
Guillaume Duchenne 1806-1875
Unter
einer Muskeldystrophie versteht man eine erblich bedingte Muskelerkrankung,
die zu einem fortschreitenden Schwund des Muskelgewebes führt und mit einer
dramatischen Erhöhung der Serum-Creatinphosphokinase (CPK) verknüpft ist.
Kennzeichnend für Patienten mit einer Muskeldystrophie ist eine
zunehmende, bestimmte Körperregionen bevorzugende, meist symmetrisch
ausgebildete Muskelschwäche. Es sind mehr als 30 verschiedene Formen der
Muskeldystrophie bekannt. Die einzelnen Formen der Muskeldystrophie
unterscheiden sich hinsichtlich des Vererbungsmodus, der bevorzugten
Körperregionen, dem Erkrankungsalter und dem Verlauf. Einige Beispiele sind
in der folgenden Tabelle aufgeführt:
|
Muskeldystrophie |
Vererbung |
Bevorzugte Region |
Alter |
|
Typ Duchenne |
x-chromosomal-rezessiv |
Becken, Oberschenkel |
3.-5. Lebensjahr |
|
Typ
Becker-Kiener |
x-chromosomal-rezessiv |
Becken,
Oberschenkel |
6.-16.
Lebensjahr |
|
Typ
Leyden-Möbius |
Autosomal-rezessiv |
Schulter-,
Beckengürtel |
2.-20.
Lebensjahr |
|
Typ
Erb |
Autosomal-dominant |
Gesicht,
Schultergürtel |
7.-25.
Lebensjahr |
Die
Muskeldystrophie Duchenne ist weltweit eine der häufigsten monogen bedingten
Erkrankungen bei Jungen und Männern. Sie tritt bei einem von 3200 Knaben auf.
Die Becker-Kiener Form ist seltener. Die Krankheit ist gekennzeichnet durch
den Muskelschwund, der an den unteren Extremitäten beginnt. Die
Muskeldystrophie vom Typ Duchenne zeigt einen nahezu gesetzmäßigen Verlauf.
Im 3.-5. Lebensjahr fällt bei den Patienten eine leichte Muskelschwäche der
Beine auf, die zu häufigem Stolpern und Fallen führt. Im weiteren Verlauf
ist das Treppensteigen nur mit Zuhilfenahme eines Geländers möglich. Die
Muskelschwäche der Becken- und Oberschenkelmuskulatur verursacht einen
watschelnden Gang sowie ein erschwertes Aufstehen aus dem Sitzen oder Liegen.
Die Kinder klettern an sich selbst hoch bzw. gebrauchen Wände und Möbel zum
Abstützen. Schon im 5.-7. Lebensjahr können Treppensteigen und Aufstehen aus
dem Sitzen oder Liegen nur noch mit Hilfe durch andere möglich sein, da die
Erkrankung auch auf die Muskulatur der Schulter und Arme übergreift. Zwischen
dem 7. und 12. Lebensjahr ist ein Anheben der Arme in die Horizontale oft
schon nicht mehr möglich. Viele Kinder sind in diesem Alter bereits auf den
Rollstuhl angewiesen, können sich aber noch eingeschränkt selbständig
versorgen. Meist besteht ab dem 18. Lebensjahr vollständige
Pflegebedürftigkeit. Infolge des Muskelschwundes kommt es zu Fehlstellungen
von Gelenken sowie zu Verformungen von Knochen. Charakteristisch für Kinder
mit Muskeldystrophie vom Typ Duchenne sind die sogenannten Gnomen- oder
Kugelwaden. Sie entstehen indem das zugrunde gehende Muskelgewebe durch Fett-
und Bindegewebe ersetzt wird. Typisch für einen Muskelschwund der Rumpf- und
Schultermuskulatur ist die Ausbildung der Scapulae alatae. Darunter versteht
man das flügelartige Abstehen der Schulterblätter vom Rumpf. Letztendlich
werden auch die Atemmuskulatur und der Herzmuskel von der Erkrankung befallen.
Die Muskeldystrophie von Typ Duchenne verläuft immer tödlich. Die Patienten
erreichen ein Alter zwischen 20 und 25 Jahren und versterben fast immer an
Herzversagen oder Ersticken. Die geistige Entwicklung und die Intelligenz der
Kinder sind bis auf einige Ausnahmen nicht eingeschränkt. Bei anderen Formen
der Muskeldystrophie, die einen langsameren und weniger schweren Verlauf
haben, muss die Lebenserwartung nicht eingeschränkt sein.
