Abstract- RNA interference is
the phenomenon of inhibition of gene expression by the binding of short 21 to 23
nt long siRNA molecules to cognate mRNA and target their destruction. This
technology can be utilized as a potential antiviral therapy. Bioinformatics can
be used to identify conserved sequences of key viral proteins and to design
siRNA targets against these regions. Our approach gives a sequential approach
in designing siRNA targets using bioinformatics as a tool. As an example, HIV-1
is taken against which potential target sequences have been identified. Finally
these are validated by comparing them with human expressed genes to confirm
that these siRNAs are incapable of inactivating human genes.
1 Introduction
RNA interference is the phenomenon of silencing a gene by preventing its expression. It was first discovered by Andrew fire while working on Caenorhabditis elegans. [1]. When a double stranded RNA is introduced into a cell it is recognized by the DICER enzyme and is cleaved to give 21 to 23 nucleotide long double stranded RNA strands with 2 nucleotide overhangs on both the strands. These are called as small interfering RNA (siRNA).[2] These RNA combine with certain proteins to form a riboprotein complex called as RNA Induced Silencing Complex (RISC). This complex recognizes the mRNA complementary to the siRNA and bind to it. The formation of this complex prevents the translocation of the ribosome and prevents gene expression. Further the siRNA bound to mRNA is either cut by DICER to give more siRNA or serves as a template for RNA dependent RNA polymerase which elongates it using mRNA as template to produce dsRNA that is further cut by DICER to create more siRNA. This starts a cascade that leads to total silencing of the gene. Some research has shown that these siRNAs further cause rearrangement of the chromosome preventing the expression of the gene itself and hence is also called as Post Transcriptional Gene Silencing (PTGS)[3,6,7]. This mechanism is part of an inherent antiviral immune response that recognizes double stranded RNA is foreign to the cell and tries to eliminate it and its source[4]. Researches have revealed that introduction of double stranded RNA is sufficient to cause gene silencing as it is processed by host systems to siRNA which are further extended by RNA polymerase to form more dsRNA[7].
This phenomenon termed RNAi has
shown good potential in antiviral therapy in-vitro. By transducing double
stranded RNA of key viral genes derived from cDNA libraries [11] silencing of
the corresponding gene has been shown for AIDS [8,10,12,15], Hepatitis and even
SARS viruses [16]. But the inherent drawback of this technology in antiviral
therapy is that viruses undergo rapid mutations in their genes to evade the
host systems. Viruses that undergo point mutations in their genomes are not
susceptible to RNAi [17]. Also introduced dsRNA are cut randomly into 21
nucleotide long siRNA which might span a region of low homology across various
HIV-1 strains. Moreover these random siRNA if homologous to any of the host
genes might result in silencing of host genes. Hence the requirement is that
the RNAi mechanism in vivo should be introduced by using target specific siRNAs
and not dsRNA. The siRNAs that bind to mRNA which were previously thought to
act as primers for RNA polymerase elongation to produce dsRNA have been proved
to act as target guides only and are incapable of elongation by RNA polymerase
[18]. Thus the chances of dsRNA and hence more random siRNA being produced from
our siRNA target is remote.
We have demonstrated an approach
for the design of siRNA targets by using conventional techniques in bioinformatics.
The steps used by us are firstly, the peptide sequences of the particular viral
protein is collected by using the BLAST program and sequences are collected
[19]. The sequences are aligned using programs that allow multiple sequence
alignment like clustal[20]. From the aligned sequences the conserved domains
are selected and are backtracked with the nucleotide sequence of the gene to
get the codons. Now, the coding sequence of this consensus sequence is selected
and complementary strand can be generated. The final output will be a 21 – 23
nucleotide long siRNA target region. Lastly, the sense and antisense strands of
this target is used to run a BLAST search against human EST and those targets
that show no or least homology/similarity are chosen as good targets. These can
be safely used in-vivo
as potential antiviral RNAi therapy. The fundamental assumption made here is
that these conserved sequenses are least prone to mutations and if the virus
attempts a mutation across this sequence it will be lethal to it. Thus a
combination of RNAi target sequences can be used in-vivo as antiviral therapy.
In this article we have taken the example of Human immunodeficiency virus 1 to
which we have generated RNAi targets and validated by matching for homology
with human ESTs.
