Sequence analysis was performed for the different sources of polygalacturonases which divides the PG into three subgroups that depends on speciation, i.e., plant, fungal and bacterial PG form different groups. Five highly conserved regions were identified in all the sequences and a few species specific residues and some other important residues adjacent to the active site regions which may play major role in specificity were noted. Structure analysis of b-helical structures in which PGs is a member were done which showed three groups depending on family and the substrate binding regions in few selected b-helical proteins are aligned suggesting functional conservation.
Three-dimensional structure prediction of tomato polygalacturonase and its inhibiting protein were carried out using homology modeling. PG was predicted using the profile-based method utilizing the multiple structure alignment of the five related PG structures. From this multiple alignment few residues that are structurally conserved and that differ in plant is inferred. Based on the model of tomato PG, it was proposed to be a processive enzyme which awaits experimental evidence.
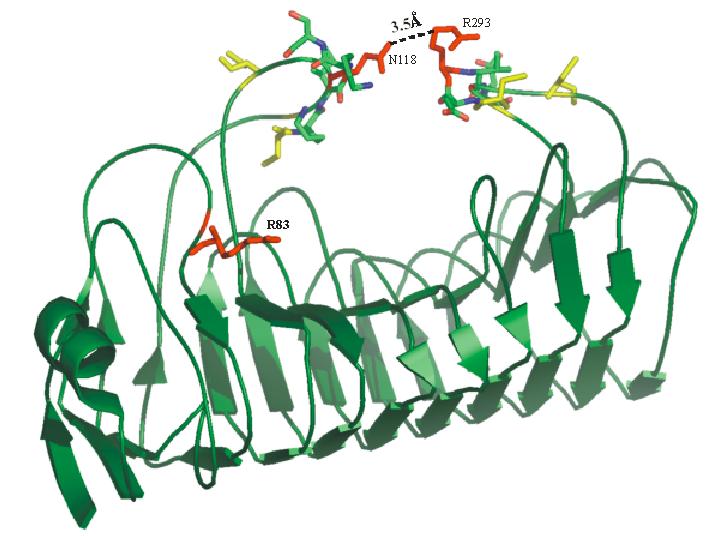
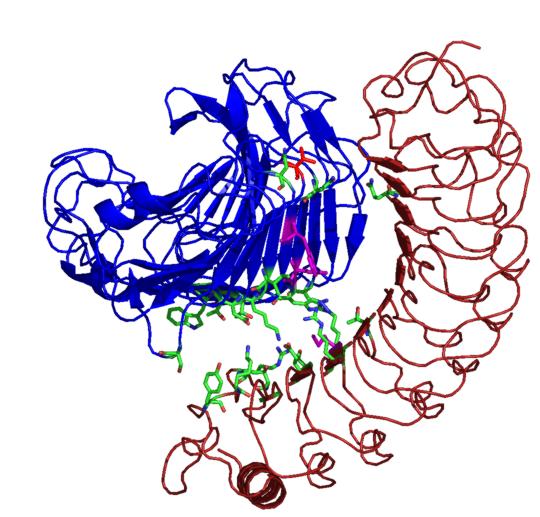
Plant polygalacturonase inhibiting protein (PGIP) is a high affinity receptor for fungal PG and not plant PG. In order to understand the molecular basis behind this, protein-protein docking between tomato PGIP with tomato PG and fungal PG was carried out. From this predictive docking the differential recognition of self and non-self PG were identified. The involvement of many biochemically identified important residues were noted in the predicted complex of fungal PG - plant PGIP. An interaction with the subsite residues explains the non-competitive mode of binding.