Go to ECOMAMA syllabus home page
Seston is the term used for all particulate matter which is in suspension in the water.
So seston is composed of all particles > 1 µm (= particulate) which cannot maintain their position against a current (= in suspension). It includes all kinds of particles and living organisms from bacteria to zooplankton.
In practice, bacteria and zooplankton are not often considered in seston studies, which concentrate mainly on phytoplankton, detritus and inorganic particles (e.g. sand).
A general quantification of the composition of a seston sample can be obtained from:
| Analysis | Quantifies | |
| - Dry weight | Total particulate matter (organic + inorganic) | |
| - P.O.C. | Particulate Organic Carbon: a measure of the amount of organic matter (living phytoplankton + detritus) | |
| - Chlorophyll | Living phytoplankton |
Water samples (usually 5-20 l) are taken with a water sampler (e.g. Niskin bottle) at the desired depth.
Division in sub-samples is done fractionally (divide a number of small volumes over each of the sub-samples).
For microscopical counting and analysis, samples are preserved with lugol's solution.
50 g I2
100 g KI in 1l distilled water
100 g acetic acid (CH3COOH)
The lugol's solution should be kept in a dark, well closed, glass bottle.
Samples of 0,25-1 l are put into glass bottles. Add 2-3 ml of lugol's solution. Keep the bottles closed and in the dark (preferably in the fridge) for at least 2-3 days.
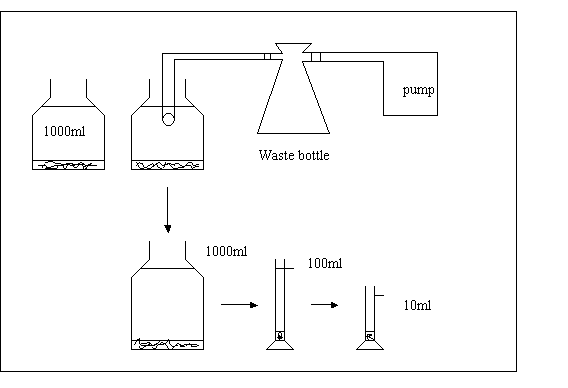
After this time, the seston is sedimented to the bottom. The supernatant can be removed by suction: under vacuum (see figure II.1.1.).
Preferably use a plastic tube that is not being used for other purposes, as lugol's solution is absorbed by plastic. Hold the tube end just below the water surface, and reduce the volume to somewhat less than 100 ml.
Shake the bottle to free the seston from the bottom, and pour the content into a 100 ml cylinder and adjust the volume to 100 ml with tap water. The cylinder is covered with aluminum foil, and kept standing (in the fridge) for minimum two days, to allow settling. Then, the volume can be reduced to 10 ml by the same procedure as described above.
These samples can be kept in closed glass containers (preferably dark) and stored in the dark (preferably in the fridge). If the samples are stored for a long period (months), formaline should be added (1 ml of 40 % formaline per 10 ml of sample). For shorter periods, addition of some extra lugol's solution should be done every two weeks, so that the color is kept dark brown.
- Reversed microscope:
Bring a few ml's of the concentrated sample into the counting chamber (depending on the type of counting chamber used). Allow to sediment for + 15 min. before analysis.
- Normal microscope:
Bring a drop of the concentrated 10 ml sample on a glass microscope slide. Cover with a cover glass.
Put the slide on the microscope table under the clamps.
Figure II.1.1. Procedure for concentrating seston samples
- Analysis: (both type of microscopes)
Use the smallest magnification (shortest objective) to get an overview of the sample.
Focus with the control knob. When the slide's contents are focused under low magnification, switch to a higher magnification.
Only use the fine adjustment knob when focusing under high power magnification.
For a good recognition of the phytoplankton cells, an objective 40 x should be used.
- Qualitative observations
In a first phase, seston analysis can be limited to the identification of the dominant phytoplankton types and the determination of the proportion of phytoplankton cells to other particles.
This is done by looking at a certain number of viewing fields (e.g. 20).
In each viewing field, the number of phytoplankton cells and the number of other particles (mostly detritus) are counted. From the total counts of all the fields, the proportion phytoplankton - detritus is calculated.
- Quantitative observation
* Counts (concentration in sample), using a reversed microscope
At a given magnification Mi a number of viewing field (Nv) are analyzed.
In each viewing field, the number of particles of interest (n) are counted.
Calculations: N = [ (n.x) / Nv ] . ( 1 / v2 ) . ( 1 / cf )
With N: concentration of particles in the original sample (particles/ml)
x: nr. of viewing fields in the cuvette at magnification
Mi = [ surface cuvette (Sc) ] / [ surface viewing field (Sv) ]
v2 volume of concentrated sample in the cuvette (ml)
n nr of particles counted in Nv viewing fields
Nv nr of viewing fields counted
cf concentration factor
to determine x:
* Measure surface of viewing field (Sv)
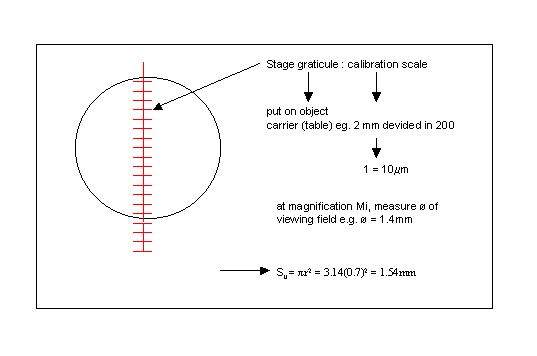
* measure dimensions of cuvette (Sc)
e.g.
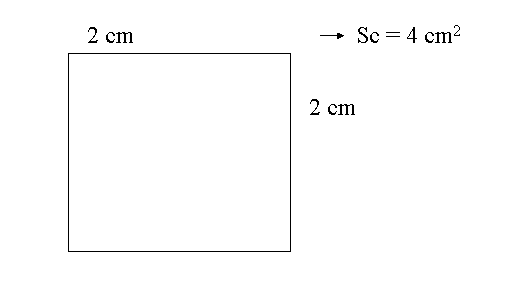
==> x = 400 / 1.54 = 260
Remark: cf, v2: can be adjusted to concentration of particles of interest in original sample.
Sometimes different combinations necessary for different types of particles in one sample.
