Cluster Reaction Chemistry
Mike Geusic, Michael Geusic, M. E. Geusic
V. GAS PHASE CLUSTER REACTION STUDIES
A. Introdution
The study of reactions, on or with, transition metals has been the subject of wide interest. Much of the impetus for these investigations comes from their behavior as catalysts which is both scientifically intriguing and economically important. Surprisingly though, for many years there have been only two main techniques used to study these reactivities. The first is ultra high vacuum single crystal experiments69,70 in which the single crystal is used as a model for the poly-crystalline bulk. The second technique approaches the reactivity from the opposite view point, that is, the atomic and molecular scale. The use of matrix isolation for these type studies has been, in fact, the only way to study these species easily.
Using this technique controlled amounts of metal vapors and reactants are co-condensed with an inert gas ( ex. Ar ) onto a cold surface allowing for low temperature ( or photochemically induced ) reaction products to be stabilized and then probed spectroscopically. The application of this technique has led to a vast literature27, 71-74concerning the reaction chemistry of transition metal atoms along with some diatomic and triatomic species. However, the extension of this technique to larger species is hindered by complications such as identification of the reactant products and multiple matrix sites. This makes the study of clusters beyond the trimer extremely difficult, if not impossible.
Recently, there has been the development of several techniques 75-77 to study gas phase reactions of transition metal clusters. One of these was developed in our lab and has been used to obtain some initial data concerning the reactivity of metal clusters as a function of size. This technique is based on the previously developed cluster source coupled to a reaction tube. A complete description of the reaction tube75b and data75c obtained will be presented in full below.
B. Experimental
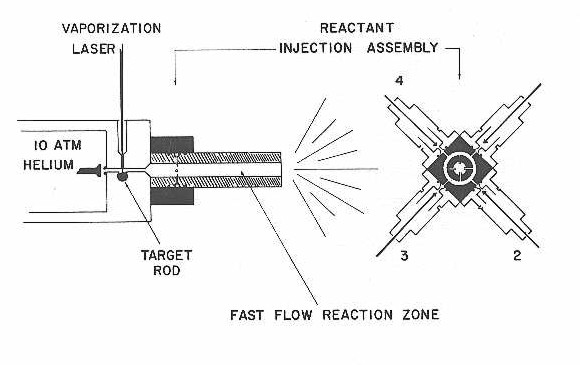
The clusters used in these studies were produced by the laser vaporization cluster source previously described,14,15,47 but modified slightly to allow the attachment of the reaction tube. Briefly, a pulsed double solenoid nozzle is used to produce a 200-400 us pulse of helium that flows down a .2 cm diameter channel passing over the target rod. As this helium pulse flows over the sample, a Q-switched Nd:YAG laser, second harmonic, focused to a .15 cm diameter spot, is fired producing a plasma which is entrained within the carrier gas. This plasma is then neutralized, thermalized, and clustered within the remaining 1.8 cm length of the .2 cm diameter channel. The helium plus clusters then flow through a diverging adaptor region into a 1 cm diameter, 10 cm long reaction tube. All cluster formation is accomplished in the .2 cm diameter channel since expansion into the 1 cm diameter reaction tube causes a 25 fold decrease in both helium and cluster density. The reaction tube design75b is shown from both a side and end-on view in Figure 19. Four hypodermic needles, positioned approximately 2 cm downstream of the .2 cm diameter channel and protruding .3 cm into the reaction tube are used to inject dilute reactants into the flowing helium plus metal clusters. These needles are equally disposed around the diameter of the reaction tube and are supplied by a common annular reservoir of .5 cm3 in volume.
Figure 19
This reservoir is fed by four independently controlled pulsed reactant nozzles (General Value, Model # 9-199-902) each being supplied by independent gas-lines. These nozzles were adjusted to deliver 3 ms wide (FWHM) , with a backing pressure of a few atmospheres. Their firing was synchronized 1 ms before the main cluster nozzle, so as to provide a steady flow of the reactant gas during the 400 us of the main nozzle pulse. The annular reservior served as a gas ballast feeding each of the four injection needles equally, thus ensuring that the flow of reactant into the reaction tube is independent of which nozzle is fired.
The overall design of this injection scheme is necessary in order to eliminate a number of possible errors. In measuring relative rates of reaction, one must make sure the results are independent of which reactant nozzle is used, eliminating systematic errors that would otherwise be present. The extension of the needles .3 cm into the reaction tube, along with their ends being filed at 45 degrees and positioned such that the flow will be towards the oncoming helium plus clusters, has been designed to ensure turbulent flow causing a complete mixing of reactants.
The reactants were diluted in helium before injection. Reagents were metered through a Hastings Teledyne (Model- NA11-100) digital flow meter and mixed with a flow of pure helium. The mixture was then passed through a catalytic purifier (Airco Model #98) which served mainly to remove molecular oxygen from the system. This mixture then entered a ballast tank used to keep the nozzle backing pressure constant.
In these experiments, although the reaction tube was designed to house four reactant nozzles, only two were actually used. The multiple nozzle scheme was designed to permit direct comparison of reactivities of a number of reactant gases. This is accomplished simply by alternating which nozzle is fired. Since all the reactants were diluted in helium, each mixture was compared to a pure helium pulse from the other nozzle. This was done in an effort to negate any error introduced by scattering of the metal clusters from the beam. The extent of reaction is best monitored by depletion of the metal clusters within the beam. Therefore, by using the alternating nozzle arrangement and comparing, shot for shot, the reactant mixture and pure helium injections, one can easily observe any resulting differences in the monitored photoion intensity of the metal clusters.
After some (approximately 175 us) time within the reaction tube, the resulting beam was then expanded from the 1 cm diameter orifice into the main chamber. The beam is then skimmed twice after which it enters the final B chamber. Once inside the photoionization region of the time of flight mass spectrometer, the metal clusters and reaction products are monitored by direct one-photon ionization using the F2 (7.98 eV) excimer laser line. The fluence of the laser was kept low enough (1014 photon/cm2) to ensure that multipohoton dissociative events would not contribute to the observed mass spectrum.
The resulting analog time of flight photoion signal was then multiplied and digitized using a fast 20 MHz transient digitizer. The entire experiment was run at a repetition rate of 10 Hz.