2 Targeting HIV-1
– An example
2.1 The HIV genome and its genes
The Human immunodeficiency virus 1 is a 9181 base pair long retrovirus that has a linear single stranded RNA genome. The HIV-1 genome is divided into 9 Open Reading Frames ORFs also called loci termed from HIV1gp1-9. These are Gag-pol (HIV1gp1), Gag (HIV1gp2), vif (HIV1gp3), vpr (HIV1gp4), tat (HIV1gp5), rev (HIV1gp6), vpu (HIV1gp7), env (HIV1gp8) and nef (HIV1gp9). The Gag-pol gene codes for the protease, reverse transcriptase and the integrase proteins. The gag gene codes for the capsid, matrix, p2, nucleocapsid, p1 and the p6 proteins. The vif gene codes for p23 viral infectivity factor which is a viral accessory protein important for virus replication in vivo. The vpr gene codes for the viral protein R, a viral accessory protein important for virus replication in vivo and viral replication. The tat gene codes for p14 transcriptional activator, a viral regulatory protein required for virus replication, interacts with transcription factors and associated with pathogenicity of the virus. The rev gene codes for p19 - a regulator of expression of virion proteins, prevents splicing of viral RNA, shuttles unspliced viral RNA to the cytoplasm for expression of viral proteins and incorporation of full length viral genomic RNA into virions. The vpu gene codes for p16 viral protein U which is a viral accessory protein important for virus replication in vivo; promotes degradation of CD4 and down-regulates cell surface expression of MHC class I proteins; helps mediate efficient virus particle release from infected cells; reported to induce apoptosis and attenuates the level of Env precursor(gp160) biosynthesis. The env gene coded for envelope glycoproteins gp120 and gp41 that are synthesized from a common gp160 precursor. The nef gene codes for p27 negative factor; a viral accessory protein important for virus replication in vivo; determinant of HIV-1 pathogenesis; down-regulates cell surface CD4 and MHC class I molecules; enhances virus infectivity through interactions with multiple cellular signaling proteins. There are 15 proteins mde all together. The Gag-pol gene is translated as a polyprotein which is later cleaved by the viral protease to individual peptides. The Env gene is translated to give gp160 precursor that is cleaved by a host encoded protease present in the golgi body to give gp120 and gp41 proteins. A point worth noting here is that integration is necessary for transcription of viral genes and transcription of viral DNA to ssRNA that are packed into virions. The transcription of the integrated sequence gives full length mRNA whose length is the size of the HIV genome.
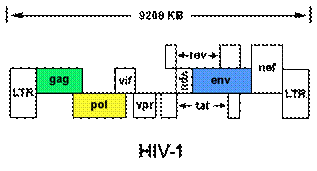
Fig 1: Pictorial representation of the HIV-1 genome
2.2 The approach
In the approach used by us, the sequences of the individual
peptides were obtained by BLAST search for nonredundant sequences of GENBANK
and SwissProt through the
Fig 2. Important genes of HIV-1
2.3 Integrase
The integrase gene belongs to the HIV1gp1 locus and encodes the integrase enzyme that is involved in the integration of the double stranded HIV genome into the host genome. The results show that a stretch of aminoacids with the sequence YNPQSQG is conserved across all strains of HIV-1. Thus this stretch is most likely to be present in a region important for the function of the protein. The codons coding for this region is determined from the nucleotide sequence of integrase gene. The codons for sequence “ PYNPQSQG” is
5’ – CCGTATAACCCGCAGAGCCAGGGC – 3’
So the siRNA should be,
5’ – UAUAACCCGCAGAGCCAGGGC – 3’
| | | | | | | | | | | | | | | | | | |
3’ – GCAUAUUGGGCGUCUCGGUCC – 5’
An important fact worth noting here is that this region shows significant homology across HIV-1, HIV-2, Simian immunodeficiency virus SIV. When Feline immunodeficiency virus FIV is also included in the MSA the region NPQSQ is conserved. The results are shown in fig 3.
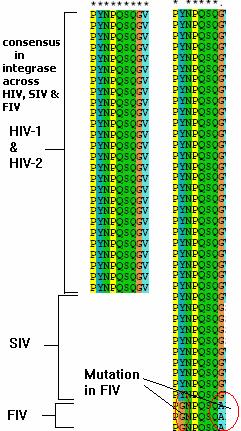
Fig3. The MSA shows that NPQSQ domain is highly conserved across HIV-1 & 2, SIV and FIV and likely to play an important role in the protein function.