Die
Erkrankung wird X-chromosomal rezessiv vererbt, d.h., Jungen und Männer mit
einem mutierten Gen erkranken, während Frauen, die ein mutiertes Gen
aufweisen, gesunde Überträgerinnen sind. Sie geben die Erkrankung
statistisch an die Hälfte ihrer Söhne weiter. Sehr selten zeigen auch
Mädchen Krankheitssymptome. Das Muskeldystrophie-Gen (DMD-Gen) liegt auf dem
kurzen Arm des X-Chromosoms und wurde 1987 identifiziert. Es ist mit einer
Länge von 2,4 Millionen Basenpaaren und 79 Exons das bisher größte bekannte
menschliche Gen und kodiert für das muskelspezifische (stabilisierende)
Protein Dystrophin. Die Mutationsrate ist sehr hoch (etwa 1/10 000), bei einem
Drittel der Patienten wird die Erkrankung durch eine Neumutation verursacht.
Bei etwa 60 % der Betroffenen kann eine Deletion, bei etwa 6 % eine
Duplikation eines Teils des Gens nachgewiesen werden. Die übrigen 34 % der
Erkrankungen resultieren aus Punktmutationen, die wegen der Größe des Gens
bisher nicht routinemäßig nachgewiesen werden können. In den meisten
Familien kann dennoch durch eine Haplotypanalyse das Überträgerinnenrisiko
der Ratsuchenden bestimmt werden.
Forscher haben das Dystrophin 1987 entdeckt. Jungen mit Muskeldystrophie vom Typ Duchenne - etwa einer von 3500 neugeborenen Jungen hat die X-chromosomal-rezessiv vererbte, rasch progressiv verlaufende Muskeldystrophie - können kein Dystrophin bilden, weil sie einen Schaden am Duchenne-Gen auf dem kurzen Arm des X-Chromosoms haben. Dystrophin ist ein Eiweiß, das ein Netz in den Muskelfasermembranen bildet. Fehlt es, werden die Membranen durchlässiger als bei gesunden Menschen. Enzyme wie die Kreatinkinase strömen vermehrt aus der Zelle aus. Deshalb haben die Kranken im Serum 10- bis 40fach erhöhte Spiegel dieses Enzyms. Infolge des gestörten Zellstoffwechsels sterben die Muskelzellen ab, zuerst im Bereich des Beckengürtels. Wenn die Krankheit bei den Kindern deutlich erkennbar wird, meist ist das zwischen dem zweiten und vierten Lebensjahr, sind bereits 40 Prozent der Muskelfasern zerstört oder in der Funktion deutlich verringert.
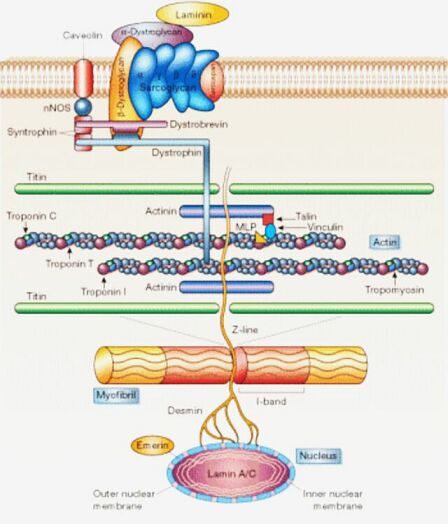
Die
Diagnose kann durch DNA-Analyse oder anhand einer Muskelbiopsie mit
immunchemischem Dystrophin-Nachweis (Histochemie oder Western-Blot Analyse)
gestellt werden. Anhand der feingeweblichen Untersuchung einer Gewebeprobe
kann die Zerstörung der einzelnen Muskelfasern sowie der Ersatz des
Muskelgewebes durch Fett- oder Bindegewebe nachgewiesen werden. Die
Muskeldystrophie Duchenne ist klinisch nicht immer hinreichend sicher von
anderen autosomal rezessiven Muskeldystrophien zu unterscheiden. Daher ist der
immunchemische Dystrophin-Test in der Regel bei sporadischen Fällen
angezeigt, wenn keine Mutation im Dystrophin-Gen gefunden wurde und auch bei
den weiblichen Familienangehörigen keine erhöhten CPK-Werte gemessen wurden,
aus denen eine X-chromosomale Vererbung ersichtlich ist.
