Pharmacology – Exam 4
|
|
||||||||
Pharmacokinetics
à Antibiotics Video
Pharmacokinetics = study of movement of drug molecules
Used to predict à
|
Drug Dissolution |
Time of dosage to disintegrate/dissolve |
|
Drug Absorption and Distribution |
Amount of drug absorbed vs. time Time of onset to target tissues Duration in target tissues |
|
Drug Excretion |
How and when the dose will be excreted |
Classes of molecules behave according to physical/chemical laws
Individual molecules behave randomly
àOverall clinical response results from total of individual random interactions
Transport across lipid membranes is the key
Path of drug once ingested:
Gut wall à Capillaries/veins/portal vein à liver à vena cava à heart (to lungs, back to heart) à systemic circulation
Liver
* May enter the Bile Duct instead (Biliary Cycle)
àfrom the bile duct to the gall bladder and then back to the duodenum
àOptions once in duodenum
1. Excretion through gut
2. Re-absorbed, then to systemic circulation
3. Re-absorbed, then to biliary cycle again
* Biliary Cycle is NOT a major route for drug excretion
the drug concentration in the gall bladder may be up to 50 mg/mL (for a 1 gram dose)
only about 200 mL of bile is produced each day
this results in only 10 mg excreted per 1 gram dose via this route
Systemic
* Drug will react with or against encountered molecules
* Drug may or may not reach target organ
* Drug may be excreted intact/unchanged/unutilized
* Crossing Cell Membranes
small molecules can pass through small pores, large molecules need help
alternatives for large molecules à
Never Cross: oral anti-fungals are designed to stay in the gut and not diffuse
Actively Transported: utilize enzyme systems (e.g., transferases)
Passively Diffuse: dissolve through membrane (lipophilic)
Rate of Transport
*Ionizability à ionized molecules CANNOT cross the lipid membrane
*Lipophilicity à molecules repelled by lipids (lipophobic) may never get in
molecules attracted to lipids (lipophilic) may never get out
lipophilic molecues will just accumulate in cell membrane or fatty tissue
*Protein Affinity à protein bound molecules are temporarily tied up and unavailable for transport
Rates are represented by KA (rate of absorption) and KE (rate of excretion)
à thus, absorption is rate-dependant
Example: Incomplete absorption in the duodenum is the initiator of side effects
diarrhea, super-infections, candidiasis, destruction of normal gut flora that may result from antibiotics
Partition Coefficient à KD
Ratio of drug concentration in lipid to drug concentration in water
Higher KD indicates a drug is more lipophilic
|
Drug A |
KD = 10 (10:1 ratio) |
Drug A is 100 times more lipophilic than Drug B |
|
Drug B |
KD = 0.1
( |
There is continual exchange across membranes (or phases)
The
rate in/across, K1, and the rate out/across,
For
Drug A, K1 is 10x faster than
For
Drug B,
At any given
time, the drug concentration on one side of a membrane depends on the rates K1
and
Equilibrium is eventually established and the ratio across both layers of a membrane will be the same
Following the examples of Drugs A and B, we can see their equilibrium concentrations from the duodenum to the blood
|
KD |
Duodenum Concentration (water phase) |
Membrane (lipid phase) |
Blood Concentration (water phase) |
Action Into Membrane |
|
Drug A = 10 |
1 |
10 |
1 |
Moves in quickly Stays in |
|
Drug B = 0.1 |
10 |
1 |
10 |
Moves in slowly Rushes out |
An ideal drug is on that is lipophilic enough to get into the cell membrane and hydrophilic enough to get out
Fortunately, there are determinants other than KD that direct molecules into the blood…
Protein Affinity à molecules are bound to blood proteins (e.g., albumin), and are unable to cross the membrane
In general, á lipophilicity results in á protein binding
Donan Effect à reduced number of available (diffusible) molecules on one side of a membrane (due to protein-bound molecules) creates a vacuum of sorts on that side, pulling more across the membrane to re-establish equilibrium
Protein-Binding creates a reservoir of potentially available drugs for tissue distribution
It does NOT inactivate the drug (as many people incorrectly think)
As non-bound molecules are used up, more molecules are released from protein to maintain equilibrium
Ionizability à drug molecules are ionized under specific conditions (remember, only non-ionized molecules can cross lipid membranes)
pH greatly determines a molecules potential to ionize
Acidic molecules do not ionize in acid (most antibiotics are acidic)
For these, the greater the pH (more alkaline, more basic), the greater tendency to ionize
*An acid antibiotic will remain intact/non-ionized/undissolved in the stomach where pH = 2
*In the duodenum where pH = 6 (only slightly acidic), the proportions of ionized and non-ionized are approximately equal
*In the blood, pH = 7.4 (slightly alkaline) which induces two things:
1à a large portion of lipophilic, non-ionized molecules are ionized upon entering the blood
2à a Donan Effect is set up (reduced non-ionized molecules in the blood compared to the duodenum pulls more non-ionized molecules into the blood)
Ionizability, similar to protein-binding, represents a reservoir of potentially available drug for tissue
Note: Drug is absorbed only from a short segment of the duodenum because of the pH. The pH of the gut gets more alkaline which increases the amount of ionization which in turn decreases membrane transport from the gut.