Magnification: M increased as particle size decreased
Nv: in principle, increase till n ~ ct
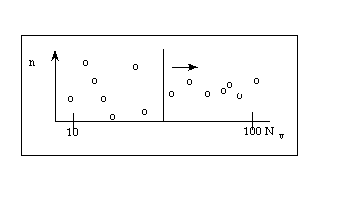
in practice n ~ 100 (at least > 20) (depends on "feasibility")
* Measuring the size of particles
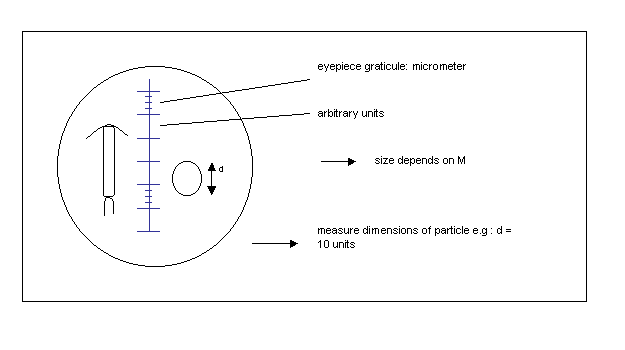
calibration:
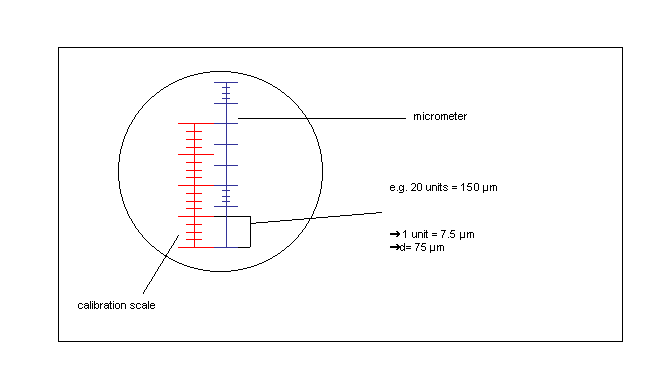
e.g. Coulter Counter, Elzone
Electronical particle counters count particles and divide them in a number of size classes (channels) according to their spheric equivalent diameter (S.E.D.).
The particle concentration can be plotted as volume distribution, by multiplying the concentrations (in number of particles ml-1) by the spheric equivalent volume (S.E.V.) of each size class (e.g. fig. II.1.2). For details see specific instructions of the apparatus at hand.
Figure II.1.2.
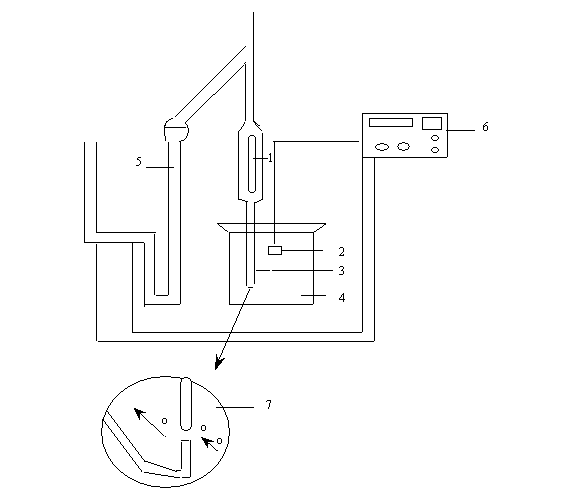
* Before analysis, a count is made (see below) with filtered seawater to check the background (electrical noise).
* A beaker (4) with + 200 ml of the sample to be analyzed is put under the measuring tube.
The aperture tube and electrode (1,2) are emerged in a beaker with + 200 ml of the sample to be analyzed (4). The aperture tube (opening of the orifice) (3), is chosen in function of the largest particles present in the samples (to be determined experimentally). Sometimes multiple tube analysis is necessary to cover the complete range of particle sizes. The volume of water sucked through the orifice can be adapted to each type of tube by the manometer (5). after each count, the results are either printed by the coulter-population count accessory (PCA) unit (6)., or stored in the computer for further data processing.
The amount of total particulate matter present in a sample can be quantified as dry weight.
Glass-fiber filters (Whatman GF/C 0.47 mm) are numbered and predryed at 60 °C during > 6 hours. The blank weight of each filter is determined on a electrobalance (see specifications for each model) after the filter has cooled for a few minutes in a dessiator.
Samples are filtered onto a glass-fiber filter (Whatman GF/C 0.47 mm) (keep suction low < 10 cm Hq vacuum), and dried at 60° C for 6 hours. Usually, 1 l samples are filtered, but the volume can be adjusted to the seston concentration at hand. As a rule of thumb, material should be clearly visible on the filter, but the filter should not clog (filtration must be fluent).
Dried filters are cooled to room temperature in a descicator and weighed on a micro-balans
(DW sample - DW blank) x 1000 = g/l if the reading of
the micro-balans is in g
volume of sample filtered (ml)
* The amount of organic material (phytoplankton + detritus) in a seston sample.
Sea water is filtered on a GF/C-filter (47 mm of diameter) under low suction (see II.1.2.).
Take care not to touch the filters with your hands when handling them: always use a pincette !
Standard, 1 l of sea water is filtered. Sample size can of course be adjusted to the concentration of seston in the samples.
For storage, filters can be deep-frozen after packing them in a clean special filter box or in aluminium foil.
The P.O.C. can be measured by:
a) a carbon analyzer
b) the wet oxidation method
As a C-analyzer is not always available, only the principle of this method is given. The wet oxidation method is described completely.
The function of this instrument is based on an automatic coulometric titration. Carbon is transformed to carbon dioxide (CO2), which is absorbed in an alkaline barium perchlorate (Ba(ClO4)2) solution. This causes a decrease of pH of the solution. With barium hydroxide (Ba(OH)2) formed by electrolysis, an automatic back titration to the initial pH takes place. A built-in electronic unit transforms the measured amount of electricity used for the back-titration.into counts displayed, of which 1 count represents 2 x 10-7 g C.
- Total Particulate Carbon (T.P.C.): all of the carbon present is oxidized to CO2 in an oxygen stream while the filter is heated to 900° C in a quartz tube.
- Particulate Inorganic Carbon (P.I.C.): all of the inorganic carbon present is converted to CO2 in an acid solution (8.5 % orthophosphoric acid (ortho-H3PO4)).
- Particulate Organic Carbon (P.O.C.)
P.O.C. = T.P.C. - P.I.C.
Carbon is determined by "wet ashing" with a mixture of potassium dichromate (K2Cr2O7) and concentrated sulphuric acid (H2SO4). The decrease in extinction of the yellow dichromate solution after it has been reduced by the organic matter is measured.
2K2Cr2O7 + 3C + 8H2SO4 , 2K2SO4 + 2Cr2(SO4)3 + 3CO2 + 8H2O
The following text describes the practical handlings for performing this method.
This method uses strong acids. Be careful !! Wear a lab-jacket !
C is oxidized by acid dichromate
2K2Cr2O7 + 3C + 8H2SO4 ==> 2K2SO4 + 2Cr2(SO4)3 + 3CO2 + 8H2O
The reaction is quantified by measuring the decrease in adsorption of the K2Cr2O7 solution spectrophotometrically.
- GF/C filters (47 mm of diameter), filtration apparatus
- an oven to reach 100-110°C
- a spectrophotometer
- Glassware: ideally stoppered graduated measuring cylinders.
50 ml Pyrex glass beakers fitted with cover-glasses.
Cleaning of the glassware: the beakers should be cleaned in hot chromic-sulphuric acid cleaning mixture and stored in a desiccator or another container where dust contamination is impossible. It is also possible to just clean the beakers thoroughly with alkaline soap and hot water, rinsing them thoroughly with aq. dest, rinse with diluted nitric acid (HNO3) and rinse them again with aq. dest.
Dissolve 4.84 g of potassium dichromate K2Cr2O7 in 20 ml of distilled water.