2.4 Reverse Transcriptase
The reverse transcriptase gene belongs to the HIV1gp1 locus
and codes for the reverse transcriptase protein. The function of this protein
is to convert the single stranded RNA of the virus into double stranded DNA to
facilitate its integration by the integrase enzyme. The enzyme is encoded as
p66 subunit that includes the RNAseH domain. Without this domain it is called
as p55 subunit. Multiple sequence alignment of their sequences gave mutable
regions but overall the region was conserved across many strains. The sequences
are “QKLVGKLNW”, “QWTYQIYQ” and “TWIPEWEF”
show conserved patterns and are amenable for targeting. The nucleotide sequence
for these regions are ”CAGAAGTTAGTGGGGAAATTGAATT”, “CAATGGACATATCAAATTTATCAAGA”
and “ACCTGGATTCCTGAGTGGGAGTTTG”
The targets are shown below,
5’ – GAAGUUAGUGGGGAAAUUGAA – 3’
| | | | | | | | | | | | | | | | | | |
3’ – GUCUUCAAUCACCCC UUUAAC – 5’
5’ – AUGGACAUAUCAAAUUUAUCA – 3’
| | | | | | | | | | | | | | | | | | |
3’ – GUUACCUGUAUAGUUUAAAUA – 5’
5’ – CUGGAUUCC UGAGUGGGAGUU – 3’
| | | | | | | | | | | | | | | | | | |
3’ – UGGACCUAAGGACUCACCC UC – 5’
2.5 Protease
The protease enzyme does the important function of cleaving the individual peptides from the nascent peptide translation product of HIV mRNA. It is coded by the protease gene located in the HIV1gp1 locus. It should be pointed out that the mRNA obtained by transcription of the integrated HIV genome is polycistronic. Multiple sequence alignment was done and a well conserved region was obtained. The sequence is “IGGIGGFI” from this sequence the target sequence is derived and I given below,
5’ – AGGGGGAAUUGGAGGUUUUAU – 3’
| | | | | | | | | | | | | | | | | | |
3’ - UAUCCC CCUUAACCTCCAAAA – 5’
MSA not shown here due to space constraints. Included at the end. (fig 4)
2.6 Rev gene
The rev gene codes for the p19 protein which is a regulator of expression of virion proteins, prevents splicing of viral RNA, shuttles unspliced viral RNA to the cytoplasm for expression of viral proteins and incorporation of full length viral genomic RNA into virions. The nucleus has inherent mechanisms to prevent unspliced mRNA from leaving the nucleus. This mechanism is taken care of by the rev protein. The rev gene is also considered as a potential RNAi target [13]. We have identified two region that is conserved given by the sequence “QARRNRRRRWR”. This might serve as good RNAi targets. The siRNA can have the following sequence,
5’ – GGCCCGAAGGAAUAGAAGAAG – 3’
| | | | | | | | | | | | | | | | | | |
3’ – GUCCGGGCUUCC UUAUCUUCU – 5’
The MSA picture is shown below,

Fig4. MSA of the rev protein.
2.7 Nef gene
The nef gene is present in the HIV1gp9 locus and codes for the p27 negative factor which is a viral accessory protein that is important for virus replication in vivo. It determines HIV-1 pathogenesis, down-regulates cell surface CD4 and MHC class I molecules, enhances virus infectivity through interactions with multiple cellular signaling proteins. Multiple sequence analysis of this peptide sequence gave two conserved regions “FLKEKGGL” and “PQVPLRPMT”. The siRNA sequences for these targets are respectively,
5’ – UUUAAAAGAAAAGGGGGGAC UG – 3’
| | | | | | | | | | | | | | | | | | | |
3’ – AAAAAUUUU CUUUUCCCCCC UG – 5’
and
5’ – UCAGGUACCU UUAAGACCAAU GA – 3’
| | | | | | | | | | | | | | | | | | | | |
3’ - GGAGUCCAUGGAAAUUCUGGUUA – 5’
The MSA analysis is shown below,

Fig6. MSA of the nef protein.
2.8 Env gene
The env gene is present in the HIV1gp8 locus and codes for the glycoprotein precursor gp160 that is further cleaved to give gp120 and gp41 proteins. These proteins are components of the envelope of HIV and have been identified as potential targets for many antiviral therapies. Multiple sequence alignment analysis identified about 9 regions that showed considerable potential for targeting. The sequences are "WVTVYYGVPVW", "TTLFCASDAK", "LKPCVKLTPLC", "VSTVQCTHGI", "VSTVQCTHGIRP", "GLLLTRDGG", "ELYKYKVV", "FLGFLGAAGSTM", "LTVWGIKQLQAR" . Out of these two regions showing considerable homology were selected which were “VSTVQCTHGI” and “ELYKYKVV”. siRNA target sequences are shown below,
5’ – GUCAGCACAGUACAAUGUACA – 3’
| | | | | | | | | | | | | | | | | | |
3’ – GUCGUGUCAUGUUACAUGUGU – 5’
and
5’ – GAAUUAUAUAAAUAUAAAGUA – 3’
| | | | | | | | | | | | | | | | | | |
3’ – UAAUAUAUUUAUAUUUCAUCA – 5’
The MSA analysis is given below,

Fig 8. MSA analysis of the env protein.