Einige
für die Muskulatur charakteristische Enzyme sind häufig schon vor dem
Auftreten der ersten Symptome der Erkrankung im Blut erhöht. Dazu gehören
die Kreatin-Kinase (CK) und die Lactat-Dehydrogenase (LDH). Beide Enzyme
kommen in unterschiedlichen Formen in den Zellen der Skelettmuskulatur und im
Herzmuskel vor. Bei einer Zerstörung der Muskelzellen durch die
Muskeldystrophie werden diese Enzyme freigesetzt und sind dann im Blut
nachweisbar. Bei der Muskeldystrophie vom Typ Duchenne können bereits im
Nabelschnurblut stark erhöhte Werte der Kreatin-Kinase nachgewiesen werden.
Ein
weiterer typischer Laborbefund ist ein verminderter Wert des Kreatinins im
Harn, wohingegen das Kreatin erhöht ist. Kreatin ist im menschlichen
Organismus vorwiegend in Muskelzellen enthalten und wird dort in Kreatinin
umgewandelt, das über die Nieren ausgeschieden wird.
Durch
beide Verfahren werden die Aktionspotentiale der Skelettmuskulatur (EMG) bzw.
der Herzmuskulatur (EKG) abgeleitet. Unter einem Aktionspotential versteht man
eine schnelle elektrische Spannungsschwankung, die durch eine Änderung des
Erregungszustandes einer Zelle, z.B. durch einen Nervenreiz oder einen
elektrischen Reiz, ausgelöst wird. Diese Spannungsschwankungen werden
graphisch als Kurvenbild aufgezeichnet und zeigen je nach Krankheitsbild
charakteristische Veränderungen. Bei Patienten mit Muskeldystrophie sind z.B.
die Dauer und die Amplitude der einzelnen Aktionspotentiale im EMG deutlich
vermindert.
Es gibt bisher keine kausale, das heißt gegen die Ursache gerichtete, Therapie der Muskeldystrophie. Die therapeutische Betreuung umfasst die Motivation des Patienten zur Bewegung, die Verhinderung von Kontrakturen und die möglichst lange Erhaltung guter Atemfunktionen durch gezielte Atemtherapie.
Die Behandlung sollte assistive, dynamische und isometrische Komponenten umfassen, dabei ist immer zu berücksichtigen, dass die Muskulatur sehr dehnungsempfindlich ist. Bewährt hat sich auch die Behandlung im Bewegungsbad. Für den Fall, daß ein Schlingentisch zur Verfügung steht, sollte dieser ebenfalls eingesetzt werden. Das regelmäßige Stehen in entsprechenden Geräten (Heidelberger Stehständer, Stehbrett) ist unerläßlich und fördert Stoffwechselfunktionen, wirkt kreislaufregulierend, begünstigt die Atmung und hat auch kontrakturprophylaktische Bedeutung. Hierbei sind aber in jedem Falle Zeitlimits einzuhalten, um organische Funktionen (z.B. Nieren, Verdauungsorgane) nicht überzustrapazieren, insbesondere auch das schwerkraftbedingte Absacken des Blutes bei nicht vorhandener Muskelpumpe.
Eine sich zwangsläufig entwickelnde Skoliose kann u.U. chirurgisch korrigiert werden.
Durch eine medikamentöse Behandlung
kann der Krankheitsverlauf zwar nicht aufgehalten aber verzögert werden. Zum
Einsatz kommen Ribonukleinsäuren, die die Muskulatur stabilisieren und den
Schwund der Muskulatur verlangsamen.
Das Ziel jeglicher Therapie besteht darin, die Lebensqualität der Patienten solange wie möglich zu erhalten. Dazu gehören selbstverständlich auch ausführliche Gespräche mit den Eltern und den Patienten selber über die Erkrankung. Darüberhinaus existieren vielerorts Selbsthilfegruppen.
Eine ausführliche Betrachtung zu Therapieformen, Hilfsmitteln, Operationen und Studien zur medikamentösen Behandlung findet sich auf einer gesonderten Seite und im Linkverzeichnis zum Thema.
zurück
zur Seite ![]() Krankheitsbilder,
Neuromuskuläre Erkrankungen
Krankheitsbilder,
Neuromuskuläre Erkrankungen