Add this solution little by little to about 500 ml of concentrated analytical quality sulphuric acid (H2SO4 p.a.) in a 1000 ml volumetric flask. Cool the mixture to room temperature and make to volume with more concentrated acid. Store the solution in a glass stoppered bottle. This solution stays stable indefinitely.
Dissolve 45 g of best quality anhydrous sodium sulfate Na2SO4 in 1000 ml aq. Dest
* filter a convenient volume of sea water or algal culture in triplate on a GF/C-filter under low suction, allow the filter to be sucked dry.
* add 2.0 ml of the sodium sulphate solution, and immediately suck the filter dry; repeat this once more.
*fold the filter double in aluminium foil, put in fridge or deep-freeze while perparing calibration and measurements reagents (if you're not performing the analysis immediatelly)
* proceed together with calibration series, see next page
- prepare a standard glucose solution: dissolve 7.50 g of pure glucose (C6H12O6) in 100 ml aq. dest. Keep this solution in the refrigerator.
If you plan to store longtime, add a few crystals of mercury (II) chloride (HgCl2).
- diluted glucose solution for calibration curve:
dilute 10.0 ml of the glucose stock solution to 1000 ml with aq. dest.
Make a new diluted solution daily.
* take 13 GF/C filters
* take 17 beakers of 50 ml: 3 blanks, 4 samples + 5 x 2 for 5 different concentrations of glucose solution.
* in 13 of them: put a filter moistured with 1 ml phosphoric acid, cover with Al. foil.
* in the 4 remaining beakers: put the filters with your samples and moister with 1 ml phosphoric acid
* heat the beakers for 30 minutes in the oven at 100-110° C
* to three of the beakers: add 4.0 ml aq. dest and 10.0 ml of the sulphuric acid-dichromate oxidant = blanks
* to 5 x 2 beakers: add 10 ml oxidant solution and an increasing number of mls of the diluted glucose solution (e.g. 0.5-1-2-3-4 ml), if necessary adjusting to 14 ml with aq. dest.
* replace the Al. foil on the beakers and reheat them all for 60 minutes in an oven at 100-110° C
* let the mixture cool to room temperature
* transfer the mixture with filter pulp to a volumetric flask of 50 ml, washing the sides of the beaker thoroughly with aq. dest
* bring to volume with aq. dest and mix
* allow most of the filter pulp to settle
* decant 10 ml into a centrifugation tube
* centrifuge for about 5 minutes at 2000 rpm
* adjust the wavelength of the spectrometer to 440 nm.
* zero the spectrometer with a cuvette filled with aq. dest
* measure the extinction of each blank solution
* measure the extinction of the calibration samples and the phytoplankton samples.
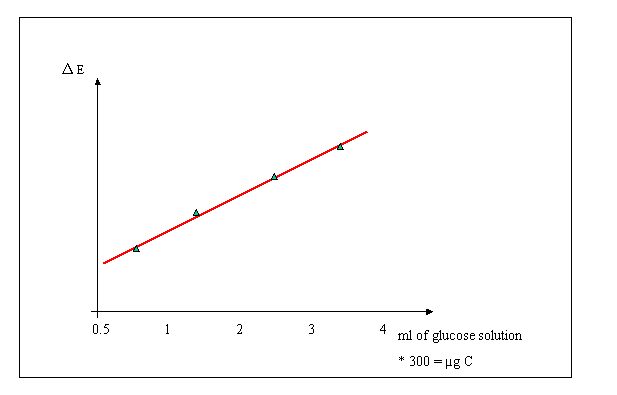
?E: mean extinction of blanks mean - extinction of calibration samples
The molecular weight of glucose (C6H12O6) is 180, of which 40 % is carbon weight.
The glucose stock solution (being 7.50 g in 100 ml) contains 40 % or 3.00 g carbon/100 ml.
The stock solution is diluted 100 times (10 ml stock diluted to 1000 ml). The diluted glucose solution contains
3 g/100 ml = 30 mg C/100 ml = .3 mg/ml = 300 µg/ml
100
Thus: 1 ml diluted glucose solution corresponds to 300 µg of carbon
Calculate, from the extinction values measured in the calibration, the linear regression between extinction and amount of carbon.
E = aC + b
Whereout: C = (E - b) / a µg carbon in the solution
Thus: C = (E - b) / aV µg C/l for V liter of sea water filtered
The only rapid chemical method known for estimating living plant matter in the particulate organic matter of sea water is to determine the characteristic plant pigments: the chlorophyll, carotenes and anthophylls.
The methods described determine the three chlorophylls commonly found in planktonic algae: chlorophyll a, b and c.
A. Spectrophotometric quantification of chlorophyll pigments
In the standard procedure, 1 l of sea water is filtered on a GF/C-filter under low suction. Sample size can of course be adjusted to the phytoplankton concentration in the sample.
If immediate measurement is impossible, the filters can be stored in a deep-freezer for some weeks after packing them in a clean special filter box or aluminium foil.
- GF/C-filters, filtration apparatus
- centrifuge tubes of 15 ml capacity, with glass or polyethylene stoppers
- centrifuge
- spectrophotometer
90 % acetone (CH3COCH3) in aq. dest. The acetone is especially purified for chromatographic and spectrophotometric purposes.
The filters are brought into 10 ml of 90 % acetone in a centrifuge tube and immediately placed in the fridge, as chlorophyll is very unstable in light.
During a period of at least 1 hour and maximum 24 hours in the refrigerator, the chlorophyll pigments are extracted out of the phytoplankton by the acetone.
Then the samples are centrifuged for 10 minutes at 3000 rpm. (use of the centrifuge: see specifications of the model at hand).
If not measured immediately, the samples are put back in the fridge or at least in the dark until measurement in the spectrophotometer.
The basis of quantification is the difference in extinction of light between the pigments and a blank at 665, 645 and 630 nm.
- Fill two cuvettes with 90 % aceton (blanks)
- Adjust the wavelength to 750.
- First zero the spectrometer by putting the blank cuvette in position.
- Measure the extinction after 2nd blank (correction for possible differences in optical properties of cuvettes).
- Measurement: fill the second cuvette with the supernatant of the centrifuged sample (be careful to avoid sturring up of filterparticles)
- Measure the extinction
- Switch to 665 nm wavelength, repeat zerowing and measurment
- Repeat at 645 and 630 nm
Calculation
The chlorophyll values are estimated following the Parsons-Strickland formulas.
- Substract reading at 750 nm from all other readings.
units : µg/ml if a 1 cm cuvette is used
C (Chl a) = 11.6 E665 - 1.31 E645 - 0.14 E630
C (Chl b) = 20.7 E645 - 4.42 E630 - 4.34 E665
C (Chl c) = 55 E630 - 4.64 E665 - 16.3 E645
The results obtained from these formulas are multiplied by a factor f, in order to obtain the chlorophyll concentrations in mg/m3
f = v / (l x V)
where: l = length of cuvette (in cm) this means the length of the path through which the light passes the cuvette
v = ml of acetone used for extraction
V = volume of sea water filtered (in l)
B. Measurement with H.P.L.C. (High Pressure Liquid Chromatograph (demonstration)
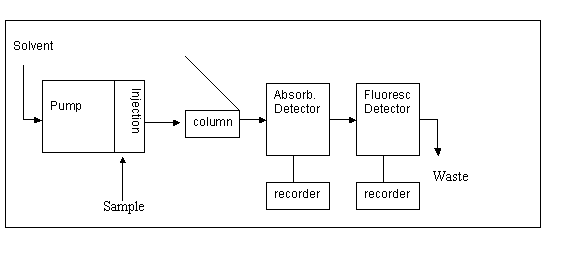
Scheme of the measurement unit:
Calculation of the Chlorophyll contents are based on standard curves.
