continúa hasta formar el compuesto tribromado:
|
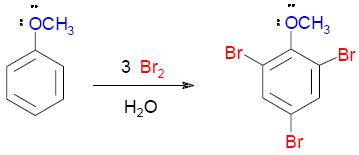 |
El grupo amino.
|
 |
Los electrones no enlazantes del nitrógeno estabilizan al complejo sigma si el ataque se lleva a cabo en las posiciones orto y para.
|
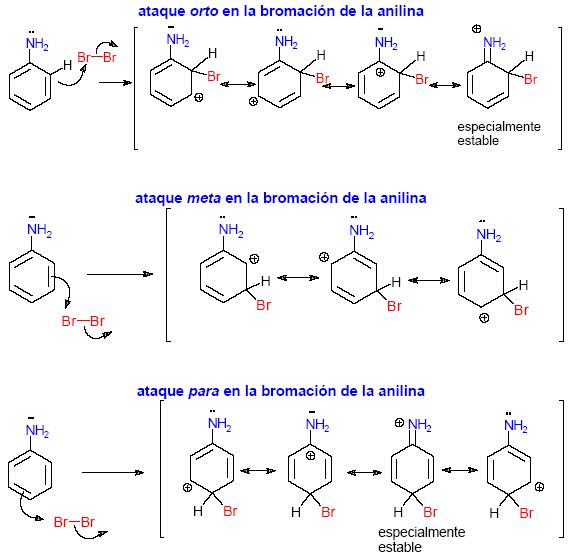 |
Muchos sustituyentes
con pares de electrones no compartidos pueden estabilizar por resonancia
al complejo sigma. Todos son poderosos activantes del anillo aromático
en el proceso de Sustitución Electrofílica Aromática
y orientan la entrada del electrófilo hacia las posiciones orto
y para. A continuación se reúnen en una tabla los sustituyentes activantes que son al mismo tiempo orto y para dirigentes.
|
 |
Sustituyentes desactivantes, directores meta.
|
 |
El grupo nitro es
un potente desactivante del proceso SEAr porque su fuerte efecto inductivo
electrón-atrayente provoca una importante disminución de
la densidad electrónica del anillo aromático y hace que
el anillo sea menos nucleofílico que el anillo del benceno. ¿Cuál será la posición atacada por el electrófilo en las reacciones de Sustitución Electrofílica Aromática del nitrobenceno? El grupo nitro se describe adecuadamente mediante dos estructuras resonantes equivalentes en las cuales siempre aparece una carga formal positiva sobre el átomo de nitrógeno.
|
 |
El ataque a las posiciones
orto, meta o para del nitrobenceno forma tres cationes ciclohexadienilo
que tienen la carga positiva repartida por tres átomos de carbono.
Sin embargo, en los ataques orto y para una de las estructuras resonantes
contiene dos cargas positivas en átomos contiguos. Como las cargas
son de igual signo se repelen y se produce una importante desestabilización
de los intermedios de
|
 |
 |
La mayor parte de los grupos desactivantes son meta dirigentes. Por ejemplo, el grupo acetilo de la acetofenona provoca el ataque mayoritario del electrófilo en la posición meta. En este caso, los cationes ciclohexadienilo correspondientes a los ataques orto y para presentan una estructura resonante que coloca la carga positiva al lado del carbono carbonílico, que contiene una carga parcial positiva. Esta es una situación desfavorable que no se da cuando el electrófilo ataca a la posición meta, que es por esta razón el isómero mayoritario en las reacciones SEAr de la acetofenona.
|
 |
 |
Por lo general, y con la sola excepción de los halógenos, que se verá más adelante, los grupos electrón-atrayentes son desactivantes y meta dirigentes. Estos grupos se reúnen en la tabla que se indica a continuación.
|
 |
 |
Sustituyentes halogenados: grupos desactivantes y orto/para dirigentes.
