Factors Influencing Fetal Growth* Department of Pediatrics, Division of Perinatal Medicine, University of Colorado Health Sciences Center, Denver, CO.
After completing this article, readers should be able to:
The principal determinants of fetal growth are fetal genotype and in utero environment. Environmental factors include maternal and paternal genetics, maternal size, and the capacity of the placenta to provide nutrients to the fetus. These environmental factors interact with the intrinsic growth pattern of the fetus, yielding a particular rate and composition of fetal growth. Most of the variation in fetal growth in a population is due to variations in environmental factors, not the fetal genome, although a genetically abnormal fetus clearly might not grow as well as a normal fetus if affected genes include those that are important for growth.
Many genes contribute to fetal growth and birthweight. Techniques in which specific genes can either be deleted ("knockouts") or overexpressed have led to greater understanding of how some of these genes regulate fetal growth. Such studies have shown that both maternal and paternal influences are present during fetal development and are passed on to the developing fetus by spermatozoa or oogonia by a mechanism called imprinting. Although the maternal genetic composition exerts greater influence than fetal genotype in the overall regulation of fetal growth, both maternal and paternal genomes are important in fetal growth and development ((1))((2)). For example, gynogenetic zygotes (two maternal genome copies) lead to underdeveloped extraembryonic tissues but well-developed embryos. The more modest regulation offered by the paternal genotype is essential for trophoblast development. For example, zygote nuclear transfer experiments have shown that androgenetic zygotes (two paternal genome copies) develop extensive trophoblast tissues but contain underdeveloped embryonic tissues. Additionally, hydatidiform moles that result from excessive trophoblast formation have primarily a paternal genetic composition ((3)). Although several genes have been described as maternally or paternally imprinted, insulin-like growth factor I (IGF-I) and IGF-II are two protein products of genes that specifically regulate the development of trophoblast cells, which form the placenta. The IGF-II gene transcript mRNA has been localized to the proliferating cells of the placental labyrinthine area in the rat and to cytotrophoblast cells that differentiate into the syncytiotrophoblast in the human placenta ((4))((5)). Gestational profiles of IGF-II, IGF-binding proteins, and IGF receptors (types 1 and 2) suggest they are involved in enhancing placental growth ((6)). Additionally, IGF-II may regulate placental growth directly, as evidenced by 60% placental and fetal growth restriction in mice lacking IGF-II ((7)), which implicates IGF-II in the proliferation of trophoblast cells. Other gene knockout experiments have shown that IGF-I and IGF-II are important regulators of both fetal and placental growth. Removal of the paternal IGF-II allele in mice produces fetal growth rates 60% of normal ((7))((8)). Additionally, the paternal IGF-II allele is required for fetal IGF-II expression, indicating imprinting of the mouse IGF-II gene. IGF actions in fetal development also have been assessed with IGF-I, IGF type 1 receptor (IGF-IR), IGF-II (p-) gene deletions, and a combination of double mutants ((4)). Deletions of IGF-I or IGF-IR yield birthweights 60% and 45% of normal, respectively. Deletion of both IGF-I and IGF-IR results in the same decrease in birthweight as IGF-IR mutants. However, when IGF-II and IGF-IR genes are deleted simultaneously, birthweight is reduced to only 30% of normal ((4)). Such experiments indicate that specific genes must be expressed normally for placental and fetal growth. Other studies have addressed how the normal expression of these genes during embryonic and fetal development can accentuate or inhibit their expression and/or action to produce regulatory proteins that control the metabolic processes that determine cell growth. For example, nutrient substrates (eg, glucose, amino acids, fatty acids) and coordinated hormones (eg, insulin, thyroid hormone, cortisol) regulate gene expression and action within the developing embryo and fetus. More specifically, it now appears that nutritional stimuli regulate placental and fetal tissue expressions of glucose transporters and hexokinase ((9)). Furthermore, gene expression of components of the fatty acid oxidation and ketogenesis systems can be altered by changes in the supply and fetal concentrations of glucose, glucagon, long-chain fatty acids, and insulin ((9)).