The ratio P.O.C./Chl a gives information about the phytoplankton part in the organic matter.
B. Primary production/Respiration
The phytoplankton population growth depends on growth coefficient (b) and biomass (B)
DB / dt = b x B mg / m3dt
The growth coefficient is determined by
k = photosynthesis rate
r = respiration rate
e = excretion rate
m = mortality rate
p = predation rate
Therefore:
dB/dt = (k-r-e-m-p) B = nett production.
Brute production = nett production + respiration + excretion + mortality + predation
Thus: population increase if: k > r+m+e+p
population decrease if: k < r+m+e+p
In lab experiments with cultures, such as carried out here, 'r' is the most important loss-factor
(r >> m + e + p)
| hv | |||
| 6 CO2 + 6 H2O | <==> | C6H12O6 + 6O2 |
- to an increase in the O2 concentration in the medium
- to an incorporation of inorganic carbon into the cells and a formation of organic carbon in the cells
The photosynthesis rate can therefore be measured as
- an increase in O2 concentration in the medium (in a closed system) during a period of time unit (e.g. 1 hour). The O2 concentrations can be measured with the Winkler method.
- the amount of "newly formed organic matter", incorporated into the cells, during a period of time.
To distinguish existing and newly formed organic matter, a radioactive tracer HC14O3- is introduced in the medium (at t0) and the amount of C14 incorporated in the cells after a period of time is measured. C14 in the cells can be measured in a scintillator counter (as desintegration part per minute (DPM)) after filtration of the samples on a GFC filter.
It is hypothesized that the tracer is incorporated to the same relative extend as the natural compound.
We therefore determine, in the case of photosynthesis, the fraction of the tracer (HC14O3-) incorporated in the cells (filtered on a GFC filter) during a time period and we multiply this by the total amount of natural HCO3- (HC12O3-) in the medium (mg/m3). This gives the total amount of HCO3- that is incorporated per time unit and thus the rate of photosynthesis.
Excretion rate: e
Excretion(exudation) of organic matter leads to a loss of organic matter from the cells and to an addition of dissolved organic matter in the medium.
The excretion rate can be measured considering this "new" dissolved organic matter. The excretion rate is therefore measured as the increase in radioactive organic material (after elimination of the inorganic radioactive material (evaporation)) in the solution (filtrate) during a period of time.
A respiration rate can be measured as
- a decrease in oxygen in the medium (closed system) after a period of time. This gives a measure of the total sample respiration rate (including respiration by phytoplankton, bacteria and zooplankton).
- a loss of organic matter from the cells, (filtered on a GFC filter) during a period of time after pre - introducing HC14O3- in the medium for a period of time, to allow the cells to incorporate C14 before the experiment starts. This value also includes loss due to excretion. One can distinguish light (= photo-respiration) and dark respiration.
Primary production and respiration of algae (micro or macro) can be measured by determining the O2 produced and respired in closed bottles during different incubation. Due to the production or respiration of the organism studied, the O2 concentration in the water will change.
- Fill 4 bottles (50 ml) with seawater or culture (using a plastic tube) and fix immediately with MnSO4 and alkaline KI to determine O2 concentration at time o (to) (see I.4.1.Winkler titration).
- Fill 'x' 50 ml bottles with (seawater) culture ( 'x': to be determined by the number of samplings, x4 replicate; i.e. 4 x 4 botles for 4 samplings )
Incubate for different periods of time (f.i. 30, 60, 120, 180 minutes) in the light incubator (light bottles, LB) and in the dark (DB), at constant water temperature. Determine O2 concentration in all bottles. The changes in O2 concentrations in the light bottles are due to photosynthesis and respiration. The changes in O2 concentrations in the dark bottles are due to respiration.
When oxygen concentration is calculated (mg/l), primary production and respiration can be calculated as
Respiration (R, in mgO2l-1hr-1) = Fo - Dbi
With Fo = oxygen concentration at time zero
Dbi = oxygen concentration in the dark bottle
T = incubation time (hr)
Netto production (NP, in mgl-1hr-1) = LB - to
With LB = oxygen concentration in the light bottles at the end of the incubation
Brutto production (BP, in mgO2l-1hr-1) = NP + R = LB - DB
When working with phytoplankton, the cells are considerated to be homogeneously distributed over the incubation bottle and the volume sampled (eg some) is considerated as a measure of 'biomass'. Production and Respiration can thus be directly converted into 'in situ' rates (eg mgO2l-1hr-1)
When working with organisms whick are not homogeneously ditributed in the water (eg seaweeds, zooplankton) one measures the effect of the production and/or repiration of these organisms on the oxygen concentration of the water only. To calculate the production or respiration of the organisms, the amound of oxygen which has been produced (respired during the incubation) has to be calculated.
Eg.
Primary Production experiment with a piece of macroalgae with a wet weight of 3g, incubated in 100ml of water. Incubation time : 6 hours.
Fo = 7 mgl-1 + or - 1 mgl-1
LB = 10 mgl-1 or 1,5 mgl-1
Difference in oxigen concentration created by primary netto production of the leaf
= 10 -7 = 3 mgl-1
Amount of oxygen netto produced in 6 hours
= (3 mg x 100 ml) / 1000 ml = 0,3 mg/6hr
Netto production = 0.3 / 6 = 0,05 mgO2hr-1 by the piece of algue = 0.05 / 3 mgO2hr-1g wetweight-1
For incorporation in the C-cycle, production and respiration can be converted into mgCl-1hr-1 (cfr. II.5.1).
From the different O2 concentrations measured we can therefore calculate:
Respiration (R) = to - DB
Netto production (NP) = LB - to
Brutto production (BP) = NP + R = LB - DB
Wear plastic gloves at all times when working with radio-activity
Do not inhale scintillation liquid vapors
* The primary production can be measured by determining the amount of C14 incorporated in the cells as organic matter after addition of a known quantity of NaH14CO3. The following time series will show a linear uptake indicating that recalculation to hourly rates does not introduce errors.
The fraction of tracer incorporated into the cells per time unit (= DPM counted/DPM added x t) is thus determined in a time series.
By mulitplying this fraction with the total amount of dissolved inorganic C in natural marine waters (25 000 mg/m3) one obtains the total amount of C incorporated into the cells per time unit.
- Fill 2 bottles of + 400 ml with a phytoplankton culture or seawater, add 20 µCi of NaH14CO3 to each bottle (see remark about DPM added), shake well. Incubate 1 bottle under light condition (incubator) and 1 bottle in the dark, at constant temperature.
- Filter immediately (to) and after different time intervals (cfr. O2 method) 3 x 10 mls of samples from the light and dark bottles on glass-fiber (GF/C.45 µm) filters (ø 25 mm). Wash the filters with an exess of seawater.