|
 |
b) Por otra parte, los halógenos tienen electrones no enlazantes que pueden donar su densidad electrónica mediante la formación de un enlace pi. Esta cesión de densidad electrónica les permite estabilizar cargas positivas adyacentes haciendo que los isómeros mayoritarios de la reacción SEAr sean los orto/para. Por ejemplo, la reacción de nitración del clorobenceno proporciona la siguiente mezcla de isómeros:
|
 |
Cuando el electrófilo ataca al clorobenceno en la posición orto el catión ciclohexadienilo se puede describir mediante cuatro estructuras resonantes. En una de ellas, los electrones no enlazantes del cloro ayudan a deslocalizar la carga positiva sobre el átomo de cloro mediante una estructura de ión cloronio. El mismo efecto de deslocalización de la carga positiva se observa en el ataque para.
|
 |
 |
Cuando el electrófilo
ataca la posición meta del clorobenceno el catión ciclohexadienilo
sólo se describe mediante tres estructuras resonantes. La reacción
en la posición meta da un complejo sigma cuya carga positiva no
está deslozalizada por la estructura del ion cloronio. Por tanto,
los ataques orto y para son los mayoritarios porque generan un catión
ciclohexadienilo más estable que el que resulta del ataque
|
11. La reacción de alquilación de Friedel-Crafts.
|
 |
Mecanismo
de la reacción de alquilación de Friedel-Crafts. 1º. Formación del carbocatión t-butilo.
|
 |
2º Reacción SEAr entre el catión t-butilo y el benceno.
|
 |
La pérdida del protón en el catión ciclohexadienilo lleva al producto de sustitución. El catalizador AlCl3 se regenera en el último paso.
|
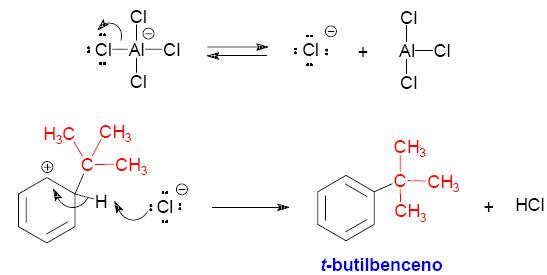 |
En la reacción
de alquilación de Friedel-Crafts se emplea una amplia variedad
de halogenuros secundarios y terciarios, que son sustratos que generan
carbocationes relativamente estables. Si se emplean halogenuros primarios
no se llega a formar el carbocatión primario porque es demasiado
inestable. En su lugar, el electrófilo del proceso es el complejo
que resulta de la coordinación entre el cloruro de aluminio y el
halogenuro de alquilo. En este complejo, el enlace carbono-halógeno
se Mecanismo de la reacción de alquilación de Friedel-Crafts con un halogenuro primario.
|
 |
La reacción
de alquilación de Friedel-Crafts tiene algunas limitaciones: 1. Sólo se
lleva a cabo con benceno, con halobencenos y con derivados activados del
benceno. Con compuestos muy desactivados como el nitrobenceno, los ácidos
bencenosulfónicos o las fenilcetonas la reacción no funciona. 2. Como en la reacción
de alquilación de Friedel-Crafts participan carbocationes es posible
la formación de productos resultantes de reacciones de transposición
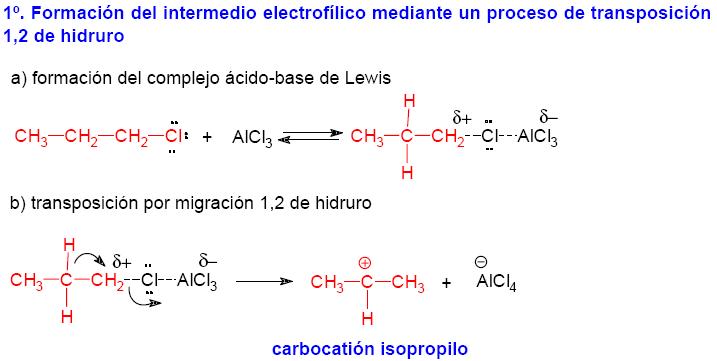
de estos intermedios. Por ejemplo, la reacción de alquilación
de Friedels-Craft del benceno con cloruro de n-propilo no proporciona
el n-propilbenceno sino el isopropilbenceno, por que el complejo ácido-base
de Lewis que se genera entre el cloruro de n-propilo y el AlCl3 experimenta
un proceso de transposición que lleva a la formación del
carbocatión isopropilo, que es un carbocatión secundario. Mecanismo de la reacción de alquilación de Friedel-Crafts con transposición de la especie electrófilica.
