Qualità in autoimmunologia
Marco Pradella
manca la rappresentazione grafica?
La natura insolita delle scale in autoimmunologia crea ancora altri problemi.
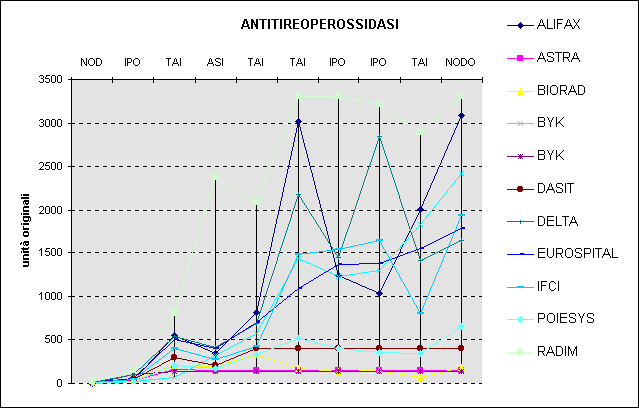
Si veda ad esempio la Figura 6 in confronto alla Figura 7.
Figura 6. Risultati di anticorpi anti perossidasi tiroidea con diversi metodi: scala originale
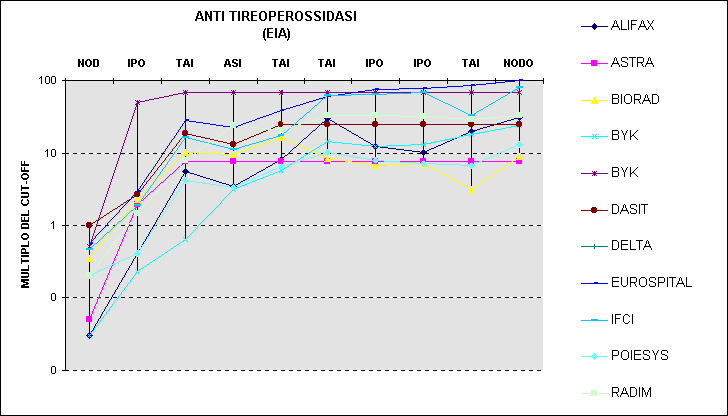
Figura 7. Risultati di anticorpi anti perossidasi tiroidea con diversi metodi: scala modificata con il calcolo del multiplo rispetto al cut-off e la trasformazione logaritmica.
Le conclusioni indotte dalla prima rappresentazione (elevata dispersione di origine casuale sui livelli più alti) sono opposte a quelle che si ricavano dalla seconda (presenza di differenze sistematiche ben distribuite tra le concentrazioni; notevole variabilità intorno al cut-off).
Ecco come l'opportuna scelta di una scala può favorire la costruzione di un grafico significativo.
Analogamente, il titolo di un siero di controllo positivo in immunofluorescenza procede per "gradini" in base al raddoppio della diluizione. Il titolo 1/40 è rispetto a quello 1/80 alla stessa distanza che separa 1/640 da 1/1280, nonostante le cifre siano, in scala naturale, molto diverse. L'uso della trasformazione logaritmica (si vedano anche le Figure 8 e 9) risolve appunto questo problema. Sarebbe anzi sufficiente considerare il titolo come una variabile (meglio un "attributo") di tipo qualitativo.
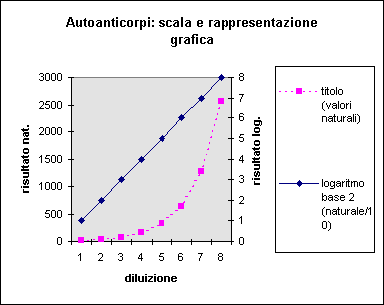
Figura 8. Effetto della scala di misura sulla rappresentazione grafica.