C. Reaction Tube Characteristics
In order for the results of these experiments to be interpretable certain critical parameters, such as, temperature, pressure, and contact time during the reaction must be reasonably known. With this in mind the next few sections will present estimates of these parameters. Although the complexity of the formation and reaction of the metal clusters using this technique may seem hopeless, it turns out that it is not. Gas dynamics78-82for flows, in the .1 to .2 atmosphere gas densities, are well developed which allows for reasonable estimates of the above parameters to be made.
D. Flow from the Main Nozzle
Of course, before one can hope to understand conditions within the reaction tube, there must be a thorough grasp of the pulsed nozzle cluster source without the reaction tube in place. From earlier studies published,47 the behavior of this source is well understood. The nozzle has been measured to deliver 1.5 torr liters of helium in a pulse of approximately 400 us duration. This helium pulse flows directly down the .2 cm diameter tube where vaporization, cluster formation, and thermalization take place with only small losses out of the vaporization laser input hole. The mass flow rate,80 F, through this sonic orifice is:
F = A.C.P
where,
A is the cross-sectional area of the orifice
C is the speed of sound at the nozzle temperature
P is the backing pressure
This can be used to calculate an average density at the .2 cm orifice. From the pulse duration and flow per pulse the average density is calculated to be 1.6 atm , at 300 K.
The helium carrier gas then expands freely from the .2 cm diameter orifice to form a supersonic jet whose flow velocity rapidly increases to a terminal velocity80 , μt
μt = (2T0Cp/M)1/2
where,
Cp is the constant pressure heat capacity
T is the temperature of the helium before expansion
M is the mo1ecular weight of the gas
From the equation it can be calculated that for helium at 300 K, the terminal velocity would be 1.77x105 cm/s. Although, if one considers the effects of the vaporization laser firing within the helium pulse, then there might be additional heating which needs to be taken into account. By measuring the time of arrival for the clusters, it is possible to estimate the effective stagnation temperature, including the vaporization process. The clusters' arrival time is seen to be 10 us wide (FWHM) centered about a total delay of 640 us. For a total distance of 122 cm travel, the 640 us flight time implies a terminal velocity of 1.9 x 105 cm/s. Using this velocity one can back calculate solving for the effective temperature of the source to be 350 K. This is approxi mately 50 K above what is expected without the vaporization laser firing which suggests that thermalization is quite efficient within the .2 cm diameter tube prior to expansion.
E. Flow with Reaction Tube
The presence of the reaction tube on the exit of the cluster source will no doubt have an effect on the previously mentioned parameters of the gas entering it. The major effects are most likely to be a reheating of the gas due to shock waves and turbulence, along with a reduction of the flow velocity to subsonic. In order to understand these effects and their influence on the experimental results, the next few sections will present discussions on temperature, time and density within the reaction tube.
Temperature
From the discussion of the main nozzle, one might suspect that the expansion from the .2 cm orifice would leave the gas in the reaction tube extremely cold. Although in considering the downstream boundary conditions,78 it is understood that some kind of shock wave will be set-up within the first couple of centimeters of the reaction tube. The most likely effect of these boundary conditions (ex: sudden inward turn of flow by reaction tube wall, pressure downstream, etc.) will be to set up an oblique shock attached to the inside corner at the end of the diverging adaptor region. In the limiting case the shock wave system will move up into this diverging region becoming a normal shock wave.
The effect of this shock wave will be to reheat and partially compress the gas. This heating however is not unbound because the flow across the shock wave is so fast that the overall process is adiabatic. Therefore, the only source of energy to support the reheating is the directed translational energy of the flowing gas. For an adiabatic expansion the enthalpy (per unit mass) of the original gas prior to expansion, h0, is related to the enthalpy of the flowing gas, h, by the energy equation:78
h0 = h + μ2 /2
where, u is the flow velocity of the gas. From this equation it can be seen that, as long as the flow is adiabatic, the enthalpy of the flowing gas can never be higher than the original static gas. For a near perfect gas the enthalpy is a function of temperature (5/2 KT per atom), so that for helium the temperature cannot be any higher than that of the static gas. In fact, since the gas is flowing it should be somewhat cooler than the source.
Once the gas has entered into the reaction tube the flow may not remain adiabatic. In fact, the reactant injection needles with their opposing flow were designed to produce turbulent mixing and boundary interactions which would introduce heat exchange between the gas and the walls of the reaction tube. Although, since the walls are at 300 K, the only effect would be to help heat the gas to this temperature.
With this information above and consideration of the vaporization process (heating) taken into account, reasonable estimate of the reaction temperature would be 300-400 °K, with the best estimate being 320 K.
Residence Time
The best way to estimate the actual residence time is to measure the cluster arrival time in the photoionization region and compare it to the arrival time without the reaction tube on. The arrival time of the latter case (no reaction tube) , was measured to be 640 us. To consider the reaction tube, a few cases were investigated in order to understand the effect of each of the elements (walls of the tube, injection needles, injected gas) separately. First of all, when the tube was put in place without the injection needles, an additional 60 us was observed. With the needles inserted but without any gas having been injected, an additional 80 us was observed as compared to without the reaction tube. Finally, if gas was also injected a total delay of 120 us was observed, giving an overall arrival time of 760 us.
The major portion of these added arrival times is most likely due to the time spent in the reactlon tube. By considering the estimated temperature of 320 K (presented above) in the reaction tube, the terminal velocity of the beam would be nearly the same as the main nozzle itself, except for a small delay (5 us)83 due to a slower acceleration to terminal velocity from the 1 cm diameter orifice of the tube. From the average velocity of 1.9 x 105 cm/s it is known that a travel of 10 cm would under normal expansion conditions take 53 us. Therefore, with the added delay of 120 us, the total residence time is 120 + 53 = 173 us.
The observed delay of 120 us can be explained in terms of two effects. The first is a slowing of the gas from terminal velocity to slightly subsonic for the 10 cm length of the reaction tube, along with a slight delay to regain terminal velocity from expansion out the 1 cm diameter orifice. The second effect is the counter current injection of the reactant gas causing a further decrease of the velocity down the tube. Taking both of these effects into account, a reasonable estimate for the residence time is between 150 and 200 us.