|
 |
 |
 |
Como consecuencia
del proceso de transposición se obtiene el isopropilbenceno en
lugar del n-propilbenceno. 3. Otro de los inconvenientes
de la reacción de alquilación de Friedel-Crafts deriva de
la capacidad activante de los grupos alquilo, que hacen que el producto
de la reacción sea más reactivo que el material de partida
y por lo tanto que sea difícil evitar las alquilaciones múltiples.
Por ejemplo, la reacción de 1 mol benceno con 1 mol de cloruro
de etilo, en presencia de una cantidad catalítica de AlCl3, genera
una mezcla de dietilbencenos, una pequeña cantidad de etilbenceno,
trietilbenceno y algo
|
 |
12. Reacción de acilación de Friedel - Crafts. Los principales inconvenientes
que presenta la reacción de alquilación de Friedel-Crafts
se pueden evitar empleado la reacción de acilación de Friedel-Crafts.
En este proceso la especie electrofílica es un carbocatión
acilo, que se genera mediante la reacción entre un cloruro de ácido
y un ácido de Lewis. Mecanismo
de la reacción de acilación de Friedel-Crafts. 1º. Formación
del intermedio electrofílico. El mecanismo de la reacción de acilación se asemeja al de la reacción de alquilación. El primer lugar, el cloruro de ácido reacciona con el catalizador AlCl3 formando un intermedio ácido-base de Lewis, que se rompe heterolíticamente para formar un catión acilo estabilizado por resonancia.
|
 |
2º. Rección SEAr entre el benceno y el catión acilo.
|
 |
El producto de la reacción de acilación es una acilbenceno (una alquil fenil cetona). El grupo carbonilo de la cetona tiene electrones no enlazantes que se complejan con el catalizador AlCl3, lo que hace que se necesite 1 equivalente de AlCl3 en la reacción de acilación. Agregando agua se hidroliza el complejo cetona-AlCl3 y se obtiene el acilbenceno libre. El electrófilo de la reacción de acilación es un complejo grande y voluminoso, probablemente R-C=O-AlCl4. Si se efectúa la reacción de acilación SEAr sobre un benceno que presente un sustituyente orto-para dirigente se obtiene predominantemente el isómero para, debido a que el electrófilo es muy voluminoso y la posición orto está estericamente bloqueada por la presencia del grupo activante.
|
 |
El producto de la
reacción de acilación está desactivado en relación
con el benceno que lo genera, debido al efecto inductivo electrón-atrayente
del grupo acilo. Debido a este efecto inductivo la reacción de
acilación, al contrario que la reacción de alquilación,
se detiene después de una sola sustitución. La reacción
de acilación supera dos de las tres limitaciones de la reacción
de alquilación: los cationes acilo no sufren transposiciones, al
contrario que los carbocationes, y el producto de la reacción se
desactiva de modo que no se producen reacciones posteriores. Sin embargo,
al igual que la reacción de alquilación, la reacción
de acilación no puede efectuarse sobre anillos aromáticos
muy desactivados. La transformación
de los acilbencenos en alquilbencenos se puede conseguir mediante la denominada
reducción de Clemmensen. La reducción se consigue tratando
al correspondiente acilbenceno con zinc amalgamado con mercurio (zinc
tratado con sales de mercurio) en HCl. Esta secuencia de dos pasos permite obtener muchos alquilbencenos que no se pueden obtener directamente por alquilación directa. Por ejemplo, el n-propilbenceno se obtiene por acilación del benceno con cloruro de propanoilo seguida de reducción de la propiofenona:
|
 |
13. Reacción de Sustitución Nucleofílica Aromática.
|
 |
El mecanismo de esta reacción se explica mediante el ataque nucleofílico del anión hidróxido al carbono del anillo aromático que soporta al grupo saliente. Esta reacción genera un complejo sigma aniónico. La carga negativa está deslocalizada sobre los carbonos orto y para y en estos carbonos se encuentran los grupos nitro que ayudan a deslocalizar a la carga negativa por efecto inductivo y resonante. La pérdida del ion cloruro en el complejo sigma produce el 2,4-dinitrofenol. Mecanismo
de la reacción de Sustitución Nucleofílica Aromática. 1º. Formación del complejo sigma aniónico
|
 |
2º. Eliminación del cloruro y formación del producto de sustitución.
|
 |
Si no hay grupos electrón-atrayentes en las posiciones orto y para no es posible la estabilización del intermedio aniónico sigma y la reacción de sustitución nucleofílica aromática no tiene lugar. El mecanismo del bencino: eliminación-adición.
|
 |
De igual manera el clorobenceno reacciona con el amiduro sódico (NaNH2) para dar lugar a la anilina (PhNH2).
|
 |
La sustitución nucleofílica de derivados desactivados del benceno se lleva a cabo mediante un mecanismo diferente al de adición-eliminación que se acaba de explicar en la síntesis del 2,4-dinitrofenol. De hecho, cuando el p-bromotolueno reacciona con amiduro sódico se obtiene una mezcla 50:50 de meta-toluidina y de para-toluidina.
|
 |
El mecanismo de la reacción de Sustitución Nucleofílica Aromática no puede explicar la formación de la meta-toluidina en la reacción anterior. Los dos productos anteriores se explican por un mecanismo de eliminación-adición que se denomina el mecanismo del bencino, debido al intermedio poco usual que implica. El amiduro de sodio, o el hidróxido de sodio en el proceso Dow, reaccionan sustrayendo un protón del anillo aromático y originando un carbanión, que contiene un par de electrones no enlazantes localizado en el orbital sp2 que antes formaba el enlace C-H.
|
 |
El carbanión expulsa al bromuro para formar una especie neutra. Cuando sale el bromuro con el par de electrones de enlace queda un orbital sp2 vacío. Este orbital se solapa con el orbital lleno vecino, dando lugar a un enlace adicional entre los dos átomos de carbono.
|
 |
El carbanión expulsa al bromuro para formar una especie neutra. Cuando sale el bromuro con el par de electrones de enlace queda un orbital sp2 vacío. Este orbital se solapa con el orbital lleno vecino, dando lugar a un enlace adicional entre los dos átomos de carbono.
|
 |
Los dos orbitales
sp2 están separados 60° de modo que el enlace no es muy efectivo.
Al intermedio que resulta de este solapamiento, que es extremadamente
reactivo, se le denomina bencino porque es posible simbolizarlo mediante
un triple enlace entre los dos átomos de carbono. No obstante,
los triples enlaces son lineales, de modo que el triple enlace del bencino
está muy tensionado y es por tanto muy El anión amiduro es una base fuerte con cierto carácter nucleofílico y puede atacar cualquier extremo del reactivo y débil triple enlace del bencino. La protonación subsiguiente produce la toluidina. Aproximadamente la mitad del producto es el resultado del ataque del ión amiduro en el carbono para y la otra mitad del ataque en el carbono meta.
|
 |
14. Hidrogenación de anillos aromáticos.
|
 |
La hidrogenación catalítica del benceno es el método comercial para la producción de ciclohexano y sus derivados sustituidos. La reducción no se puede detener en alguna etapa intermedia, como ciclohexeno o ciclohexadieno, porque estos alquenos se hidrogenan más rápidamente que el propio benceno. 15. Reducción de Birch.
|
 |
El mecanismo de la
reacción de reducción de Birch es semejante a la reducción
de alquinos a alquenos trans con sodio en amoníaco líquído.