Distribution to tissues depends largely on pH, as most tissues have a different pH range.
It also depends on blood flowà poor flow = poor distribution
Excretion depends on the same variables.
Protein-bound molecules will not be filtered out in the glomerulus, thus remaining in the blood
Only small, non-bound (meaning non-lipophilic and ionized) molecules are filtered out, into the tubule
àthis represents unused molecules that were not distributed during circulation through the blood
àthe protein-bound reservoir still remains, although the bound amount will â as equilibrium is re-established
From the tubule, the drug may be excreted or resorbed (recycled)
pH of the urine determines the amount of drug that is recycled
àDiets high in acidifying foods (oranges, vitamin C, etc) or high in carbohydrates will lower the pH of the urine and prevent/reverse ionization of antibiotics in the tubule and thus allow for resorption/recycling of the filtered drug
àDiets high in protein or in alkalizing agents will raise the pH of the urine and enhance ionization of antibiotics in the tubule and prevent resorption (more excretion of drug and less recycling)
Penicillins are actively removed from the blood by transport enzymes
When the enzymes are transporting penicillin they are not transporting the normal intended molecules
For penicillin, probenecid competes for the active site on the enzyme
àthis
reduces the amount of penicillin excreted in this manner
Some drug is excreted through the feces due to higher pH (8 or 9) in the lower gut
Weight in grams (g or gm)
Volume in liters (l or L)
|
giga (G) |
109 |
= billion |
|
mega (M) |
106 |
= million |
|
kilo (k) |
103 |
= thousand |
|
deci (d) |
10-1 |
= tenth |
|
centi (c) |
10-2 |
= hundredth |
|
milli (m) |
10-3 |
= thousandth |
|
micro (m or mc) |
10-6 |
= millionth |
|
nano (n) |
10-9 |
= billionth |
|
pico (p) |
10-12 |
= trillionth |
Rules for conversions
If the unit changes to a larger unit (milli to deci) the number gets smaller (100 mm = 1 dm)
If the unit changes to a smaller unit (milli to nano) the number gets larger (1 mm = 1,000,000 nm)
1 kg = 2.2 lbs
1 lb = 0.454 kg
1 L = 1.06 qt
1 gal = 4 qt
1 pint = ½ qt
1 mL = 1 cm3 = 1 cc
1 mL of water = 1 g (1 L = 1 kg, 1 pint = 1 lb)
Concentration Units
% à a weight to volume (w/v) ratio
defined relative to density of pure water à 100% = 1 g/mL
thus, 40% = 40 g / 100 mL à 0.4 g/mL or 400 mg/mL
may also be written as 1:100 which equals 1%
1:100,000 = 1g/105mL, which is the same as 10-5g/1mL, which is the same as 10mg/mL or 0.001% or 1mg%
Dose
Response Curves and Population-Response Curves
Some definitions:
Dose à total concentration of drug present in the body, or amount administered to patient
Association constant à or drug-receptor affinity constant or equilibrium binding constant, Keq
Higher Keq gives stronger receptor-binding, lower Keq gives less receptor-binding
Dissociation constant à Kd = 1/ Keq. Kd is the value of [D] at which 50% of the receptors are occupied by drug
Receptor-occupation curve à shows the percent of receptors occupied as a function of the amount of drug present
Dose-response curve à shows the pharmacological response of a subject as a function of the amount of drug present
A pharmacologic response is directly proportional to the number of receptors that are bound to/occupied to/complexed with the drug molecules
ED50 à the dose that produces a half-maximal response and equals Kd for the receptor mechanism
At low drug concentrations, fewer receptor sites are occupied, thus a decreased response
At higher drug concentrations, more receptor sites are occupied, thus an increased response
There is a maximum saturation point of receptors above which increasing the concentration has no increased effect
![]() Binding
is reversible and described by the equation
Binding
is reversible and described by the equation
D = drug molecule, R = receptor, and DR = drug-receptor complex
The affinity for the drug to bind to the receptor is indicated by the drug-receptor affinity constant, Keq (equilibrium constant)
A higher Keq indicates that D binds very strongly to R (more DR)
A lower Keq indicates that D does not bind very well to R (less DR)
Keq is described by the following equation:
![]()
It is always true that the total number of receptors in the body, [R]total = [R] + [DR], as some receptors are bound and others are not.