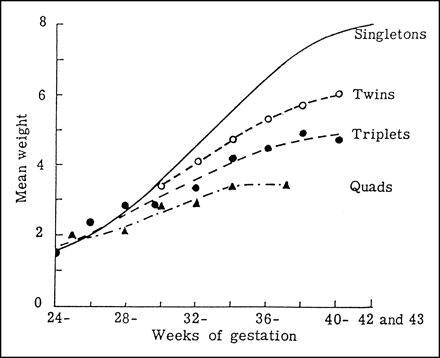
Uterine Size Under usual conditions, fetal growth follows its genetic potential, unless the mother is unusually small for her species and limits fetal growth by a variety of factors considered collectively to represent "maternal constraint." Maternal constraint results primarily from a limited uterine size and, thus, the capacity to support placental growth and nutrient supply to the fetus ((2)), not to any particular genetic factor. The nongenetic maternal nature of this effect is demonstrated by embryo transfer and cross-breeding experiments. For example, a small-breed embryo transplanted into a large-breed uterus will grow larger than a small-breed embryo remaining in a small-breed uterus. Conversely, embryo transfer of a large-breed into a small-breed uterus will result in a newborn that is smaller than would be seen in its natural large-breed environment. An elegant set of experiments performed by Walton and Hammond ((10)) demonstrated the effect of maternal size on fetal growth when they crossed small Shetland ponies with much larger Shire horses. The offspring from Shire mares crossed with either Shire or Shetland pony stallions had similar birthweights, while Shetland mares produced foals normal for that breed. Thus, despite identical genetic makeup, the principal determinant of fetal growth rate and birthweight was the size of the mother. Another clear example of maternal constraint is the reduced rate of fetal growth of multiple fetuses in humans ((11)), who optimally support only one fetus (Fig. 1
Fetal growth constraint from the maternal environment is a physiologic process that includes the maternal-specific capacity of uterine size, placental implantation surface area of the uterus, and uterine circulation, which together support the growth of the placenta and its function. Obviously, small fetuses of small parents or large fetuses of large parents do not reflect fetal growth restriction or fetal overgrowth, respectively. Their rates of growth are normal for their genome and for maternal size. Unless maternal constraint is particularly prominent, such fetuses would not grow faster or to a larger size if more nutrients were provided. Pattern of Gestational Growth The length of gestation is more strongly correlated with growth of neural tissue (range, 0.015 to 0.033 g1/3/d�a 2.2-fold range) than with growth of the fetal body (range, 0.033 to 0.25 g1/3/d�a 7.6-fold range). The physiologic significance of this relationship is not known, but intrauterine development of a large brain/body mass ratio in humans is favored in a single fetus and is made possible by a slow rate of somatic growth. The latter allows a steady increase in fetal cerebral metabolic demand while the total metabolic demands of the conceptus are kept within limits that the mother can easily support without stress on her own metabolism. At the end of gestation, fetal mass varies considerably among mammals, reflecting both differences in fetal growth rate and length of gestation ((12)). Including marsupials, the range in newborn weight from the smallest marsupial weight of 10 mg to the largest eutherian mammal (the blue whale, weighing 2,000 kg) is 200 million-fold. Among eutherian mammals, total newborn weight is inversely related to adult animal size and directly related to the length of gestation.
Normal variations in maternal nutrition have relatively little effect on fetal growth because they do not markedly alter maternal plasma concentrations of nutrient substrates or the rate of uterine blood flow, the principal determinants of nutrient substrate delivery to and transport by the placenta. Human epidemiologic data from conditions of prolonged starvation as well as nutrition deprivation in experimental animals indicate that even severe restrictions in maternal nutrition only limit fetal growth by 10% to 20% ((15)). Calorie and protein intakes must be reduced to less than 50% of normal for a considerable portion of gestation before marked restrictions in fetal growth are observed ((16)). Such severe conditions often result in fetal loss before late gestation fetal growth rate and fetal size at birth are affected. Similarly, fetal macrosomia is only common in pregnancies complicated by gestational diabetes mellitus in which maternal plasma hyperglycemia and hypertriglyceridemia combine with fetal hyperinsulinemia to produce excessive fetal adiposity.