Put filters (+ 1 blank: either a dry filter, or a filtered on which non radioactive culture has been filtered) in scintillation-bottle.Let the filter dry, and add a few drops of H2SO4 (10 %) let evaporate (to CO2). Add 10 ml of scintillation liquid. Mix thoroughly and put in the scintillation counter for analysis after at least 2 hours.
Use of scintillation counter: see instructions on apparatus at hand.
With a modern apparatus, counts per minute (CPM) are automatically corrected for noise and the readout gives desintegrations per minute (DPM).
- Take 3 x 10 ml samples out of each bottle at time intervals : 30, 60, 120, 180 minutes
DPM values of blank filters have to be subtracted from these.
PP = (DPM/10 ml x 25000) / (DPM added/10 ml x t) (mg C/m3/hr)
Where:
DPM: readout of the scintillation counter, corrected for blank value
t: incubation time
25000: concentration of dissolved carbon in sea water (mg m-3)
Remark: Be careful on the units used !
DPM: Radio-active NaH14CO3 is obtained in glass containers which usually contain (1 mCi) of RA liquid. The usual procedure to divide this amount into smaller amounts of e.g. 50 µCi, is to dilute the 1 ml to 20 ml with distillated water. So a stock is obtained in which 0,4 ml ª 20 µCi. The exact amount of radioactivity added has to be determined for each set of measurements. To keep the read out of the undiluded trace measurement within the range of the machine, put a small amount (eg 0,2 ml ˜10 µci) in a scintillation bottle). Add 10 ml of scintillation liquid. Analyse in scintillation counter after at least two hours.
The PP at different light intensities (necessary to integrate PP over the water column, different depths and thus different light intensities) can be measured by incubating a series of bottles (200 ml), covered with light filters (0-100 % transparency) over a period of time (usually 3 hours).
The transparency of the bottles can be measured beforehand with a lux meter.
To determine PP in situ in the water column in situ incubation can be done at several depths:

Total production of the water column can than be calculated as:
example : PP (mg C m-3 hr-1)
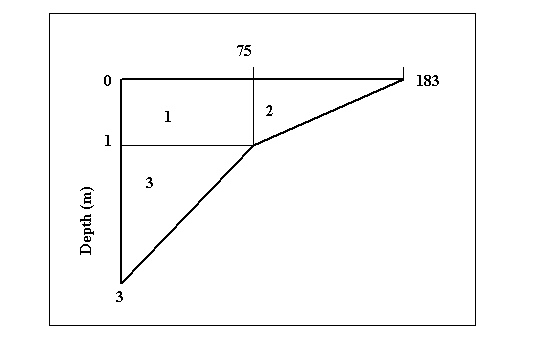
PP measured at surface:
183 mg C m-3 hr-1
PP measured at 1 m depth:
75 mg C m-3 hr-1
PP measured at 3 m depth:
0 mg C m-3 hr-1
Total PP over the water column:
1) 75.00 x 1
= 75
2) [(183 - 75) x 1] / 2
= 54
3) (75.00 x 2) / 2
= 75
Total:
= 203.91 mg C m-3 hr-1
Average:
203.91 / 3 = 67.97 mg C m-3 hr-1
For zooplankton sampling a known volume of water (50-200 l) is filtered through a 50 or 65 µm net. The concentrated zooplankton is rinsed into a small container, and preserved with formaline. Final concentration 4 %; so for example 10 ml formaline (40 %) is added to 90 ml of sample. The concentration of formaline has to be exact, if the samples will later be used for weight determinations, because dry weight of organisms are affected by formaline.
For sampling of larger zooplankton organisms, which have a lower in situ abundance larger volumes of water have to be filtered. This can be done with a high-speed sampler, pulled behind the ship.
The volume of water sampled is then measured by means of a flow-meter, attached in the mouth of the net. The readout of the meter is noted before and after the tow.
The opening area of the plankton net must be known or has to be calculated.
The water volume passed through the plankton net is determined as follows:
indicated number of revolutions x 0,3 x net opening area (m2) x 1000 = water volume (l).
The Plankton Net Model 438 030 has a diameter of 40 cm, i.e. the opening area is 0,125 m2. If the number of revolutions associated with a tow is 266 (noted from the flow-meter counter), the water volume passed through the plankton net is
266 x 0,3 x 0,125 x 1000 = 9.975 liters
- Samples are concentrated to a small volume over a 50 µm net, and all organisms are counted on a grid-petri plate under binocular.
- If the samples contain too many organisms (takes too long to count), the sample is brought in a beaker and the volume is adjusted to a certain value (e.g. 100 ml). The sample is mixed with a stirrer on a magnetic turning table, and with an automatic pipet a 2 - 3 ml sample is taken and counted.
- The volume of sample in the beaker and the sub-sample taken can be adjusted according to the concentration of animals in the sample. Sometimes different dilutions or sub-samples have to be taken for different stages (e.g. nauplii/adults of copepods) which can differ greatly in abundance within one sample.
Use of the ovens and balances: see specific instructions.
- When determining dry weight of marine organisms care has to be taken to remove salt particles adhering to the organisms. Rinse in fresh water.
- The weight of the "recipient" in which the organisms are weighed (e.g. aluminum foil), must be as low as possible to allow an accurate weighing. If filters are used, a series of blanks must be treated as the sample (e.g. rinsing with fresh water, drying) to determine an average blank-value.
- To obtain a reliable weight measurement for a type of organisms, one has to obtain a sample that is "representative" of the population in question.
If the organism is large enough to measure its weight individually, several specimens can be weighed one by one.
If the organisms are too small, a sample containing a (large) number of individuals has to be weighed.
To determine what is the minimum size of the sample to be representative, series of different sized samples (e.g. 10 animals; 50 animals, 100 animals sample-1) can be analyzed in replicates.
Sample size is sufficient if the average weight measured obtains a constant value.
Figure II.2.1. Weight of copepods from the North Sea (Van Gijsegem, 1979)
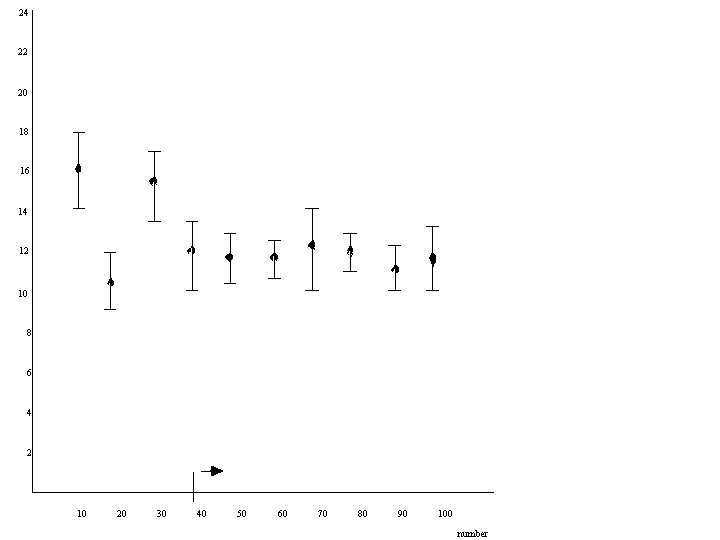
- When samples are stored prior to dry weight analysis a preservative is added. For crustaceans a formaline solution of 4 % is used. The addition of this preservative however causes a certain loss of dry weight.