Gas Density
From the known flow rate and pulse length of the main nozzle it is easy to place a lower limit of 40 torr for the effective stagnation pressure of the helium gas near the exit of the reaction tube. This limit is set by considering the area change upon entering the larger diameter reaction tube. Although a better estimate can be calculated using the average flow velocity (.59 x 105 cm/s), the mass flow rate (3000 torr liter second), and temperature (320 K) of the gas within the reaction tube. This leads to an average density of 65 torr for static helium.
Due to viscous drag effects on the walls and injection needles along with the counter directed flow of reactants, it is likely that a pressure gradient along the tube should exist. Reasonable estimates for the pressure at the entrance, middle, and exit of the reaction tube, therefore are 100, 65, and 40 torr, respectively.
F. Results
The first few examples of these metal cluster reactions will focus on the reaction of a few simple small molecules such as N2, CO, and D2 with both niobium and cobalt clusters. These metals are particularly good subjects for these studies because both have only one naturally occuring isotope which makes the interpretation of their reaction product mass spectra more informative. A presentation of similar reactions with copper, nickel, and iron clusters will also be made, although since these metals have multiple isotopes their reaction product mass spectra are somewhat less revealing.
Carbon Monoxide Chemisorption
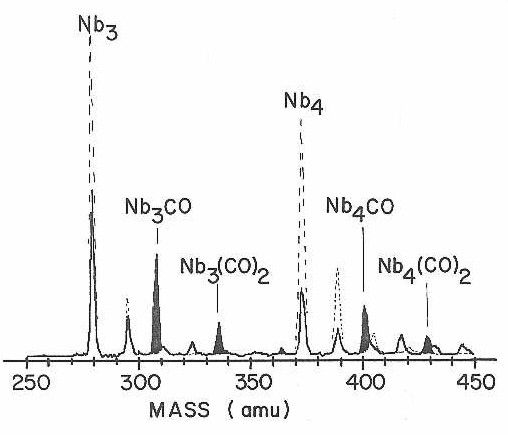
A reasonable place to begin is with the chemisorption of carbon monoxide since it is in most respects the simplest reaction to be considered. With every metal we have investigated (including most of the metals discussed in this thesis) carbon monoxide was found to readily chemisorb on all clusters with more than two atoms. For example, Figure 20 displays a limited region of the time of flight mass spectrum for niobium clusters reacting with CO. The figure shows both the mass spectrum of the metal cluster beam with only pure helium injected through the reactant needles (the control spectrum) as well as the spectrum when CO was injected into the reaction tube. As shown in the figure, both the niobium trimer and tetramer react readily, producing a depletion of the parent cluster signal. In the case of CO chemisorption the ionization signal lost in the metal cluster channel reappears in higher channels in the form of Nbn(CO)m reaction products with m = 1-3. Although not shown in Figure 20, all higher clusters of niobium were found to react with CO in a similar manner. With the limited mass resolution it was impossible to follow the successive additions of CO's to each cluster beyond three, but, at least up to this extent of reaction, all niobium clusters showed the same tendency to chemisorb multiple CO molecules.
In contrast with this facile reaction for the higher clusters, the Nb atom and dimer were found to be remarkably inert. Even with far higher concentration of CO than used in Figure 20, no significant reaction of Nb or Nb2 was observed. In reactions of atoms and dimers, however, we do not claim this lack of apparent reaction is necessarily significant.
Figure 20
These are gas phase addition reactions where thermalizing collisions with the helium buffer gas are essential to take away the energy of reaction and stabilize the reaction product. The 50-100 torr, 320 K helium buffer gas conditions of the reaction tube provide thermalizing helium collisions with reaction products on the order of once every few nanoseconds. In the case of the larger clusters, the unimolecular decomposition time of the reaction complex is certainly much longer than this, and the required bath gas collisions will always occur prior to decomposition. In the case of clusters wlth only a few atoms, on the other hand, the absence of observable reaction products may simply mean that the reaction complex is too short lived to survive until the final expansion region where cooling occurs. In the case of Nb2 + CO, a rough estimate of the lifetime of the Nb2CO reaction complex (with an energy in excess of the 1 eV binding energy by kT) shows the observed absence of reaction here is on the verge of being significant: the calculated lifetime (RRK)84 is on the order of 10-7 second. Since so little is presently known with certainty about the critical parameters that go into such a lifetime calculation for a metal cluster --CO complex, we prefer to stay on the conservative side, and not draw any firm conclusions as to the reactivity of the atom and dimer for any of the metals and reaction partners considered in this study.
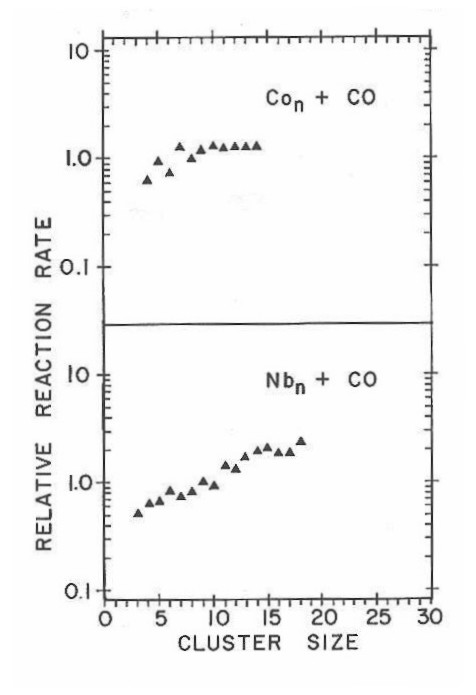
The chemisorption of CO on metal clusters was found to be generally facile, with very little change as a function of cluster size. This can be seen in Figure 21 which plots a coarse estimate of the relative reaction rates for CO chemisorption as a function of cluster size for both niobium and cobalt, obtained from a series of mass spectra taken at various CO concentrations. These relative rate data are based on the following simple picture. The chemisorption reaction for Nbn was assumed to proceed in a single elementary step of the form
Nbn + CO ----kn -----> NbnCO
for which the elementary rate expression is84
d[Nbn ] / dt = kn [Nbn][CO]
Assuming that CO is in such excess that its concentration change is negligible, integration gives an expression for the rate constant, kn , in terms of the measured fraction of Nbn left unreacted (Dn ), CO concentration ([CO]), and reaction time (t):
kn = -( [CO] t )-1 ln Dn
Figure 21
where
Dn = [Nbn]reaction/[Nbn]control
From these initial survey studies of cluster chemisorption reactions, we have only a rough knowledge of the average CO concentrations and reaction times appropriate to the reaction tube. Although it is possible to make reasonable estimates of these parameters and arrive at a coarse measure of the absolute reaction rates, kn , we have far more confidence in taking the ratio of the reaction rate for the n-th cluster to that for some reference cluster measured in the same mass spectrum. For this reason the rates plotted in this thesis for the various studied chemisorptions are relative reaction rates,
R = In Dn / In Dr
where Dn is the measured unreacted fraction of the n-th cluster, and Dr is the corresponding unreacted fraction of a reference cluster.