The placenta modulates the environment for fetal development and growth by mediating the maternal systems to both recognize and support pregnancy. Endocrine and architectural characteristics of the placenta provide an adequate supply of nutrients for the developing fetus, a site for nutrient uptake and waste removal, and a line of defense against pathogens ((17))((18))((19)). A diverse group of hormones, growth factors, and cytokines can be produced by the placenta and/or other endocrine or nonendocrine organs ((18)). True placental hormones (hormones produced only by the placenta) have been discovered in eutherian mammals, but their physiologic functions remain to be fully elucidated. True placental hormones include the conceptus interferons ((20)), chorionic gonadotropin (hCG) ((18)), and members of the growth hormone (GH)/prolactin (PRL) gene family ((18)). Members of the GH/PRL gene family produced only in the placenta include placental lactogen (PL), growth hormone (human (h)GH-V), and prolactin-related proteins (PRP) ((21)). PL is believed to have a pivotal role in the growth and development of the fetus by coordinating metabolic and nutrient supply from the mother to the developing fetus to promote growth ((22)). The placenta exerts strong control over fetal growth by providing nutrients directly or in metabolically altered forms and amounts. Naturally and experimentally, placental growth precedes fetal growth, and failure of the placenta to grow is directly associated with subsequent decline in fetal growth ((23)). Placental control of fetal growth can vary considerably. For example, experimental placental growth restriction in pregnant sheep reduces fetal weight, but not proportionately ((24)). This indicates either an adaptive increase in the capacity of the smaller placenta to transport nutrients to the fetus or fetal development of an increased capacity to extract nutrients from the placenta and direct them to growth. Placental insufficiency in sheep reduces the capacity of the placenta to transport glucose to the fetus, which is exemplified by an increased maternal-to-fetal glucose concentration gradient, but the net uptake of glucose by the fetus is normal per fetal body weight ((25))((26)). These data suggest a reduction of glucose transporters at the maternal-fetal interface. Currently, no data on the abundance of placental glucose transporter expression and/or activity exist in placental insufficiency models, although data from a chronically hypoglycemic pregnant sheep model show that reduced glucose concentrations decrease placental GLUT 1, and to a lesser extent, GLUT 3 ((27)). Therefore, the evidence suggests that changes in glucose transporter expression might alter glucose transport capacity within the placental insufficiency model. Similarly, although both experimental and human clinical data show reduced concentrations of amino acids in growth-restricted fetuses ((28)), and there is some evidence in humans that selected amino acid transporter expression is reduced under these conditions ((29)), no evidence yet demonstrates a functional correlation between decreased placental amino acid transporter expression and net amino acid transport to the fetus. Characteristically, therefore, limitations of placental transfer
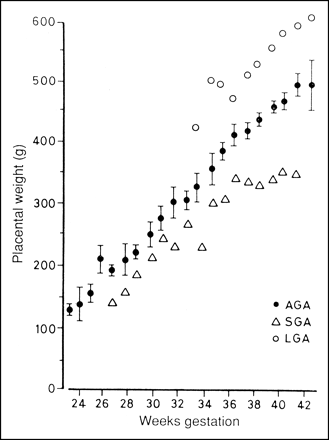
of nutrients to the fetus directly limit fetal growth. A direct
relationship between fetal weight and placental weight in humans
indicates that large-for-gestational age (LGA), average- or
appropriate-for-gestational age (AGA), and
small-for-gestational age (SGA) infants are directly associated
with LGA, AGA, and SGA placentas (Fig. 2
In general, decreased rates of fetal growth represent an "adaptation" to inadequate nutrient supply. Accordingly, intrauterine growth restriction (IUGR) that results from decreased nutrient supply can be interpreted as a successful, if not perfect, adaptation to maintain fetal survival. Fetal undernutrition also appears to affect rapidly growing fetuses more than slowly growing fetuses, who may have been programmed to grow more slowly for genetic or embryonic and early fetal pathologic reasons ((31)). Reintroduction of nutrients can return fetal growth to normal in those fetuses whose rapid growth rate was decreased by undernutrition, but too rapid an introduction of nutrients has often caused fetal pathology, including hyperlactatemia and occasionally even acidosis and hypoxemia. Fetal Glucose Supply, Metabolism, and Regulation of Fetal