A correction factor fresh-dry weight versus preserved-dry weight has to be determined in these cases. This factor is also influenced by the time the samples are kept in formaline solution.
Wear plastic gloves at all times when working with radio-activity
Do not inhale scintillation liquid vapors
The herbivorous activity of an animal is quantified by measuring the uptake of radio-active plant material.
In plankton experiments the phytoplankton (culture or natural sample) can either be prelabeled (A) or labeled during the experiment itself (B).
To 1100 ml of algal suspension, add 50 µCi of NaH14CO3. Incubate for 12-24 hours under light conditions.
Filter 2 x 50 ml of the prelabeled culture onto GFC (.45µm) filters. Rinse filters with a few ml's of filtered seawater, and put them in a scintillation bottle.
Add zooplankton (e.g. + 25 adult Artemia l-1).
Incubate for 0.5 hrs.
Remove zooplankton (filter over a net (50 µm)).
Filter 2 x 50 ml's of the filtrate on GFC filter. Rinse with a few ml's of filtered sea water.
Put filter in scintillation bottle.
Isolate the animals in e.g. 5 sets of 5 individuals. Put in scintillation bottles with as little water as possible. If animals have to be stored prior to analysis, put them in deep freezer (e.g. in a plastic container with a little bit of filtered sea water), or preserve with formalin (4 % concentration). Also put one blank filter.
To all of the scintillation containers add 10 ml of scintillation liquid. Count in scintillation counter after 24 hours.
For use of the scintillation counter: see instructions on the apparatus at hand.
Bring 200 µCi l-1 of Na214CO3 into the algal suspension on which grazing activity is to be measured.
Bring zooplankton into the grazing bottle.
Animals can either be brought in individually (after isolating them under binocular) or concentrated on a 50-100 µm net. If natural zooplankton concentrations are high, concentration is not necessary. As a rule of thumb + 25 adult copepods are necessary for measuring radio-activity. Incubate for 1 hour under light conditions.
Further procedures as in a.
Calculations
F = [(DPMzoo at time t)/[(DPMphyto to + DPMphyto t)/2] x (1/t)
(ml animal hour-1)with :
F: clearance rate.
DPMzoo: desintegrations per minute in zooplankton (DPM animal-1)
DPMphyto: desintegrations per minute in phytoplankton (DPM ml-1)
t: incubation time (h)
n: number of animals used
F = [(DPMzoo at time t)/[(DPMphyto t)/2] x (1/t) (ml animal hour-1)
The counting method measures the feeding activity of an animal on suspended particulate matter by quantifying the difference in particle concentration between a grazing bottle (with animals) and a control bottle (without animals). Determination of the particle concentration can be done by microscope (cf II.1.1) or by an electrical counter (Coulter Counter) (cf II.1.1).
* Fill 9 glass bottles with the particle suspension on which grazing is to be measured to about halfway the bottle volume (usually 200 ml bottles are used). When working with natural samples, these have to be pre-filtered through a 100-150 µm net to remove zooplankton. Take care to fill the bottles gradually (dividing sub-samples of water over the different bottles).
Isolate 3 x N* Artemia in a small volume of algal suspension (e.g. 20 ml). Add the Artemia to 3 of the bottles (grazing bottles).
Fill all bottles upto 200 ml exactly with suspension, close them and put them in a rotating incubator (2 rpm.), preferably in the dark. Keep 3 samples for time 0 (t0) sampling.
From each to bottle take subsamples for the following analysis. Shake bottle well to hologenize before sampling
- 50 ml + lugol ==> microscopic count
- 50 ml ==> Coulter Count
- 2 x 25 ml ==> Chl a
At the end of the experiment, (t ~ 6 h) take sub-samples from each bottle for analysis as explained above for to.
Calculations
Ct = Co . ekt
Czt = Co . e(k-g)t
k = (1/t) ln (Ct / Co)
g = (1/t) ln (Ct / Czt)
With k: growth constant (hr-1)
g: grazing constant (hr-1)
Co: particle concentration (in control-and grazing bottles) at time o
Ct: particle concentration in control bottle at time t
t: incubation time (hr)
Czt: particle concentration in grazing bottle at time t.
F = (V.g) / n
With:
F: clearance rate (ml animal-1hr-1)
V: volume of water used (ml)
n: numbers of animals in the grazing bottle
l = [F Co (e(k-g)t -1)] / (k-g) t
(particles animal-1 h-1)With: I = ingestion rate.
When counting particle concentration with an electronical counter, these formulae are applied to each of the size classes. Total ingestion rate is calculated by summing over all size classes.
The respiration (R) of aquatic animals can be measured by enclosing them in a (dark) BOD bottle (50-100 ml) and measuring the difference in oxygen concentration between control bottles without animals and respiration.
It is important that the exact volume of the respiration bottle is known: use calibrated BOD bottles or determine the exact volume by filling the bottle completely with tap water, closing it with a given glass-stopper, and afterward pouring the water into a measuring cylinder.
Fill 6 bottles of 100 ml with algal suspension (or filtered sea water), using a plastic tube to avoid air bubbles.
Fill 3 bottles halfway. Isolate 3 x N* Artemia in a smal volume of algal suspension (e.g. 20 ml) with a pipette. Add N animals to each of the halfway filled bottels. Fill bottles completely with algal suspension, close all bottels.
Note time o. Fix 3 to bottles (with algal suspension only) to determine O2 concentration at time o (see I.4.1. Winkler method). Incubate 3 control (with algal suspension only) and 3 respiration bottles (with Artemia) at room temperature during 4-6 hours. Note time end; fix all bottles and calculate:
- O2 concentration at to (mean), te control (mean) and te (respiration) for each respiration bottle
For each respiration bottle :
DO2 = (O2 control te) - (O2 (resp. te):
use mean value for control, calculate DO2 for each respiration bottle seperately.
Total amount of O2 used = (DO2 x v) / 1000 mg O2
with: v volume of water used (ml)
R = (DO2 x v) / (1000 x t x n) mg O2 animal-1hour-1
With : t : incubation time (hours)
n : number of animals used in each bottle.
O2 to - O2 control te: measures O2 produced or consumed by algae and bacteria in the controls.
In experiments where a process is quantified by measuring differences in concentration of a variable (i.g. difference in O2 concentration in a respiration experiment) care has to be taken to some methodological conditions.
- To measure a decline in O2 concentration, the experiment has to be carried out in a closed system, so there is only a given amount of oxygen present
(O2 = {O2} x volume flask).
- To be able to measure respiration, one has to put enough animals in the flask so that the difference in {O2} is large enough to be detected with the measuring device used (Winkler method, O2 electrode).
- On the other hand, one cannot put too many animals in one flask, because their respiration will be influenced by stress, and by the available oxygen.
- The factor incubation time can influence the results in a similar way as the concentration of test animals.
Water is usually collected by means of a water sampler (cf abiotic factors, seston) and brought into sterile glass bottles before further treatment.