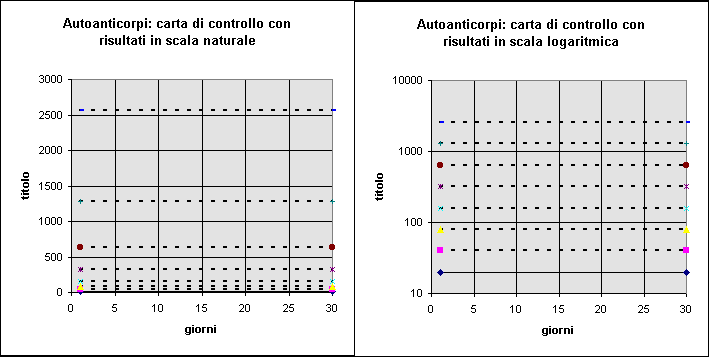
Figura 9. Effetto della scala di misura sulla rappresentazione grafica di una carta di controllo
Capita sovente di osservare che l'operatore di laboratorio diventa recalcitrante all'applicazione del controllo di qualità perchè il materiale a disposizione non ha un "valore assegnato".
Sembra quasi che si intenda il monitoraggio della qualità come un continuo confronto con la "verità", un gioco a scommettere quanto il nostro sistema analitico (reagenti più eventuale calibratore) questa volta sia andato vicino al "giusto".
Questo non è controllo di qualità.
Vale la pena di ricordare la descrizione operativa del CQI: analizzare più volte campioni stabili oppure campioni di Pazienti appartenenti a popolazioni con caratteristiche stabili. Il "valore atteso" di queste analisi non è assegnato da altri che il laboratorio stesso, il quale deve stabilire, in una iniziale fase di "avvio" del sistema, ad identificare la propria "media di processo" (che non necessariamente è costituita da una media aritmetica).
Il "valore vero" di un materiale ha una funzione in altri campi del sistema di qualità, precisamente quelli della "prevenzione" (v. Figura xx). Il valore del calibrante, ad esempio, deve essere tale da ottenere valori vicini al valore vero (cioè accuratezza) nei campioni dei Pazienti. Il valore vero, simato con metodi definitivi, costituisce l'anello di partenza della catena della tracciabilità in una attività di standardizzazione.
manca un intervallo di accettabilità?
Un'altra osservazione frequente è quella di attività di monitoraggio della qualità costituite unicamente dalla ripetizione dell'analisi di materiali di controllo. I risultati, poi, vengono magari riportati in un grafico, ma l'interpretazione pratica viene lasciata all'intuizione o all'estro dell'operatore, in qualche caso sorretto da una generica regola "12s".
Le decisioni conseguenti al monitoraggio della qualità, invece, dovrebbero essere basate su criteri ben più oggettivi.
Oggi possiamo descrivere un processo molto preciso per la costruzione delle decisioni, un processo in cui assumono particolare valore gli obiettivi analitici.
Possiamo descrivere il processo in 6 fasi:
Le scuole di pensiero sul "goal" analitico sono essenzialmente due: una fondata sulle "opinioni" ed una sulla "variabilità biologica".
Non entriamo qui nel merito della disputa tra le due scuole, anche se va riconosciuta la netta prevalenza negli ultimi anni della seconda.
Un criterio di partenza è comunque necessario. Da questo indice (Errore Massimo Accettabile, EMA) si possono ricavare parametri da utilizzare concretamente nel controllo di qualità.
Si distinguono infatti nell'EMA tre componenti: errore sistematico stabile (Bias, B), errore sistematico instabile (D SE), imprecisione (D RE).
Il primo è caratteristico del metodo e si osserva in tutte le serie per tutti i campioni.
Il secondo è caratteristico di una serie e si osserva per tutti i suoi campioni.
Il terzo è caratteristico del singolo campione.
Le quattro grandezze sono legate dalla seguente relazione:
EMA = B + D SE + z D RE
dove z è la variabile casuale normale per il livello di confidenza prescelto (1.65 corrisponde all'errore a di 0.05, ossia alla probabilità del 95%).
Ipotizzando che B sia pari a 0, il calcolo di D SE e D RE è facile. D SE e D RE vengono applicati nelle curve di potenza di ciascuna regola per verificarne l'efficacia.