La disolución de Na en NH3 líquido genera una disolución
azul de electrones solvatados que son los que inician el mecanismo de
reducción generando un anión radical. Este intermedio se
protona mediante una reacción ácido-base con el alcohol.
A continuación, tiene lugar
|
 |
 |
Los dos átomos de carbono que se reducen pasan por intermedios aniónicos. Por tanto, con anillos bencénicos que contengan sustituyentes electrón-atrayentes, que estabilizan a los carbaniones, la reducción de Birch tiene lugar sobre los átomos del anillo bencénico enlazados a estos sustituyentes.
|
 |
Por el contrario, si el anillo aromático contiene grupos donadores de electrones, la reducción de Birch tiene lugar sobre los átomos de carbono del anillo bencénico que no están enlazados a este tipo de sustituyentes.
|
 |
16. Reacciones de la cadena lateral en los derivados del benceno.
Un anillo aromático
imparte estabilidad adicional al átomo de carbono más cercano
de sus cadenas laterales. El anillo aromático y un átomo
de carbono de una cadena lateral pueden sobrevivir a una
|
 |
b) Reacciones de halogenación radicalaria de la cadena lateral. Los alquilbencenos participan en reacciones de halogenaciones por radicales libres mucho más fácilmente que los alcanos, porque la sustracción de un átomo de hidrógeno de la posición bencílica genera un radical bencilo estabilizado por resonancia. Por ejemplo, el etilbenceno reacciona con bromo, en ausencia de ácidos de Lewis, bajo irradiación fotoquímica para formar el a-bromoetilbenceno.
|
 |
La formación
de este compuesto se explica mediante un mecanismo radicalario. En la
etapa de iniciación el bromo molecular se escinde homolíticamente
para formar dos radicales bromo. En la etapa de propagación el
radical bromo abstrae un átomo de hidrógeno de la posición
bencílica originando un radical bencílico, que está
estabilizado por resonancia con el anillo aromático. El radical
bencílico reacciona con el bromo molecular para dar lugar al alfa-bromoetilbenceno
y a un radical bromo que
|
 |
c) Sustitución Nucleofílica Unimolecular (SN1) en la posición bencílica. Las reacciones SN1
sobre los haluros bencílicos son muy rápidas porque transcurren
a través de
|
 |
El mecanismo SN1 que explica la reacción anterior se inicia con la escisión heterolítica del enlace C-Br, lo que provoca la formación del carbocatión 1-feniletilo. Este carbocatión es secundario pero además es capaz de deslocalizar la carga positiva por el anillo aromático, de manera que su estabilidad es similar a la de un carbocatión alquílico terciario. El agua reacciona rápidamente con el catión formando el alcohol.
|
 |
La estabilidad de un carbocatión bencílico aumenta al aumentar el número de grupos fenilo unidos al centro catiónico. Por ejemplo, la gran estabilidad del carbocatión trifenilmetilo permite la obtención de algunas sales estables, como el fluoroborato de trifenilmetilo, que se puede almacenar durante años sin que sufra ninguna descomposición.
|
 |
d) Sustitución Nucleofílica Bimolecular (SN2) en la posición bencílica. Al igual que los halogenuros alílicos, los halogenuros bencílicos son unas 100 veces más reactivos que los halogenuros de alquilo primario en reacciones de tipo SN2.
|
 |
Cuando un halogenuro bencílico participa en un proceso SN2, el orbital p que enlaza parcialmente al nucleófilo y al grupo saliente en el estado de transición, se solapa con los electrones pi del anillo aromático. Esta conjugación estabilizadora disminuye la energía del estado de transición aumentando la velocidad de la reacción.
|
 |
|