Marine bacteria can be grown and the colonies visually counted on petri dishes:
- 22,4 gr. marine agar powder is diluted in 400 ml distiled water using a 500 ml jar. The bottle is covered with cotton and Aluminum foil and sterilized for 20 minutes in an autoclave. The air pressure of the autoclave is dropped to take out the bottle and + 15 ml agar is poured onto the petri dishes (don't put the cover on the table and sterilize the bottle mouth between filling 2 petri dishes), leave for 24 hours.
- 0.05 ml seawater is added to the petri dishes using sterile pipets (3 hours, in oven at 180° C), sterilizing the mouth of the pipet holder after taking a pipet.
The seawater drop is spread on the dishes with a bacterial spreader (sterilized with alcohol and heating (above the flame)).
The dish walls may not be touched !
- The same routine is applied with serial dilution series of 10-2, 10-4 and 10-6 (10-2 = 0,1 ml in 10 ml sterile seawater). The sterile seawater is made by putting 10 ml of seawater in test tubes, closing them with metal covers and sterilizing in the autoclave for 20 minutes.
Two dishes per dilution is usually taken as a minimum.
- The petri dishes are left (reversed) for x days and counted
(10 days for marine bacteria).
- Considering the number of colonies counted, the dilution factor and the volume of sea water added to the petridish one can than calculate the estimated average number of colonies/m3.
In a similar way, abondance of bacteria such as coli's (e.g. E. coli) or streptococs can be measured on special agartypes after incubation at specific temperatures.
In these cases, dilution of the sample is usually not necessary.
Acridine Orange Direct Count (AODC)
* Black polycarbonate nuclepore filters (Ø=25 mm, pore size=0.22 µm)
* 0.1% Acridine Orange (filtered through 0.22 µm GFC filter)
* Non-fluorescent immersion oil
* glass slides
* glass cover slips
* Samples of + 20 ml are fixed with 40 % formalin (2 % final concentration) and stored at 5 to 10° C in the dark.
* Place a black polucarbonate nuclepore filter on a filterstand. If only white filters are available, these can be put in irgalan black solution for a few hours and rinsed with deionized water before use.
* Place 2 ml of water sample into the filtering apparatus. Add 0.2 ml acridine orange (final concentration 0.01%). Filter after 3 minutes. Use low suction (<20 cm Hg)
* If the bacterial numbers per viewing field are too high, dilutions can be made. If bacterial numbers are too low, the volume to be filtered can be increased up to 10 ml, but with the proportionate amount of acridine orange (final conc. 0.01%)
* Put a drop of oil on a glass slide. Transfer filter from the filter stand to the glass slide. Place another drop of oil on top of the filter and cover with a cover slip. Add one more drop of oil on top of the slip.
* Counting is done under an epifluorescene microscope using blue light excitation (470 nm wavelength) at 1250 x magnification.
* Bacterial cells will fluoresce bright green in a black background. Count fluorescing bacteria in 10 viewing fields. Measure the diameter of a viewing field using a calibration slide.
* Prepared slides must be kept in the dark (use provided slide tray) and can be stored at 4°C (fridge) for a few days.
NOTE : When using the microscope, please do not change any settings except for focusing purposes. Clean the objectives after use.
Microscopic Calculation
number of bacteria/ml = (N x a) / (c x b x d)
With n: number of bacteria counted in c viewing surfaces
a: area of the filter
b: area of the viewing-field
c: number of viewing fields counted
d: volume of sample filtered.
Biomass calculations
Biomass is expressed in mgC/m3 and is calculated by :
number of bacteria/ml x 20 x 10-15 gC/cell(Lee and Fuhrman, '87)
If it is possible to measure length and wdth of a bacterial cell, the conversion factor of 5.6 x 10-13 gC/µm3 can be used (Bratbak, '85).
|
+ sun energy |
photosynthesis | |
|
6 CO2 + 6 H2O + nutrients |
<==================> | C6H12O6 (organic matter) + 6O2 |
|
+ energy |
respiration = Heterotrophic Activity |
The planktonic respiration or the amount of O2 consumed can be calculated after dark incubation (see primary production).
The amount of "easily degradable" organic material can be measured by the biological oxygen demand method (B.O.D.5): B.O.D. for 5 days: (B.O.D.5) at 18° C is often taken as a standard setup.
50 ml BOD bottles are filled completely (with a tube), closed (no air !) and kept at constant water t° in dark. The O2 is fixed at 0, 30, 60, 90, 120, 180 hours and 5 days (see primary production) (use triplicates).
The uptake rate of a substrate (glucose (vg) can be determined by the use of a radioactive tracer (C14 -glucose)
Theoretically as:
[(C14 taken up by bacteria)(concentration of glucose natural + C14 glucose added)] / [(C14 added). t]
Vg = [dpm filter (natural glucose concentration (g C/l) + g C14/l added)] / dpm added . t
= g C / 1 hour
- As the uptake of glucose is regulated by an active transport system, the uptake rate (Vg) increases with the water glucose concentration until saturation (vmax).
- As the glucose incorporated into the bacterial cells is respired after a certain time, the C14 glucose on the filter (A) in function of time is only linear for a certain period. The initial uptake rate from the filter is therefore calculated as dpm/h or % 14C glucose incorporated/hour.
- As the natural substrate concentration is often not known, the bacterial activity is expressed as a turnover rate T = [Radio-activity added (dpm)] / [initial uptake rate (dpm/h)]
The uptake rate can also be calculated from the addition of the 14C cells (A) and the 14CO2 respired back into solution and measured in the filtrate (B)
250 ml of seawater is poured into a sterile 250 jar, 0.1 µCi glucose C14 is added and the solution is mixed. The sample is kept at water temperature, in dark. A sub-sample (10 or 20 ml) is taken at time 0, 20, 40, 60, 120, 180 minutes on a 0.2 µm filter, (under vacuum).
A) The filter is washed with 5 ml saline water (under vacuum) and dropped into a scintillator flask. Add 10 ml scintillation liquid to the flask and measure the DPMS after 2 hours in a scintillation counter.
The curve of radioactivity (C14) on the filters in function of time allows one to calculate the initial rate of uptake of C14 glucose (DPM/h). The turnover time of glucose (t) is than:
t(h) = [Radioactivity added (DPM)] / [initial uptake rate (DPM/h)]
B) the filtrate is collected and the respired CO2 in the filtrate immediately bubbled into a 10 ml scintillator liquid with addition of 0.2 ml H2SO4.
The radioactive CO2 can directly be measured in the scintillator (Don't forget blanks).
The respiration curve: * 14CO2 in function of time can be determined. The curve has a lag. time (the original, non radioactive CO2 is eliminated).
III.4. Benthos
Benthos are the animals that live in or on the ocean bottom; The ocean has a great diversity of physical and nutritive conditions evolving in different densities, with different taxonomic representatives of the benthos in different areas (eg different sediments).
There are different ways of studying benthos. This practical exercise emphasis on methodology, biological diversity and dispersion (zonation).
Benthos can be studied according to oceanic biozones
littoral -------- sublittoral --------- bathydal ----------- abyssal
LT-HT 0-200m 200-4000m 4000-6000m
Benthos can also be studied according to place of occurrence and mesh size of the sampling sieves
Most common classification:
Endobenthos (living in bottom)
Macrobenthos (> 1mm)
Meiobenthos (42 µm < xx < 1mm)
Nanobenthos (2 µm < xx < 42 µm)
Epibenthos (2 cm < xx < 40 cm and living near bottom)
Hyperbenthos (> 1mm and living in the lower part of the water column)
…
For each type of benthos a specific sampling methodology exists.