La descrizione teorica del meccanismo non aiuta molto l'operatore del laboratorio clinico, specie in autoimmunologia.
Facilmente verrà obiettato che nessuno ha stabilito obiettivi analitici per le ricerche di autoanticorpi e che spesso i risultati sono "qualitativi" o appena "semiquantitativi" (!).
Green e collaboratori hanno recentemente suggerito una soluzione al problema, prendendo esempio dal dosaggio qualitativo dell'antigene del virus dell'epatite B. Essi partono dalle seguenti osservazioni:
Abbiamo verificato l'approccio di Green in autoimmunologia prendendo come oggetto dello studio il dosaggio immunoenzimatico degli anticorpi anti nDNA.
I risultati sono descritti nella Tabella VI e nella Figura 10.
Tabella VI. Analisi di 5 serie per il dosaggio degli anticorpi anti nDNA con il metodo di Green e coll. (LDL = limite di sensibilità analitica)
| dose | ass1 | ass2 | ass3 | ass4 | ass5 | media ass | CV% |
0 |
18 |
27 |
19 |
32 |
26 |
24 |
24% |
20 |
73 |
169 |
139 |
377 |
331 |
218 |
60% |
90 |
313 |
536 |
481 |
1269 |
963 |
712 |
55% |
200 |
704 |
992 |
910 |
1968 |
1766 |
1268 |
44% |
350 |
1229 |
1621 |
1007 |
2544 |
2190 |
1718 |
37% |
| cut off | 40 |
IU/mL | |||||
| ass cut off | 359 |
ass | |||||
| cv cut off | 58% |
cv ass | |||||
| ass LDL | 42 |
ass | |||||
| TAE | 88% |
ass | |||||
| dSEcrit | -0,1383 |
s ass |
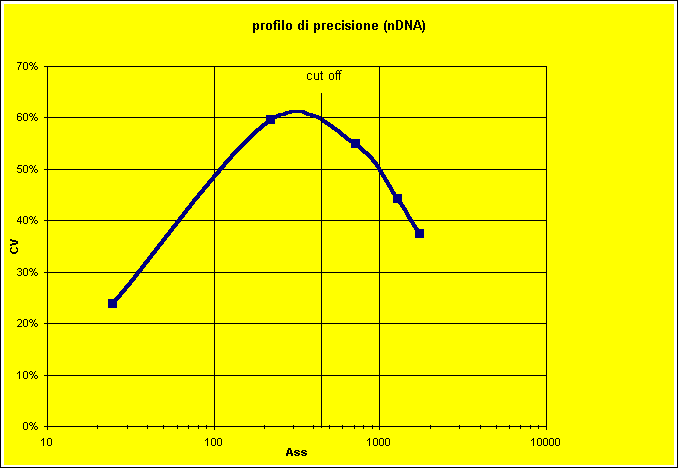
Figura 10. Profilo di precisione di alcune serie per il dosaggio degli anticorpi anti nDNA, dal valore delle assorbanze strumentali
L'analisi così condotta consente di rilevare che il profilo di precisione peggiora proprio laddove meno sarebbe necessario, ossia intorno al cut-off.
L'errore massimo accettabile (un valore positivo, oltre il cut-off, che si confonde con il negativo, sotto la soglia di sensibilità analitica) è pari all'88% dell'assorbanza al cut-off. Il valore dell'imprecisione a questo livello (58%) è talmente alto che non è possibile ottenere una soglia di D SE che garantisca il 95% dei risultati entro il limite: il valore ottenuto è infatti negativo.
C'è chi potrebbe facilmente obiettare che il valore strumentale (assorbanza) viene convertito in concentrazione dai calibranti, proprio al fine di compensare le variazioni analitiche che si riflettono nell'assorbanza. Per stimare la variabilità del metodo, allora, dovrebbero essere utilizate non già le risposte analitiche originali, ma il "ricalcolo" (o backfit) delle concentrazioni degli stessi calibranti. Operazione che spesso i sistemi analitici computerizzati, al fine di infondere sicurezza nell'operatore, svolgono gratuitamente.