For the data in Figure 21, Co8 and Nb9 were picked as the reference clusters for the top and bottom panels, respectively. The measured relative rates are seen to increase quite gradually with increasing cluster size, the rate of increase differing a bit between the two metals.
There is little in these CO chemisorption data to suggest that anything particularly dramatic is changing as the size of the metal cluster increases. Either there is not much difference among this range of small clusters, or CO chemisorption is not a very discriminating probe. One indication that CO is not very discriminating can be found in Figure 20 where careful examination of the small peaks near the baseline shows that the extent of CO chemisorption for the various niobium cluster oxides is much the same as the corresponding niobium clusters. These oxides are formed in the high temperature plasma of the laser vaporization cluster source from trace oxygen contaminants in the helium carrier gas. Certainly the addition of an oxygen atom to a small metal cluster like Nb3 would be expected to produce a major reorganization of the electronic structure. Nevertheless CO chemisorption occurs as if nothing had changed.
Dissociative Chemisorption of D2 on Cobalt Clusters
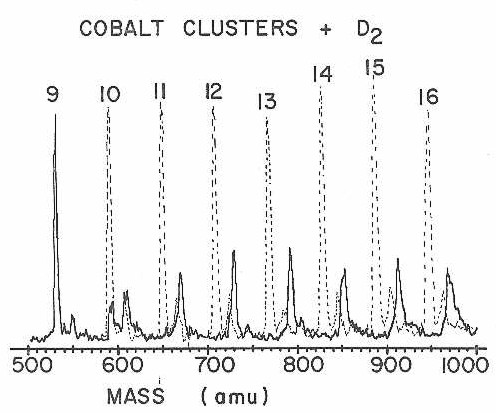
Despite the above indication from CO chemisorption that there was little difference between one cluster and another, there are other reactions for which different sized clusters are actually very different. The first indication that this is true came in a study of the chemisorption of D2 on clusters of cobalt, which appeared as a communication eariler.75a Figure 22 shows a more extensive series of mass spectra for this important reaction. As seen in the figure, the clusters display very dramatic size dependencies in their reaction with D2.
Figure 22
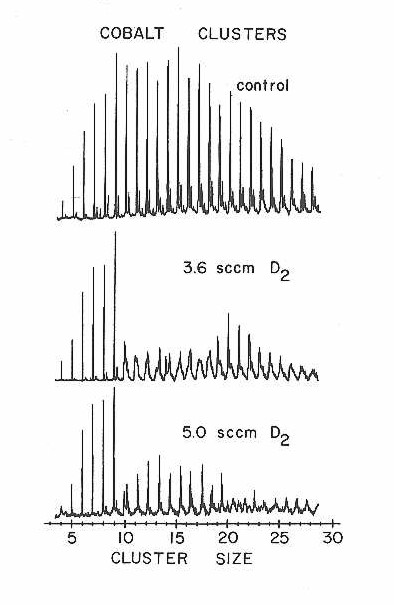
Note particularly the rapid onset of high reactivity on going from 9 to 10 atoms per cluster. At the time it was first discovered, this was the most abrupt change ever observed in any property of a metal cluster as a function of size.
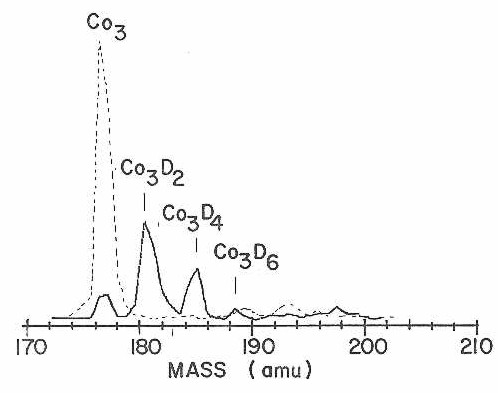
As with CO chemisorption, reaction with D2 was not found to occur on the cobalt atom or dimer. Again, it is not clear whether this is a simple kinetic effect associated with the short lifetime of the reaction complex or whether these species are inherently unreactive. The cobalt trimer, however, does react strongly. Figure 23 shows this trimer region in sufficient detail to resolve individual reaction products produced by the successive addition of up to three D2 molecules.
Figure 23
The bottom panel of Figure 22 taken with a fairly high D2 concentration is misleading. It looks as though the clusters in the 10-20 atom size range have returned, as though the reactivity were less at higher concentrations than at lower ones. The sharp peaks that look like the clusters are actually shifted to higher mass because the clusters have picked up a certain number of D2 molecules and then stopped reacting. This titration-like85 behavior is shown in more detail in Figure 24 where the control and D2 reaction mass spectra are superimposed for clusters in the 9-16 atom range. Here the mass shift for each cluster is clear enough to permit a measure of the number of D2 molecules required to "titrate" the cluster: 5 for Co11-12, 6 for Co13-14, and 7 for Co15-16.
There is also some indication in this reaction data that D2 chemisorption on cobalt clusters is auto- catalytic. As shown in Figure 22, there is only a very small concentration range over which clusters with intermediate amounts of chemisorbed D2 are observed. This may be due to an increase in the effective reaction rate after adsorption of the first D2 molecule. This point should be checked in more detail when later, more quantitative versions of these cluster chemisorption experiments are available.