The values for [DR] and [R] may vary depending on the amount of drug present, but [R]total remains constant
The relationship between [DR] and [D] of a given drug will indicate how the pharmacological response relates to the drug dose.
Manipulating the above equations will allow for the formation of the following equation:
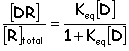
from which a receptor-occupation curve and a dose-response curve is plotted (see handout for actual plots)
At low [D], receptor occupation goes to zero
At high [D], receptor occupation saturates at 100%
When [D] = 1/Keq, then half of the receptors are bound
Strong binding (high Keq) reaches 50% occupancy at lower [D] (less of a dose)
1/ Keq is also referred to as the dissociation constant, Kd
50% saturation, and 50% dissociation is achieved when [D] = Kd
The effective dose for 50% of maximal response, or ED50 is the value of [D] that causes 50% saturation
More definitions:
Potency à relative measure of the dose of a drug required to produce a given response, and is inversely proportional to the dose
The higher the required dose, the less potent the drug
The lower the required dose, the more potent the drug
Measured on the x-axis of the dose-response curve (log[D] values)
á Kd results in â potency
increasing Kd shifts the curve to the right on the x-axis
this indicates that more drug is needed to achieve half-maximal response
since more drug is needed, it is less potent
Efficacy à relative measure of the response produced when a drug occupies a receptor
Also called the intrinsic activity of the drug à does it do what it is supposed to do?
Measures maximal response when all receptors are occupied (ceiling of activity)
Measured on the y-axis of the dose-response curve
A “higher” curve along the y-axis indicates a greater maximal response and thus a greater efficacy
Population-Response Curves
The susceptibility of a patient to a drug varies among individuals (varying Kd, intrinsic activity, etc.)
Because of biological variation, a population displays a log-normal distribution of drug sensitivities
These curves are quantal, in that they represent an all-or-none response (whereas the dose-response curves are graded)
Median effective dose, ED50, is that dose required to produce the specific effect (unconsciousness) in half the population
Other doses, ED10 and ED90, represent the same response for different percentages of the population
The closer these values are to the ED50, the less variation there is in response of the population to the drug (á predictability)
ED50 is also called the median effective therapeutic dose
Another quantal effect is death and is measured as the median lethal dose, or LD50
Drug safety is determined by comparing LD50 to ED50
The higher the LD50 is compared to ED50, the safer the drug is…
The ratio LD50/ ED50 is called the therapeutic index (TI)
A higher TI is preferred à generally around 10
Drugs with lower TI (general anesthetics are around 2) require special monitoring procedures
In some instances ED90 will overlap with LD1, and while 90% would lose consciousness, 1% would die
The certain safety factor (CSF) is the ratio LD1/ ED99 describes this overlap region
It is desirable to have a CSF > 1 which indicates little or no overlap and suggests the drug is safe
More definitions:
Graded-response curve à response continually changes as the dose changes
Also known as
a dose-response curve
Quantal-response curve à change in the dosage within a population to elicit the same response
Also known as a population-response curve
ED50 à mean effective dose; dose required to produce a given effect in 50% of the population
Can also have ED10 and ED50
LD50 à median lethal dose; dose of the drug that will kill 50% of the population
Therapeutic Index àTI; LD50/ED50
Certain Safety Factor à CSF; LD1/ED99
Effect of pH
on Drug Kinetics
Strong acids (proton donors) are always fully ionized
Weak acids and weak bases are only partially ionized
The following reactions describe the behavior of acids and bases
![]()
![]()
When considering the available form of a drug it is the non-ionized (or free) form
AH is the free form of the acid
B is the free form of the base
These are also known as the permeant forms (able to cross the lipid membrane)
An á in pH (less H+ in the solution) causes a shift to the right for both equations à these are basic solutions
The effect is more free (available) base, and less free (available) acid
A â in pH (more H+ in the solution) causes a shift to the left for both equations à these are acidic solutions
The effect is less free (available) base, and more free (available) acid
Ka is the dissociation constant for each acid and base
Measures how easily a molecule can donate protons (acids) or accept protons (bases) à become ionized
pKa is the log value of Ka
the lower the pKa of an acid, the more it is ionized (unavailable)
the higher the pKa of a base, the more it is ionized (unavailable)
Henderson-Hasselbalch Equations
These are extremely important in pharmacology
![]() à