La verifica sui dati da noi esaminati porterebbe infatti l'imprecisione al cut-off al livello del 2% ed il livello di D SE a 44 IU/mL. L'analisi del profilo di precisione, tuttavia (Figura 11) mostra il contrasto con il valore ottenuto per un materiale di controllo inserito in ogni serie.
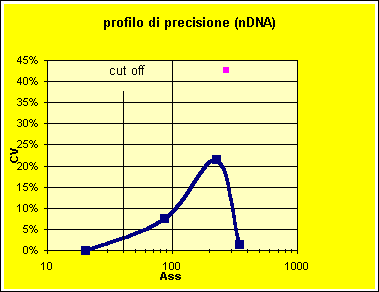
Figura 11. Profilo di precisione di alcune serie per il dosaggio degli anticorpi anti nDNA, dal valore dei calibranti ricalcolato, confrontato con l'imprecisione del campione di controllo.
Si può quindi concludere che il ricalcolo delle concentrazioni dei calibranti fornisce una stima non corretta dell'imprecisione del metodo. I materiali di controllo inoltre dovrebbero avere livelli di concentrazione vicini al cut-off, per essere utili al monitoraggio della qualità analitica.
Le regole di controllo sono dei mezzi oggettivi per prendere delle decisioni. La decisione da prendere nel nostro caso riguarda due azioni antitetiche: l'accettazione o il rifiuto di una serie analitica o di uno o più risultati in un programma interlaboratorio.
Le regole di controllo vengono abbreviate con la sigla: AL, in cui A indica il numero di osservazioni sui campioni di controllo utilizzati, oppure alternativamente il parametro statistico utilizzato, e L indica i limiti oltre i quali scatta la regola. Si possono portare alcuni esempi:
13s: significa che una osservazione (1) supera il limite dato dal valore medio ± 3 volte la deviazione standard (3s)
31s: significa che tre osservazioni consecutive superano lo stesso limite, che è il valore medio - 1 s o il valore medio + 1 s
7x –: sette osservazioni consecutive cadono sopra o sotto la media
7T (T = trend): sette osservazioni consecutive mostrano una tendenza all'aumento o alla diminuzione
R4s: la differenza (Range) di due o più osservazioni supera il valore pari a 4 volte la deviazione standard.
E' sorprendente notare quanto spesso si trascura nei laboratori la possibilità, anzi la convenienza ad usare regole di controllo diverse, anzi più regole di controllo combinate insieme. D'altro canto, è comprensibile che in campi "difficili" come l'autoimmunologia, lo sforzo per arrivare ad una qualche forma di controllo di qualità sia tale che l'intepretazione dei risultati venga intesa in qualche modo come acessoria.
Solitamente viene scelta la semplice regola 12s, che è caratterizzata, come è noto, da un tasso elevato di falsi positivi (almeno il 5%).
Lo standard NCCLS C41 riporta invece chiaramente quali regole vanno usate al fine di evidenziare una variabilità sistematica e quali per la variabilità di tipo casuale o random (Tabella VII).
Tabella VII. Regole per rivelare la presenza di errore sistematico e di errore casuale
| errore casuale | errore sistematico |
| 13s R4s |
22s 41s 7x - 12x - |
Green e collaboratori, ad esempio, nel contributo già citato, applicata la loro procedura di individuazione dell'errore critico, hanno individuato la regola 13s come quella conveniente per il metodo dell'HBsAg utilizzato.
mancano criteri per l'accettazione dei risultati (qualora le regole siano violate)?
Le regole di controllo presentano falsi negativi e falsi positivi.
Non solo, anche in presenza di un vero positivo (ossia di una deviazione significativa dal comportamento atteso del metodo in termini di precisione o di accuratezza) potrebbe essere difficile adottare il provvedimento previsto dal controllo di qualità, cioè rigettare l'intera serie dei risultati.