Chemisorption of D2 is particularly interesting in that there is only one plausible way for the D2 to bind to the cluster tightly enough to survive passage through the reaction tube and be observed in the molecular beam: D2 must dissociatively chemisorb. Remember that the reaction products, whatever their nature, are exposed in the reaction tube to 50-100 torr helium buffer gas at a temperature of 320 K. In the last section it was argued that any reaction product will be thermalized to the buffer gas temperature within a few microseconds. Under these highly collisional conditions at room temperature only quite strongly chemisorbed species will remain on the clusters. Their rate for desorption should be well approximated by an Arrhenius expression84
kdesorption= A exp(-Ea/RT)
where Ea is the heat of desorption, R is the gas constant, and T is the cluster temperature (320 K). This is the approach used in the surface chemistry technique of thermal desorption, where a large body of data has been accumulated in the application of this equation to wide ranges of real molecular desorptions. It is usually found that the pre-exponential factor,86 A, is on the order of 1014 to 1015 s-1 for surface desorption. This value is 10-100 times larger than usually encountered in simple gas phase molecular decompositions, a fact that Zeiri, Redondo, and Goddard87 have ascribed to hindered rotations of the chemisorbed molecule on the bulk surface. In the case of small clusters with only 2 to 30 atoms, we are not yet in the bulk surface limit, so an intermediate pre-expoential factor of 1014 seems reasonable.
For many of the clusters shown in Figures 22,23 and 24 the extent of reaction with D2 is nearly complete, for essentially all the cluster signals have been replaced by reaction products. Since the reaction tube has a residence time of 150-200 microseconds, the typical reaction product we see in the beam must have survived in the reaction tube for at least 80 microseconds without having the D2 desorb off the cluster.
Figure 24
The desorption rate must therefore be slower than 104 s-1. The Arrhenius equation and this maximum desorption rate together imply that the minimum heat of desorption for a reaction product to be stable in the reaction tube is 13 kcal mole.-1
Hydrogen chemisorption is one of the best known of all surface chemistries. There are known molecular chemisorption states on some transition metal surfaces, but these are very weakly bound and are stable only at cryogenic temperatures. All known room temperature chemisorption states of H288,89 on transition metal surfaces are dissociative, with the two hydrogens well separated at different binding sites on the surfaces.
So what is measured here in these gas phase reaction experiments with D2 on metal clusters is the activation barrier on the entrance channel to dissociative chemisorption. This is a reaction that requires the concerted weakening of the strong H-H bond while simultaneously making two new bonds to the metal cluster. Transition metals are uniquely capable of such chemistry, and here we are beginning to see the onset of the behavior on the surface of small clusters.
Dissociative Chemisorption of N2 on Cobalt Clusters
A parallel to these D2 chemisorption studies is a related study using N2. Figure 25, shows a characteristic mass spectrum for a control experiment along with spectra for two different concentrations of N2 in the reaction tube. Without a magnifying glass and a ruler it is a bit difficult to see the details, but it is clear that the reactivity pattern obtained here with N2 is similar to that observed in Figure 22 for reaction with D2. However, the sharpest change in reactivity is clearly, in the vicinity of Co19 and Co20.
The similarity is quite clearly seen in Figure 26 Which plots the measured relative reactivities as a function of cobalt cluster size for both the D2 and N2 chemisorption reactions. These relative reactivity plots were obtained from measured depletion ratios for each cluster as seen in a variety of experiments at different concentrations, similar to those described previously for the CO chemisorption experiments. Although we have endeavored to make these as accurate as possible, the reader is cautioned not to take these relative rate measurements too literally. We have not plotted the estimated error bounds in the relative rate figures because these errors are highly correlated and inclusion in the figure simply makes it more difficult to see the reactivity pattern.
Figure 25
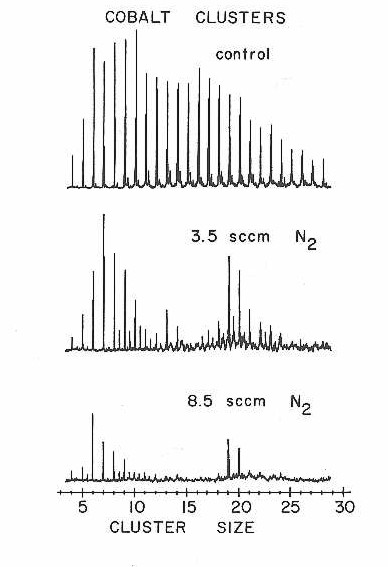
Figure 26
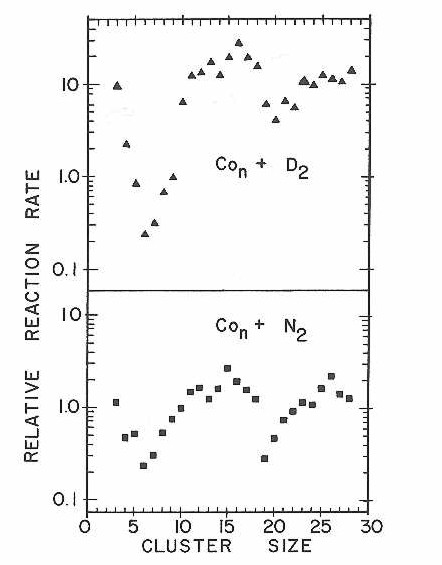
The estimated error bounds range from + 20% for relative rates near unity to over 100% for some of the extreme points corresponding to clusters with either far faster relative rates, or far slower as compared to the reaction rate of the reference cluster. The effect of these errors has been to smooth out the extremes. When more accurate reaction rate data is obtained we expect the patterns seen here to be even more pronounced.
The striking similarity in reactivity pattern seen in Figure 26 for D2 and N2 chemisorption on cobalt clusters is particularly impressive when one recalls how little variation in reactivity was seen in CO chemisorption on these same clusters (see Figure 21). Without question, the mechanism of N2 chemisorption must be more similar to the dissociative chemisorption of hydrogen (deuterium) than it is to the (presumably) molecular chemisorption of CO.