Growth Fetal Amino Acid Supply, Metabolism, and Regulation of Fetal
Growth Because the concentration of amino acids within the trophoblast cells (and perhaps some of the transport into the fetal plasma) is energy-dependent, it is not surprising that experimental evidence shows reduced amino acid transport to the fetus following decreased energy supply to the placenta. This is especially true for oxygen deficit, either from primary hypoxemia or from reduced uteroplacental blood flow, and glucose deficit from chronic maternal and fetal hypoglycemia ((37))((38)). Of course, hypoxemia and hypoglycemia could reduce fetal growth independently of reduced amino acid transport, such as by limiting anabolic hormone and growth factor production or decreasing energy supply, both of which are required for protein synthesis and limitation of protein breakdown in fetal tissues. The importance of amino acid and energy supplies for fetal protein
and nitrogen balance and for fetal growth is illustrated by experiments
in fetal sheep over the second half of gestation that
compared fractional protein synthesis rates derived from tracer
amino acid data with fractional body growth rates derived from
carcass analysis data (Fig. 3
Fetal Lipid Supply, Adipose Tissue Development, and
Regulation of Fetal Growth
Changes in maternal circulating growth hormone and growth hormone-like peptides such as PL, which increase during pregnancy, have combined effects that induce maternal insulin resistance and lead to higher circulating concentrations of glucose and lipids. Gestational changes of the maternal nutrient pool allow for increased glucose transport via facilitated transport systems and maternal use of mobilized fat stores. The increased supply of glucose to the fetus stimulates fetal insulin, promotes fetal adiposity (or macrosomia, as in the infant of the diabetic mother), and limits fetal protein breakdown ((42))((43)). In postnatal life, growth hormone (GH) stimulates cell differentiation and proliferation indirectly by mediating IGF-I and can directly affect cellular metabolism ((44)). However, during prenatal growth, its effects are minor. For example, GH deficiency, seen naturally in anencephalic infants or produced experimentally in animal models by removal of the pituitary, has no effect on fetal growth ((45)). Supplementation of GH to the fetus stimulates IGF-I from the liver and pancreatic beta-cells ((46))((47)), suggesting that GH and PL have overlapping effects in prenatal life. Nevertheless, GH does not appear to be necessary during fetal development. The postulate that many of the somatogenic effects of PL are mediated through IGFs ((22))((48)) is supported by several lines of evidence. First, when oPL is infused into the vasculature of fetal sheep for extended periods, fetal serum IGF-I concentration increases significantly, but serum IGF binding protein-2 concentrations are not affected ((49)). Second, IGF-II concentrations from rat fetal fibroblasts increase with PL treatment in vitro ((50)). Third, fetal hypophysectomy does not change IGF-II concentrations ((51)) and has no major detrimental effect on overall fetal growth. Therefore, IGF-II production appears unlikely to be affected by GH. Fourth, IGF-I concentrations in human maternal circulation increase dramatically during the third trimester when the greatest fetal growth occurs ((51))((52))((53)). Fifth, PL stimulates IGF-I production in hypophysectomized immature rats and pregnant sheep ((22)). Finally, IGF-I has anabolic effects on carbohydrate metabolism in rat fetal hepatocytes ((54)). Accordingly, PL appears to play a role in promoting fetal growth, possibly by stimulating IGF production. However, short-term (24 h) infusion of recombinant PL into the fetus does not result in an increase in IGF-I. Rather, amino nitrogen concentrations in fetal blood decrease while other metabolic parameters remain unchanged ((55)). At the same time, IGF-binding protein-3 in the fetus declines 30% ((56)). Therefore, PL may indirectly alter IGFs or the binding proteins to elicit an action on fetal