Ai fini pratici, specialmente quando sono in gioco il tempo di risposta, il costo dei materiali ed altri elementi di delicata importanza, lo standard NCCLS stabilisce che vengano stabiliti in modo formale, pena la loro adozione selvaggia, dei criteri per accettare i risultati dei Pazienti anche se alcune regole sono state violate. Questi criteri, stabiliti dal direttore del laboratorio, sono basati sulla variabilità biologica, sull'utilità medica del test, sul tipo di regola violata e sull'entità della violazione (Tabella VIII).
Tabella VIII. Criteri proposti per il calcolo dei limiti di accettabilità clinicamente utili
| Livello costituente | Criteri (scegliere il più basso) | |||||||
| inferiore a LLN + 1/4 NRI | NRI | LLN | 0.7 D | DRI | ||||
| fino a 1.5 ULN | NRI | 0.7 D | DRI | |||||
| superiore a 1.5 ULN | NRICV | 0.7 D | DRI | |||||
LLN = limite di riferimento inferiore per il normale; ULN = limite di riferimento superiore per il normale; NRI = intervallo di riferimento per il normale (ULN - LLN); D = differenza tra due valori successivi non dovuta alla variabilità analitica: in pratica ± 3 s del metodo; DRI = intervallo diagnostico (per esempio, l'intervallo terapeutico); NRICV = NRI espresso come % del ULN.
In autoimmunologia non è facile applicare questi criteri. Nel caso in cui il metodo abbia una certa componente quantitativa (come per il dosaggio dell'anti-nDNA descritto in precedenza), si potrebbe intendere come ULN il valore del cut-off e come NRI la differenza tra il cut-off ed il limite di sensibilità analitica.
manca una procedura per indagare sui risultati non accettabili?
Non abbiamo molte indicazioni sul "che fare" una volta che le regole di controllo hanno dato un allarme.
Il gran parlare e discutere di qualità che si é fatto di recente non sempre ha realmente aiutato gli operatori della medicina di laboratorio a migliorare le loro prestazioni. Si prendano ad esempio i requisiti minimi inseriti nelle linee guida recentemente emanate dal Ministero della Sanità. In questi requisiti è prescritto la partecipazione a programmi di valutazione esterna di qualità, ma non si dice nulla sulle modalità con cui questi programmi dovrebbero svolgersi né su come i partecipanti debbano comportarsi in presenza di risultati non accettabili.
D'altro canto, nemmeno gli standard per l'accreditamento inglesi, americani e italiani, a cui quei requisiti minimi si sono largamente ispirati, sono molto più dettagliati.
Con il suo proverbiale pragmatismo, invece, l'organismo statunitense per la standardizzazione delle procedure nei laboratori clinici (NCCLS) sta riuscito a condensare in poche pagine ed alcune tabelle quanto serve per imboccare decisamente la via del miglioramento della qualità, almeno al fine di cavarsela con il problema della partecipazione ai programmi di valutazione esterna della qualità (proficiency test o prove di abilità quando hanno anche qualche caratteristica fiscale, punitiva, come quelli previsti dal CLIA).
In un precedente rapporto si è descritto le basi da cui proviene la concezione della gestione della qualità globale e del miglioramento continuo in medicina di laboratorio, o almeno quelle individuate dal sottocomitato NCCLS costituito a questo scopo.
Il documento G22 si é poi evoluto, modificando anche la parte relativa alla gestione dei risultati errati, che è diventata più chiara e praticabile.
Qui diamo una descrizione di questa parte del documento G22.
A. I risultati non accettabili di una VEQ: valutazione descrittiva
Ogni laboratorio prima o poi, non fosse che per ragioni statistiche, si trova ad avere risultati di VEQ non accettabili. Nei regolamenti del CLIA '88 si ritengono risultati non accettabili quelli che superano i limiti appositamente previsti per ciascun esame, riportati nella Tabella IX.