As with D2, chemisorption of N2, is known to be largely a dissociative process on transition metal surfaces at room temperature. Single crystal data is not yet available for N2 adsorption on cobalt, but a great deal has been learned recently about the adjacent elements in the periodic table: iron and nickel. The tightest bound molecular form of N2 now known on an iron single crystal surface is the s adsorption state90 on the Fe(lll) surface. This s state is bound by 31 kJ mole-1, has a markedly reduced N-N vibration frequency (1490 cm-1 as compared to 2194 cm-1 for free N2), and is believed to be p-bonded parallel to the surface. This molecularly bound s state partially desorbs at 170 K, the remainder dissociating to form a surface nitride. In the case of nickel,91 recent angle resolved photoemisson data have shown that the g adsorption state of N2 on Ni(lOO) is molecularly adsorbed perpendicular to the surface. This also is too weakly bound to survive at room temperature. It desorbes at only 140 K. Even though no data is yet available in the case of cobalt, it is reasonable to expect that it too will exhibit only weak binding for molecular N2. For these reasons, the 320 K adsorption data for N2 shown in Figures 25 and 26 quite likely refer to a dissociative chemisorption process.
Chemisorption of D2 and N2 on Niobium Clusters
Results from the extension of these D2 and N2 chemisorption experiments to niobium clusters are presented in Figures 27-30. Once again dramatic changes in reactivity were observed as a function of cluster size. For both the D2 and N2 chemisorptions the clusters with 8,10, and 16 atoms are far less reactive than clusters with only one atom more or less. Figure 29 presents a particularly clear view of these sharp reactivity differences for N2 chemisorption on niobium clusters in the 3-9 size range.
Figure 27
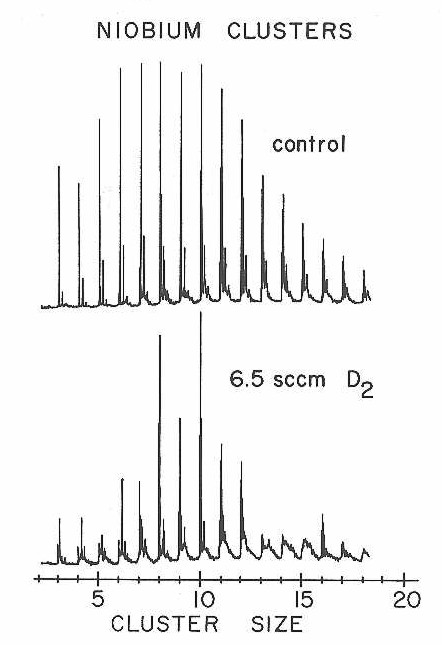
Figure 28
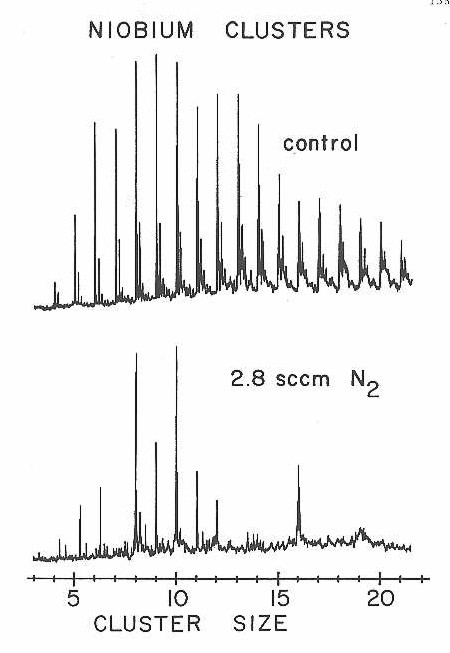
Figure 29
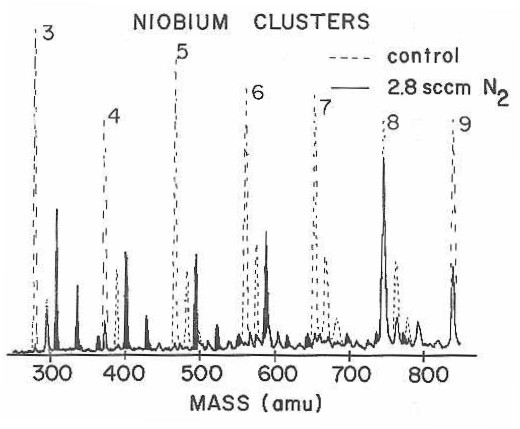
Figure 30
Note, for example, that the niobium trimer has nearly disappeared from the mass spectrum, the missing ion signal reappearing in peaks corresponding to Nb3N2, Nb3N4, and Nb3N6. In sharp contrast, the Nb8 cluster has been largely unaffected by the N2 reactant.
Also note that the reactivity of the corresponding cluster oxides is often sharply different than the metal cluster. Nb3O, for example, is seen in Figure 29 to be far less reactive than Nb3, while Nb8O is far more reactive than Nb8. In direct contrast to the observations for CO chemisorption, the pattern of reactivity of the niobium cluster oxides for D2 and N2 chemisorption is quite different from that of the clusters.
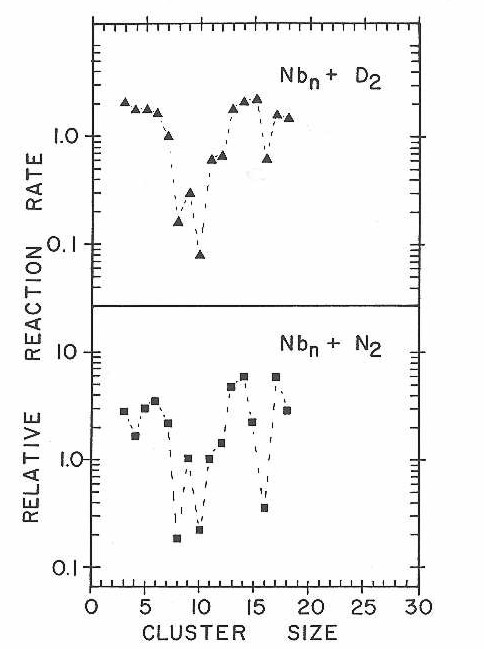
The relative reaction rates measured as a function of cluster size for D2 and N2 on niobium are presented in Figure 30. Again we emphasize that the errors in these measured relative rates are considerable. Particularly for the very fast and very slow relative rates, the measured values are likely to be biased due to the difficulty in measuring very large and very small depletions. Very slow relative rates will tend to be over estimated, very fast rates will tend to be under-estimated. However, the gross pattern of reactivity, is well determined by the current measurements.
