|
Tandem repeats finder : a program to analyze DNA sequences.
This
program has detection and analysis components employed a set of
statistically based criteria to find candidate tandem repeats and produced
an alignment for each candidate.
Software can run
on website
(http://tandem.bu.edu) or download to
your computer.
FASTA file
Compatibility
|
|
Software was downloaded from
http://tandem.bu.edu website and installed on MS-Windows.
|
| |
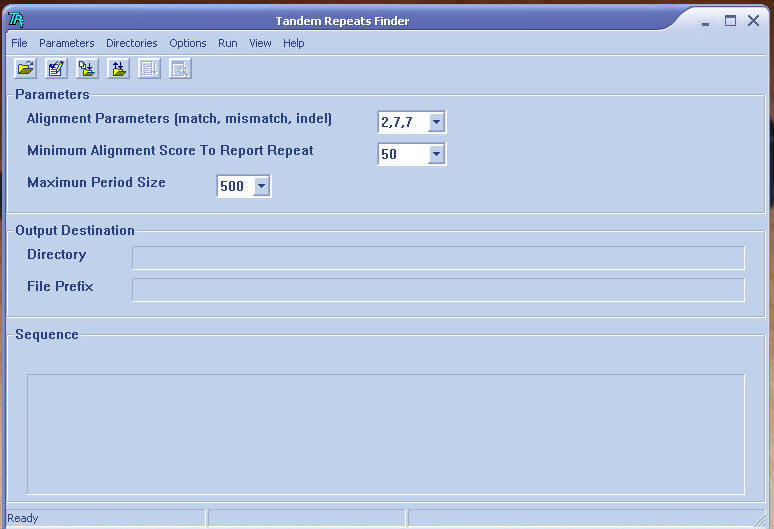
Skin of software
Input parameters consist of
Alignment weight for match, mismatch
and indel (1) default set at 2, 7, 7
Minimum alignment score to report
(2) default set at 50
Maximum period size (3) default set
at 500
|
| |
|
Sequence analysis
|
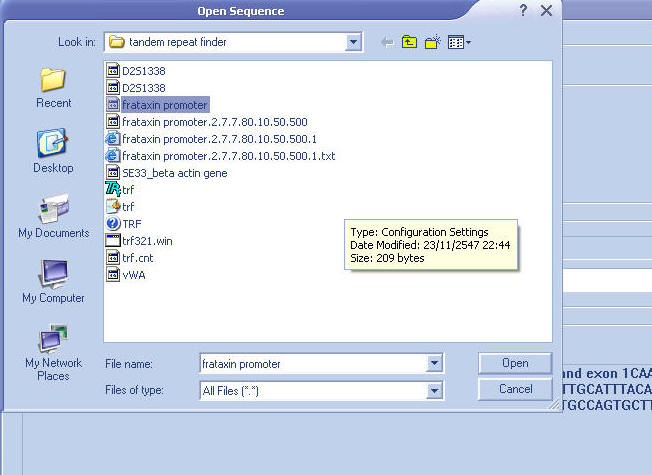
Open sequence (FASTA file only)
: frataxin promoter
|
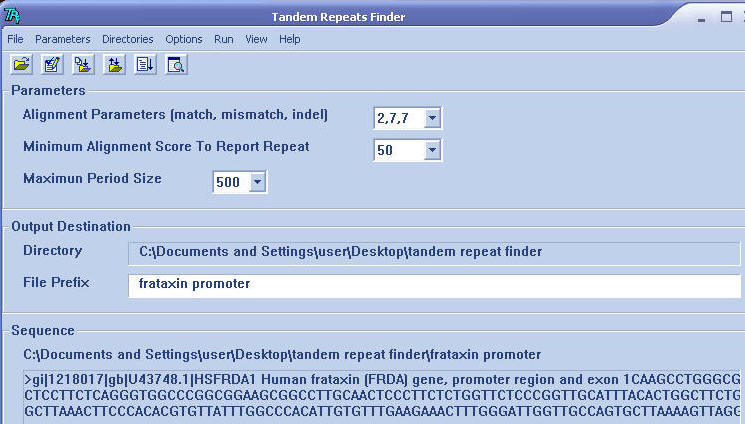
-
Output Destination window shown
directory and file suffix for report files. File output destination can be
changed by select directory menu bar and select any destination that you
want.
