What do they come to make genes there?
Normally with his birth, a child inherits particular characteristics,
like fair hair, blue eyes, which makes it resemble to his/her father or to his mother.
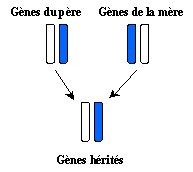
All the characteristics which we inherit our parents are the information contained
inside the cells of our body. These stocks of information are named: genes ,
and the genes are contained in the ADN of our cells . They were copied
starting from genes of the father contained in the sperm and starting from genes of the
mother contained in the ovule. Thus, there will be a gene couple made up of a paternal
gene and a maternal gene to control each inherited characteristic.
In general a gene is enough to control an activity well defined in a cell, so that if a gene inherited the one of the parents is defective , it will be replaced by healthy gene coming from the other relative, thus making it possible the cell to normally function . This is completely normal, and it frequently happens that a gene can comprise anomalies .
The ph�nylc�tonurie is a genetic disease , because the patient inherits this anomaly his parents at the time of the transmission of the genetic code.
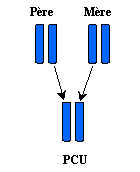
In this case, it is the gene of the phenylalanine-hydroxylase which is in question.The problem is that the parents of the phenylcetonuric child are not reached by the disease , because each relative carries 2 genes inherited his own parents: a healthy gene and a sick gene carrying an anomaly on phenylalanine-hydroxylase. In this case, it is the healthy gene, that which does not have an anomaly which will take over and will make function the cell normally, then making it possible to the relative not to suffer from this disease. One calls that, an operational carrier : because it carries the disease in its genes, but it does not suffer from it.

And well, if a healthy carrying father, meets a healthy surrogate mother, and that they
decide to make children together, there will be 4 possible cases:

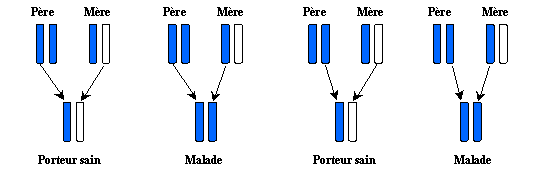
In the worst of the 4 cases, the child will inherit 2
genes carrying the disease, there will be no solution of replacement, and it
will be sick . This offers a probability of 1 out of 4 of putting at
the world a sick child of the PCU, against 2 out of 4 for an operational carrier, and 1
out of 4 for a healthy child of any anomaly concerned with Phenylalanine-hydroxylase.
With each new pregnancy of the mother, this probability is repeated.
The combination of 2 genes transmitted to the child is made in a random way ,
it is thus possible that a family can have all these sick children, or carrying healthy,
or even without anomalies.
Concerning the descent of a child PCU , the transmission repeats same
manner. All will depend on the spouse:
The probability is of:
If the spouse is phenylcetonuric : 4 out of 4 to have children PCU
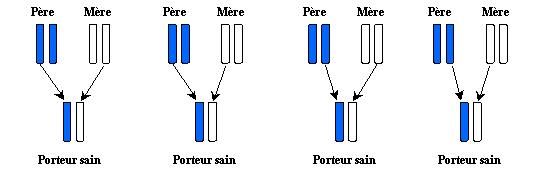
If the spouse is carrying healthy : 2 out of 4 to have children PCU and 2 out of 4 to have healthy carrying children
- If the spouse is not touched : 4 out of 4 to have healthy carrying
children
The gene of phenylalanine-hydroxylase is in the area q22-q24.1 chromosome 12 which is
composed of 90 000 nucleotides.
More than 150 transfers or modifications different were described on this
gene. It appeared that there was a relation between the genotype and the phenotype, i.e.
between the transfers observed and the residual activity of the enzyme.
This raises the question to know if it is necessary to seek by genetic study the transfers
at all the patient having a ph�nylc�tonurie.
A better knowledge of the transfers for a given patient would make it possible to
differentiate at the new-born baby the ph�nylc�tonurie from the hyperphenylalaninemy and
would make it possible to propose a mode from the start adapted better to each patient,
like deciding frequency of controls.
However, it is advisable to notice that in all the epidemiologic studies of prediction of
the intelligence quotient the quality of the mode remains the prevalent factor
and this quality of the mode is independent of the genotype.
Nevertheless, there are great variations of the phenylalanine rates among the
carriers of a same transfer what for the moment limits the utility of the analysis of
the transfer, to the antenatal diagnosis.
These new concepts of assumption of responsibility all in the direction of a strict
control of the Phenylalanine rates in order to will ensure an optimal development of the
patients.
Experiments of genic therapy practised in the animal are encouraging, but
do not allow for the moment of the long-term corrections of the defective activity of the
enzyme. While waiting, this processing which tackles the cause of the disease, the
dietetic approach is the only one which allows a harmonious development of these patients.