Copper Clusters -A Check for Hydrogen Chemisorption
Dissociative hydrogen chemisorption is a surface chemistry that is peculiar to transition metals with open d-shells.92 Copper is an interesting test case since here the 3d band is fairly narrow and situated below the Fermi level. Molecular hydrogen93 is known to chemisorb on copper surfaces, but this process is known to possess an activation barrier in the range of 3 to 5 kcal/mole, depending upon the crystal face. Calculations94 of the electronic structure of copper clusters indicate the 3d band remains narrow and filled, lying well below the Fermi level, so it is reasonable to suspect that copper clusters will be unreactive toward H2.
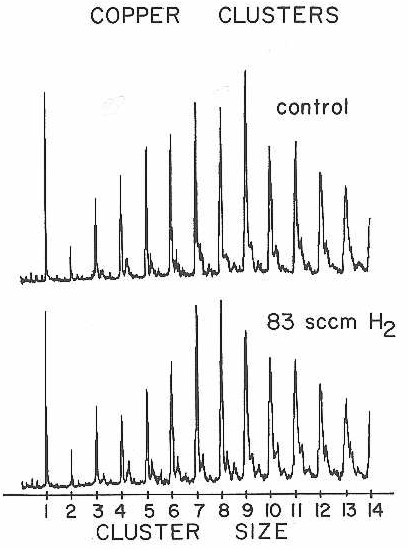
As an added check on our interpretation of the D2 chemisorption experiments with cobalt and niobium clusters, we therefore extended the hydrogen reaction studies to copper clusters. Figure 31 shows the result of this experiment. As expected, no evidence of reaction was observed with H2 (which should react faster than D2) at a concentration over an order of magnitude higher than used in the previous experiments with cobalt and niobium.
Figure 31
Nickel and Iron Cluster Chemisorption Experiments
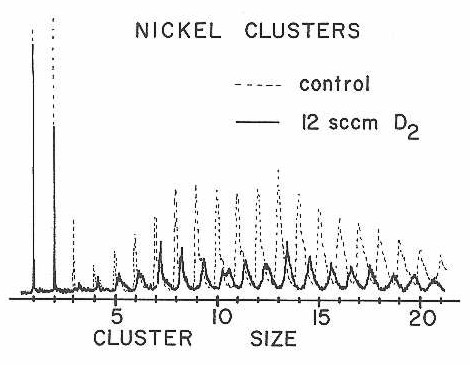
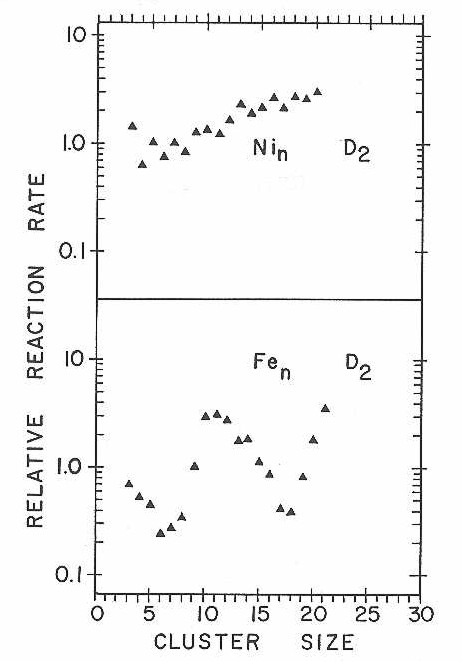
Nickel is a particularly important transition metal for detailed cluster beam studies since it is by far one of the easiest of the open d-shell transition metals to handle in quantum chemical calculations.94 Figure 32 shows the results of a D2 chemisorption experiment on nickel clusters using the new fast-flow reaction tube. Deuterium is found to chemisorb readily, but displays only a mild, nearly monotonic increase in reaction rate as a function of cluster size. In light of the dramatic effects seen with cobalt and niobium clusters, smooth reactivity variation with cluster size is rather surprising.. Due to time constraints on the molecular beam apparatus, no other chemisorption experiments were done for nickel.
Figure 32
Iron completes the list of metals we have explored in these first chemisorption experiments with the new apparatus. Figure 33 presents mass spectra from a D2 chemisorption experiment. Once again, a highly size dependent reactivity pattern is found for dissociative D2 chemisorption. Relative rates abstracted from experiments such as these have been plotted along with similar data from the nickel chemisorption experiments in Figure 34.
An extensive attempt was made to measure the relative reactivity patterns for N2 chemisorption on iron clusters. Unfortunately, the iron clusters turned out to be almost inert to N2 chemisorption. Figure 35 shows the results of one experiment where an order of magnitude higher N2 concentration was used than in the experiments with cobalt and niobium clusters.
Figure 33
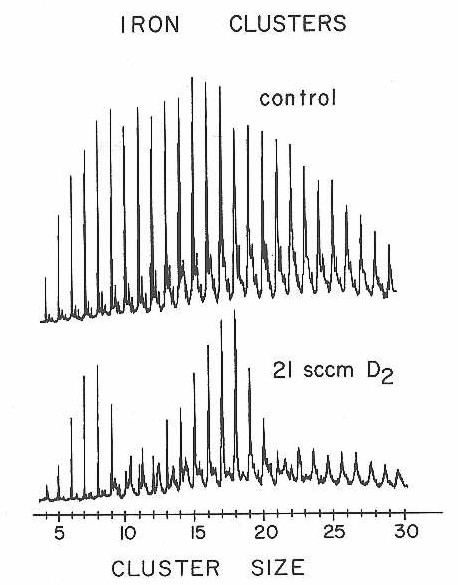
Figure 34
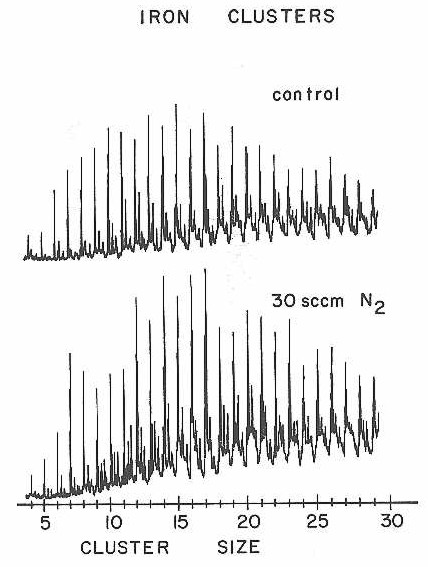
Figure 35
Even at this high concentration there is only a slight indication of reaction (Fe11, for example does show a weak but clear Fe11N2 feature growing at 28 amu to high mass in the lower spectrum of Figure 35).