-
Sequence window shown file location
of analyzed sequence and nucleotide sequences of frataxin promoter (FASTA
format)
|
| |
Tandem repeat finding was
started after click run subsequent start search.
|
| |
Current Analyzed sequence was
found at view menu bar.
|
| |
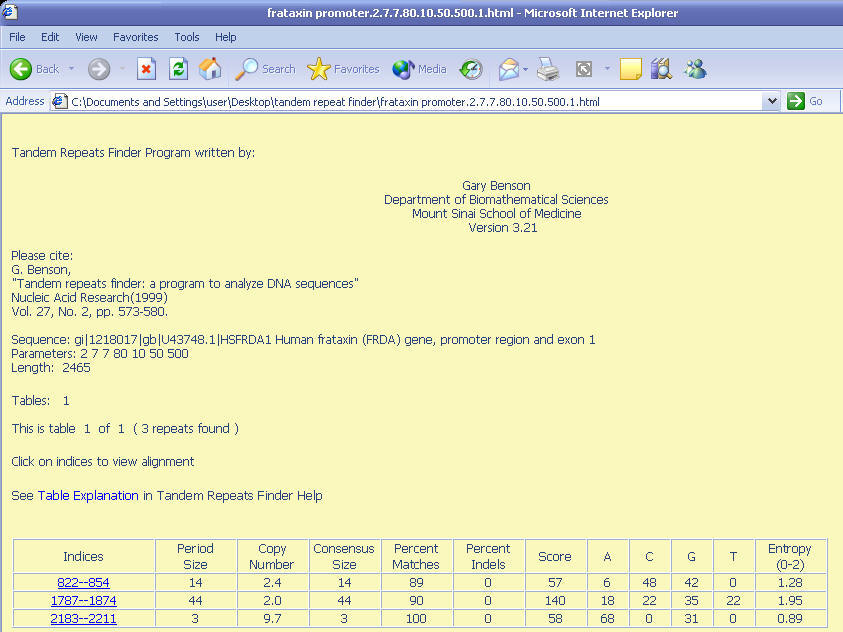
The first result that was shown in
HTML format is a summary table describing the location and statistical
properties of tandem repeat found. It described three repeats were found in
frataxin promoter and the table described
The repeat table file includes the following information:
1. Indices of the repeat relative to the start of the sequence. Each index
can click to see explanation (HTML format).
2. Period size of the repeat.
3. Number of copies aligned with the consensus pattern.
4. Size of consensus pattern (may differ from the period size).
5. Percent of matches between adjacent copies overall.
6. Percent of indels between adjacent copies overall.
7. Alignment score.
8. Percent composition for each of the four nucleotides.
9. Entropy measure based on percent composition.
|
 |
|
Figure A
|

Figure B |
| |
|
Figure A anb B are the result of the
first index (822-854) which were shown in the same window. They described
1. In each pair of lines, the actual sequence is on the top and a consensus
sequence for all the copies is on the bottom.
2. Each pair of lines is one period except for very small patterns.
3. The 10 sequence characters before and after the repeat are also shown.
4. Symbol * indicates a mismatch.
5. Symbol - indicates an insertion or deletion.
6. Statistics refers to the matches, mismatches and indels overall between
adjacent copies in the sequence, not between the sequence and the consensus
pattern.
7. Distances between matching characters at corresponding positions are
listed as distance, number at that distance, and percentage of all matches.
8. ACGT count is percentage of each nucleotide in the repeat sequence.
9. The consensus sequence.
10. If chosen as an option, 500 characters of flanking sequence on each side
of the repeat are shown.
Each of remained result are described as the first index. |
| |
|
|
| |
|
Patiya Pengon 4737611 SCFS/M
Forensic Science Grad. Prog.,
Science; Mahidol University
[email protected]
[email protected] |