L'équipe del laboratorio dovrebbe utilizzare i risultati dei VEQ come un indicatore per verificare la qualità delle prestazioni e pianificarne il miglioramento.
I risultati della VEQ non sono utili solo quando inaccettabili secondo i criteri ufficiali. Esiste una certa probabilità di ottenere risultati inaccettabili anche in assenza di problemi identificabili, semplicemente a causa della variabilità probabilistica connessa all'imprecisione ed all'inaccuratezza dei metodi. D'altro canto, anche quando i risultati delle VEQ sono accettabili, la loro analisi può rivelare l'insorgere di problemi, come un errore di tipo sistematico (tutti i risultati da un lato del valore centrale) o un aumento di imprecisione.
Per la valutazione di un risultato quantitativo devono essere disponibili gli elementi riportati in Tabella X.
Tabella IX. Intervalli di accettabilità secondo il CLIA-88
| Immunologia generale | |
| analisi | criterio di accettabilità |
| alfa-1-antitripsina | valore assegnato ± 3 SD |
| alfa-fetoproteina | valore assegnato ± 3 SD |
| anticorpi antinucleo | valore assegnato ± 2
diluizioni positivo o negativo |
| antistreptolisina O | valore assegnato ± 2
diluizioni positivo o negativo |
| anti-HIV | reattivo o non reattivo |
| Complemento C3 | valore assegnato ± 3 SD |
| Complemento C4 | valore assegnato ± 3 SD |
| Epatite (HBsAg, anti Hbc, HBeAg) | reattivo o non reattivo |
| IgA | valore assegnato ± 3 SD |
| IgE | valore assegnato ± 3 SD |
| IgG | valore assegnato ± 25% |
| IgM | valore assegnato ± 3 SD |
| mononucleosi infettiva | valore assegnato ± 2
diluizioni positivo o negativo |
| rosolia | valore assegnato ± 2
diluizioni immuno o non immune positivo o negativo |
Tabella X. Elementi necessari per la valutazione di un risultato quantitativo di VEQ.
| Componenti | Tipi di valore di confronto (target) | Tipi di intervallo di accettabilità |
• valore di confronto • intervallo di accettabilità |
• medie di altri gruppi o generali • valori derivati da altra fonte (consensus di laboratori di riferimento o metodi definitivi / di riferimento |
• percentuale fissa ( es. ± 10% del valore target) • combinazione di intervallo e percentuale (es. : ± 6 mg/dL o 10% del valore di confronto, se magggiore) • intervallo basato sulla deviazione standard di gruppo (es. ± 2 SD) |
Il modo più semplice per tenere sotto controllo le prestazioni é un grafico di bias (differenza tra risultato e valore target sulle ordinate, valore target sulle ascisse). Con questa rappresentazione, oltre alla valutazione puntuale sui singoli valori, è possibile effettuare una valutazione di linearità sull'intero intervallo analitico. Questo metodo è ideale per esercizi di VEQ singoli (as esempio, cinque campioni inviati tutti insieme).
Nei programmi di VEQ protratti nel tempo con spedizioni multiple, può essere interessante avere indicazione di modifiche progressive di accuratezza. E' allora opportuno rappresentare i risultati in sequenza temporale in una carta di Shewart o Levy-Jennings, applicando però una trasformazione per aggiustare l'intervallo di accettabilità alle diverse concentrazioni. La trasformazione consiste nel calcolare dal risultato ottenuto la percentuale di scostamento ammesso come segue:
% scostamento ammesso = * 100
B. I risultati non accettabili di una VEQ: procedura investigativa
Il laboratorio deve avere procedure scritte che descrivano dettagliatamente le singole fasi necessarie per identificare, interpretare e correggere qualsiasi problema riscontrato. Le fasi sono cinque: recepimento e revisione, classificazione del problema, valutazione dei risultati dei Pazienti, conclusioni ed azioni, documentazione (Tabella XI).