tissues. There are clinical reports of human pregnancies lacking hPL and/or hGH-V during gestation due to a deletion in both chromosomal alleles. In one such a pregnancy, the baby suffered severe growth restriction (10th percentile) but was otherwise normal ((57)). At week 33, no hPL was detected in maternal serum, IGF-I concentrations were below normal, and GH was within the normal range. Additionally, no hPL mRNA capable of translating hPL was identified in placental tissue, but low levels of hPL-1 pseudogene were found ((58)). The pregnancy was complicated with a single umbilical artery and mild pre-eclampsia, which may have contributed to the reduced birthweight ((57)). Postnatally, the baby grew normally, and no malformations were seen. These data suggest that hPL expression during gestation has a significant effect on fetal growth and development. However, other clinical reports in which genetic alterations within the human GH/PL locus have been identified have noted no changes in fetal growth ((59))((60))((61)). These data suggest substantial ambiguity in the functions of PL during pregnancy and a need to study an in vivo model that lacks PL to identify clearly its action on fetal growth and development. Insulin produced by the fetus has important, but not absolute influence over fetal growth. Pancreatectomy in late gestation fetal sheep limits fetal growth rate by 40% to 50% ((62))((63)). Human pancreatic agenesis ((64))((65)) ((66))((67)) produces severely growth-restricted fetuses who weigh 30% to 50% less than normal near term. Mice that have a mutation in the insulin gene are small at birth ((68)). Insulin infusions into the fetus and excessive fetal insulin secretion enhance fetal glucose utilization and produce increased adiposity, but only a 10% to 15% increase in fetal nonfat growth. Such hyperinsulinemic conditions also limit protein breakdown, leading to increased protein accretion. The dominant regulator of fetal protein balance, however, is fetal plasma concentrations of amino acids. Insulin, by limiting protein breakdown, actually decreases these plasma concentrations, thereby limiting protein synthesis. It is not clear, therefore, how increased fetal insulin enhances or its deficiency limits protein accretion. The primary action of insulin may be to promote glucose and lipid utilization and, in turn, enhance protein accretion by providing more energy substrate to fuel protein synthesis and to substitute for amino acids that fuel oxidative metabolism. For example, infusion of insulin into fetal sheep together with glucose and amino acids to maintain normal glucose and amino acid concentrations leads to an increase of about 30% in fetal amino acid utilization over 2 to 3 hours ((69)). Longer studies are needed to determine what adaptive metabolic mechanisms develop to modulate or even prevent such large potential increases in fetal protein accretion and fetal growth. Furthermore, removal of insulin from the fetus increases fetal glucose concentration and the transfer of glucose from mother to fetus via the placenta, which reduces net fetal carbon accretion ((70)). Similar reductions in fetal growth, as occur with fetal insulin deficiency, result when glucose supply is restricted by chronic sustained or repetitive maternal hypoglycemia. Insulin also may enhance production of IGF-I by promoting intracellular nutrient availability.
Fetal growth results from interactions among maternal, placental, and fetal factors and a mix of environmental influences through which the fetal genotype is expressed and modulated. The single most important environmental influence is fetal nutrition, and its principal determinant is the size and nutrient transport capacity of the placenta. Placental size is determined by the size of the uterus and, thus, the size of the mother. Increased nutrient supply to the fetus subsequently increases fetal tissue and plasma concentrations of anabolic hormones and growth factors. Together, the variable supply of nutrients, anabolic hormones, and growth factors in the fetus modulate the expression and/or action of growth-promoting genes and their gene products, leading to variation in fetal growth. Deficiencies in any one of these factors can limit fetal growth and produce intrauterine growth restriction.
|