The stations during this exercise are chosen for their different representation of macrobenthic communities, mainly due to the different sediments (different locations: slope and ridge of the sandbank
Samples will be taken with the ‘Van Veen’ grab (at the sea) and cores (along the beach). Replicates will be taken at the different sampling stations. Other sampling devices, such as the Reineck box core (typical for meio-benthos) and the beach sledge (hyper-benthos) will be demonstrated. (pictures available)
The sorting of samples will be done in the field, including filtering with appropriate mesh sized sieve (1mm- macro benthos); conserving the samples with 8 % formaldehyde and staining them with the indicator Bengale Pink.
The different benthic components:
Meiofauna: very small organisms (in µm), mainly Nematoda, Copepoda…Sampled with a Reineck Box corer.
Macrofauna: organisms > 1 mm, mainly Polychaeta, Bivalvia, Gastropoda, Amphipoda, Echinodermata. Sampled with a Van Veen Grab.
Epifauna: organisms living on the bottom in a narrow association with the sediments, like Sea stars, demersal fishes, crabs… sampled with a Beam trawl (not demonstrated).
Some typical macrobenthic communities:
The Abra alba – Mysella bidentata community
- High densities
- High species diversity (No) and high Shannon wiener index (N1)
- Prefer fine sandy sediments with some mud content
- Common species are: Abra alba (Bivalvia), Lanice conchilega (a tube building polychaete that form ‘riffs’, station 5), Owenia fusiformis (Polychaeta), Nephtys spp. (Polychaeta), Actinaria spp. (Anthozoa), a lot of Gastropoda,…
- Sampled in station 3 and 5 (with Lanice conchilega riffs)
The Nephtys cirrosa community
- Low densities
- Low species diversity (No) and low Shannon wiener index (N1)
- Prefer well sorted medium sand
- Common species are: Nephtys spp. (Polychaeta), Gastrosaccus spinifer (Mysidacea), Echinocardium cordatum (Echinodermata), Donax vittatus (Bivalvia), …
- Sampled in station 4
The Ophelia limacina – Glycera lapidum community
-Very low densities
- Low species diversity (No) and relative high Shannon wiener index (N1)
- Prefer coarse sand
- Common species: Ophelia limacina (Polychaeta), Echinocardium cordatum (Echinodermata), Glycera lapidum (Polychaeta), …
- Normally to be sampled in station 2, which is situated in the coarse sandy area in the Westdiep gully, but only sample 1 of station 2 consist of coarse sand. The other 2 samples consist of finer sandy sediments.
The Barnea candida community
- Low densities
- Low species diversity (No) and relative high Shannon wiener index (N1)
- Prefer compact mud
- Common species: Barnea candida (Bivalvia), Ensis spp. (Bivalvia), Nephtys spp. (Polychaeta), …
- Sampled in station 1
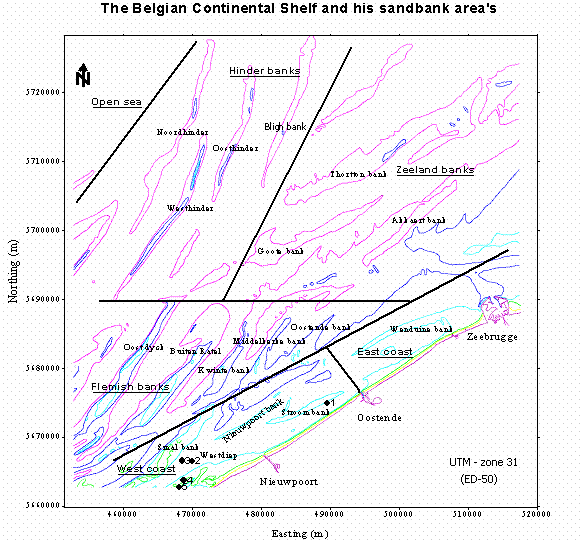
the map indicate the different zones and sandbanks and the five stations sampled on the cruise.
Sedimentology
- There are different classes of sediment: muddy, fine sand, medium sand and coarse sand
- Top of a sandbank consist mainly of well sorted medium to coarse sandy sediment (station 4)
- The flank of a sandbank consist of fine to medium sandy sediments (station 5)
- The gullies between sandbanks consist mainly of fine sandy sediments (station 3). In the Westdiep gully an area of coarse sediment exist (station 2).
- Patches of compact mud are found in the gully along the Stroombank (station 1)
Diversity can be calculated in different ways in ecological studies, mostly serving different purposes. Hence giving different information.
The most simple is:
SPECIES RICHNESS: a count of the number of different species found in a sample.
Problem: doesn’t give information on e.g. species abundance.
Another very popular approach is the SHANNON-WIENER DIVERSITY INDEX. This index combines the information of the species richness and abundance data.
Problem: some assumptions – individuals in the samples are coming from an infinitely large population
- all species are represented in the sample
H= - ∑ (pi lnpi)
pi = ni / N
N= total number of individuals observed in all species
Other information, not given in the Shannon-Wiener Index is e.g. the degree to which species in our samples are evenly distributed.
Therefore we can use EVENESS.
E= H / log S
S= number of species in the sample
E is always a number between 0 and 1.
A number close to 0 means that your species abundances differed greatly.
A number close to 1 means they were almost evenly distributed.
Shannon-Wiener Diversity index exercise
We have 5 different species (I = 1 - 5) in our sample:
No = the number of individuals of each species present in the sample
pi= the proportion of total sample represented by species I
The natural logarithm ln of the proportion pi
Definition of the Shannon-Wiener Diversity Index H = - ∑ (pi ln pi)
Species No pi ln pi pi ln pi
1 60 0.6 -0.51 -0.31
2 10 0.1 -2.30 -0.23
3 25 0.25 -1.39 -0.35
4 1 0.01 -4.61 -0.05
5 4 0.04 -3.22
-0.13
H = - ∑ (pi ln pi) = -{(-0.31)+ (-0.23) + (-0.35) + (-0.05) + (-0.13)}= 1.07
Note that rare species carry less weightNote that rare species carry less weight
Eveness E = H/ ln S
Ln S = Hmax and S is the number of species in the sample
E = 1.07/ 1.61 = 0.66
a) as a function of salinity
growth of phytoplankton can be measured as:
- Chl (and Chl/POC)
and - microscope or Coulter
? Compare the results obtained by two methods
b) as a function of light intensity
c) as a function of temperature
- as a function of different light intensities (production/light curves)
- in function of incubation time (Kinetics)
- compare results obtained by plate count and ACOD method
- determine the heterotrophic activity of bacteries in North sea Sample
b) A algal culture sample using the counting method (Coulter) or microscope or both.
c) Study the grazing activity of Artemia on a flagellate culture at different algal concentrations using the C14 method.
+ your own suggestions
* N: depends on size of Artemia (e.g. 10 adults, 50 nauplii) see also remark about density of experiental animal on p. 53
* N depends on the size of the Artemia (e.g. 10 adults, 50 nauplii)