they allow a calculation of how much of the drug is permeant (non-ionized) and
how much is non-permeant (ionized), as long as the pKa and pH are
known
à
they allow a calculation of how much of the drug is permeant (non-ionized) and
how much is non-permeant (ionized), as long as the pKa and pH are
known
For acids:
![]() For
bases:
For
bases:
A more useful quantity is the fraction/percent of total drug that is in one form or the other
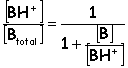
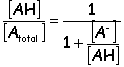 Acid: Base:
Acid: Base:
For a weak acid…
If pH > pKa, then the ionic (non-permeant) form dominates
If pH < pKa, then the neutral (permeant) form dominates
If pH = pKa, then the forms are in equilibrium
Remember, for the permeant form of an acid, the pH must be lower than the pKa (more acidic)
For a weak base…
If pH < pKa, then the ionic (non-permeant) form dominates
If pH > pKa, then the neutral (permeant) form dominates
If pH = pKa, then the forms are in equilibrium
Remember, for the permeant form of a base, the pH must be higher than the pKa (more basic)
It does not matter what the actual values are, but only the difference between pH and pKa
Keys to solving problems:
1) know whether the drug is an acid or a base
2) know the pKa of the drug
3) know the pH of the body fluid where the drug is located
Equilibration of drugs across membranes follows this ruleà drugs accumulate where they are more ionized (non-permeant)
Weak acids accumulate where it is more alkaline
Weak bases accumulate where it is more acidic
This is purely a passive processà no energy required (driven only by the pH gradient)
Kinetics
of Drug Accumulation and Disappearance
Elimination of a drug is due to redistribution, metabolism, and excretion, and follows either zero-order kinetics or first-order kinetics
Zero-order à drug is eliminated at a constant amount over time
C(t) = C0 – k0t represents zero-order kinetics
C(t) is the concentration at any time, t
C0 is the peak concentration
k0 is the rate constant
t is the amount of time since administration of the drug
Alcohol is an example of a drug that is eliminated at a constant rate
First-order à drug is eliminated at a constant fraction over time (exponential decay)
C(t) = C0e-kt
represents first-order kinetics
C(t) is the concentration at any time, t
C0 is the peak concentration
k is the first order rate constant
t is the amount of time since administration of the drug
The time at which the drug concentration is one-half of its initial value is known as the biologic or plasma half-life (t1/2)
*Each drug has a unique t1/2
*Pharmacologic half-life is similar and refers to the time required for one-half of the pharmacologic response to decay
For reversible-receptor mechanisms, pharmacologic t1/2 = plasma t1/2
*Method of drug administration affects the magnitude of C0, but does not affect t1/2
*t1/2 is independent of the initial concentration
*The first-order rate constant, k, can be derived from the exponential decay equation…
k = 0.69/t1/2
Multi-dosing and Drug Accumulation
Often a dose is administered repeatedly at equal time intervals (td is the dosing interval)
The drug accumulates in the plasma as the concentration rises further above C0 with each dose
The concentration will eventually reach a maximal value (does not increase with additional doses)
As C
increases so does the amount eliminated during td (first-order
kinetics)
Eventually, the amount eliminated during td is equal to the single dose (no further accumulation)
This is known as the maintenance or plateau state
The concentration
will have the same maximum and same minimum as the doses continue
Dose = Cmax(1-e-ktd)
If any three of the four variables are known, the fourth can be found
A special case exists when td = t1/2
Dose = ½Cmax
Time to
The Plateau Principle shows that the accumulation half-time is equal to the elimination half-time
The amount of time to reach the plateau for therapeutic purposes is generally considered to be 4 half-lives
Loading Dose
An initial dose (loading or priming) which is larger than the maintenance dose in order to achieve plateau state more quickly
Only indicated when the TI is high (large margin of safety)
Example: Suppose tetracycline is to be loaded in two equal doses with td = 6 hours. What are the loading and maintenance doses, if a peak maintenance level of 500 mg is desired?
In this case
td = t1/2 = 6 hours, therefore the maintenance dose = ½Cmax
or ½(500) = 250 mg
At the initial dose, the concentration is CL
After td, the concentration is ½ CL, at which time the second loading dose is applied for a total of 3/2 CL (½CL + 1 CL)
The purpose of loading doses is to reach Cmax quickly, so then here 3/2CL = 500 mg
Solving for CL gives a loading dose of 333 mg
COPD à chronic obstructive pulmonary disease
Includes chronic obstructive bronchitis and emphysema
Characterized by airflow limitation that is not fully reversible
Symptoms include
shortness of breath, cough, wheezing, and increased sputum production
Anti-Parkinson’s
Disease Drugs