At the relatively high concentrations of injected, N2 reactant used in the experiment of Figure 35, the cluster arrival time at the detector was observed to increase by 20 to 40 microseconds (to a total flight time of 800 microseconds). An experiment with argon injected instead of N2 revealed this time shift to be a kinetic effect associated with the heavier mass of the reactant gas as compared to the normal helium carrier. We do not fully understand as yet the detailed mechanism for this delay. Even at the highest reactant concentrations only a few percent of the total gas in the reaction tube was due to the injected heavy reactant --not enough to produce a significant lowering of the terminal beam velocity. Since the increase in cluster arrival time was greatest for the heavier clusters, the effect may be due to the heavier reactant gas being more effective in slowing and redirecting the heavy clusters in the post-shock region at the entrance of the reaction tube, thus increasing the effective residence time prior to the main supersonic expansion.
Whatever the cause of the extra delay in the arrival time of the clusters at the detector, it made it difficult to be precise about the extent of reaction, of iron clusters with N2. However, it is clear that the reaction rate is far slower than seen in the cases of cobalt and niobium at much lower N2 concentrations.Furthermore, those few cases where FenN2 reaction products are seen in the highest concentration studies are just the clusters that were found to be most reactive in D2 chemisorption.
These low reactivity results for N2 dissociative chemisorption on iron clusters are perfectly in accord with the extensive single crystal studies of Ertl and co-workers95 in their studies of N2 dissociation as the rate limiting step in ammonia synthesis on iron catalysts.
G. Conclusion
From the data presented within the last section it was shown that the reactivity of metal clusters depends on the reactant, cluster size, and metal. These initial studies have obtained the first data demonstrating that for certain reactants the reactivity as.a function of cluster size changes dramatically. For example, the relative reactivity for N2 chemisorption on niobium clusters shows that Nb16 is over an order of magnitude less reactive than Nb15 or Nb17.
As also shown in the last section certain restrictions can be placed on the reaction products due to the conditions in the reaction tube. It was estimated that the minimum binding energy of these chemisorption reaction products is at least 13 kcal/mole in order to be observed. In the case of molecular hydrogen (or D2), there is only one type of chemisorption having such a strong binding energy, that is, it must dissociatively chemisorb. Also in the case of N2 these requirements support the argument that dissociative chemisorption is involved for its reaction on clusters. This is due to the fact that the strongest known N2 molecular chemisorption state on a transition metal (N2 on rhodium)96 is completely desorbed by 300K. The D2 and N2 chemisorption reactions reported here are therefore almost certainly dissociative processes producing strongly bound surface hydrides and nitrides.
For this type of dissociative chemisorption, consensus is that the critical interaction involves electron donation from the surface of the metal into the anti-bonding molecular orbitals of the species, weakening the binding energy of the reacting molecule. Since this process involves partial electron transfer from the metal to the diatomic, it would seem reasonable to suppose that as the ionization potential of the cluster is decreased, the reactivity should increase.
In a recent publication by Whetten et al,97they investigated the reaction of H2 and D2 on iron clusters and have, in fact, pointed out such a correlation. They have measured the reactive rates of dissociative chemisorption of D2 and H2 on iron clusters in the range of Fe8 to Fe25, and with ionization threshold data previously obtained,98 have correlated the local minimum in D2 chemisorption reaction rate to a local maximum in the ionization threshold for Fe17 and Fe18. In this paper they show that the reaction rate seems to correlate well with the ionization potential for clusters of iron in the range of 8 to 25 atoms which seems to support the mechanism of electron donation from the metal to an anti-bonding orbital of H2 (or D2). Although in the data presented here, as well as that of Riley et al,66,85 there are two minima in the D2 reaction rate dependence on cluster size for iron.. The first minimum occurs near Fe6 ( a region not reported by Whetten et al.) and this minimum does not correspond to a maximum in the ionization threshold of iron clusters which suggests that there may also be other factors involved.
The reaction rate of D2 on copper clusters also indicates that ionization potential alone is not the only factor involved in determining the relative rate for these reactions. Copper clusters show an inability to dissociatively chemisorb D2 (see Figure 31), although the ionization potential of copper clusters (1-25) has been shown by Powers et al99 to be just as low as those for the highly reactive iron clusters. From the evidence, it can concluded that orbital electron donation into the anti-bonding orbital of D2 (or H2) is definitely not the only factor involved in determing the reaction rate on these clusters.
The dissociative chemisorption of molecular hydrogen on metal surfaces has been considered a chemistry unique to transition metals. This suggests that the surface d-orbitals must be the critical factor involved in these reactions. Recent ab initio calculations by Siegbahn et al100 have examined H2 chemisorptlon on various surface sites of small nickel clusters both with and without the use of 3d orbitals at the chemisorption site. Their results predict that the 3d orbitals are crucial in lowering the activation barrier to dissociative chemisorption. Therefore, it can be suggested that these studies are actually monitoring the "d" character of the frontier orbitals on the surface of the metal cluster. Although, as of this time, no definite conclusions can be drawn concerning exactly what the parameters are which control these relative reactivities. In comparing the relative reactivities of D2 and N2 on clusters of cobalt and niobium it can be seen that there are some differences however, they are slight and the overall patterns are actually very similar. Therefore, it is suspected that whatever factors control the D2 dissociative chemisorption rate also control it for N2.
In contast, the reaction of CO on cobalt and niobium clusters shows a drastically different pattern of reactivity from that of H2 or N2. This pattern is a more gradual monotonic increase of reactivity with increasing cluster size. As mentioned in the last section, CO is thought to be molecularly chemisorbed, which in fact may explain its reactivity pattern as compared with H2 and N2 suggesting that molecular chemisorption is a much less constrained process.