Il punto più delicato è indubbiamente la classificazione del problema (Tabella XII). Non va dimenticato tuttavia cha ai fini dell'accreditamento è essenziale la documentazione accurata dell'intero processo. La figura 12 riporta un esempio di modulo per l'istruttoria relativa ad un risultato inaccettabile di VEQ.
Lo standard G22 del NCCLS, da poco disponibile per tutta la comunità mondiale dei laboratori clinici, è un tipico esempio di come i criteri standard dell'accreditamento debbano essere integrati ed esplicitati con regole professionali per diventare concretamente praticabili.
La semplice partecipazione ad un programma di VEQ, anche se accreditato, è inutile ai fini della qualità delle prestazioni se non si accompagna a procedure fermamente stabilite e regolarmente applicate per investigare le cause dei risultati non accettabili. Con poca fatica ma molta determinazione è possibile non solo ottenere la documentazione sufficiente per superare con successo l'ispezione degli incaricati per l'accreditamento, ma soprattutto garantirsi la correttezza dei risultati forniti per la diagnosi dei Pazienti.
Tabella XI. I risultati non accettabili di una VEQ: procedura investigativa
| 1. recepimento e revisione | 2. classificazione del problema |
| Rivedere
tutta la documentazione relativa al risultato
inaccettabile (comprese le stampe strumentali, i piani di
lavoro e le registrazioni computerizzate). Assicurarsi di
effettuare: • la verifica di errori di trascrizione • la verifica del controllo di qualità interno, lo stato di calibrazione e gli allarmi strumentali • ripetere analisi e calcoli, se possibile; eventualmente richiedere una nuova aliquota • rivedere le prestazioni precedenti per quel costituente |
•
trascrizione • metodologico • tecnico • del materiale di VEQ • della valutazione dei risultati • inesplicabile |
| 3. valutazione dei risultati dei Pazienti | 4. conclusioni ed azioni |
| valutare eventuali conseguenze a carico dell'assistenza e documentare le azioni intraprese | se
la causa del risultato viene identificata, porre in
essere e documentare le azioni correttive appropriate; es.: accorciare la scadenza dei calibranti; cambiare la loro conservazione; rivedere un vetrino con l'interessato; introdurre un programma mensile per revisioni di preparati microscopici o un programma di formazione permanente con una istituzione esterna |
| 5. documentazione | |
| utilizzare moduli standardizzati | |
Tabella XII. Esempi di classificazione del problema
| 4.
problema del materiale di VEQ • effetto matrice (elevate differenze tra gruppi) • materiale non omogeneo (elevato CV interlaboratorio) • contaminazione batterica o emolisi • campione non vitale (microbiologia) • campione non rappresentativo • reazione debole (immunoematologia) • anticorpo rilevabile ma non identificabile (immunoematologia) • interferenza da Coombs diretto positivo |
5.
problema della valutazione dei risultati di VEQ • gruppo di metodo non appropriato • valore di confronto (target) inaffidabile • registrazione errata 6. problema inspiegabile • risultato grossolanamente errato da causa inspiegabile. Attenzione evitare la manomissione di un sistema senza aver identificato il problema (Deming) • altro |
Figura 12. Modulo per documentare l'istruttoria relativa ad un risultato inaccettabile di VEQ
Laboratorio _____________________________________
Istruttoria per risultato inaccettabile di valutazione esterna di qualità (VEQ) secondo lo standard NCCLS G22
| Data dell'indagine: |
| Identificativo del campione di VEQ: |
| Data dell'analisi: |
| Risultato inaccettabile: |
| Risultato atteso e intervallo di accettabilità: |
| Risultati inaccettabili precedenti o trend per questo esame: |
| Revisione trascrizioni, fase postanalitica: |
| Indagini sulla fase analitica: |
| Conclusioni: |
| Effetto sui risultati dei Pazienti: |
| Classificazione
del problema: trascrizione - fase post-analitica metodologico tecnico del materiale di VEQ della valutazione nella VEQ inspiegabile |
| Azioni correttive: |
| Approvazioni supervisore data direttore data |