3 Validation of targets
The last step in the approach outlined is the validation of these targets. The target sequences designed should not be homologous to human genes as there is a potential danger of inactivating human genes. This is confirmed by checking the sense as well as the antisense strands of the target and run a blast search against human expressed sequences to filter out those targets that show homology. For example validation of target for the integrase gene was done and the results are shown. The match showing highest homology was 16/21 and 15/21 for the sense and antisense strands respectively for the human genome database and 16/21 and 16/21 for the human expressed sequence database. This length is too short to create stable mRNA-siRNA hybrid and incapable of targeting and silencing human genes.
The sense and antisense strands of the putative siRNA target is subjected to BLAST analysis against human genome and human expressed sequence tag EST database
And the following results were obtained.
3.1 BLAST analysis against Human Genome database
Sense strand:
Homo sapiens chromosome 2 clone bac91a19 map 2p13 - 16 identities
Query: 6 aacccgcagagccagg 21 ||||||||||||||||Sbjct: 7271 aacccgcagagccagg 7286 Antisense strand: Homo sapiens chromosome 15, clone RP11-1001M11 - 15 identities Query: 7 ggcgtctcggtcccg 21 |||||||||||||||Sbjct: 200285 ggcgtctcggtcccg 200299 3.2 BLAST analysis against Human EST database
Sense strand:
56023117J1 FLP Homo sapiens cDNA 16 identities
Query: 6 aacccgcagagccagg 21 ||||||||||||||||Sbjct: 521 aacccgcagagccagg 536
Antisense strand: AGENCOURT_10545077 NIH_MGC_107 Homo sapiens cDNA clone 16 identities Query: 6 gggcgtctcggtcccg 21 ||||||||||||||||Sbjct: 424 gggcgtctcggtcccg 409
Thus we can conclude that the homology with human genes is only 16/21 = 76% and hence can be used safely as an RNAi target.
4 Conclusions
Thus, we believe that Bioinformatics can play a vital role in designing target specific siRNAs for RNA interference that can be used safely without any possibility of silencing of human genes. The work highlighted above outlines the series of steps to be followed to design validate RNA interference targets. Further work should be done in the laboratory by designing custom tailor made siRNA targets either by chemical synthesis or by reverse transcription from cDNA libraries, transfecting them inside the cell by using lipofection, electroporation or viral mediated transfection. Then the efficiency of gene silencing can be evaluated using suitable assays like following the specific protein expression profile of the target protein. The phenomenon of RNA interference can later be extended safely to in-vivo testing to evaluate the efficiency of therapy. Antiviral therapies can also use a combination of siRNAs that target key viral proteins so that there is a synergistic result.
Acknowledgments
We thank Dr. Sharmila Anishetty and Dr. Geetha Muthukumaran for their invaluable suggestions, our colleague Mr. Pradeep Kota for helping us out with hardcore programming approaches. We would like to dedicate this article to our mentor and guide Dr. P. Gautam.
Bibliography
1. Fire,
A. et al. Potent and specific genetic interference by double-stranded
RNA in Caenorhabditis elegans. Nature 391, 806–811 (1998).
The First paper that highlighted the
phenomenon of RNAi
2.
Zamore, P. D., Tuschl, T., Sharp, P. A. & Bartel, D. P. RNAi:
double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23
nucleotide intervals. Cell 101,
25–33 (2000).
The
mechanism was outlined and it was first shown that siRNA produced by processing
longer dsRNA direct mRNA cutting
3.
Sharp, P. A. RNA interference—2001. Genes Dev. 15, 485–490 (2001).
Review article that gives information on
RNAi
4. Waterhouse, P. M.,
Wang, M. B. & Lough, T. Gene silencing as an adaptive defence against
viruses. Nature 411, 834–842 (2001).
5.
Elbashir, S. M. et al. Duplexes of 21-nucleotide RNAs mediate RNA
interference in cultured mammalian cells. Nature 411, 494–498
(2001).
Breakthrough
paper that describes in-vitro silencing of genes by siRNA.
6. McManus, M.T, Sharp, P.A.
Gene silencing in mammals by small interfering RNAs. Nat Rev Genet. 2002 Oct;3(10):737-47.
7. Hammond, S. M., Caudy, A.
A., Hannon, G. J. Post-transcriptional gene silencing by double-stranded RNA.
Nat Rev Genet. 2001 Feb;2(2):110-9.
8. Michael T.McManus, Phillip A. Sharp. Gene
silencing in mammals by small interfering RNAs. Nature
Reviews Genetics, Vol3, 737 – 747(2002)
9. Park WS, Miyano-Kurosaki
N, Nakajima E, Takaku H. Specific inhibition of HIV-1 gene expression by double-stranded
RNA. Nucleic Acids Res Suppl.
2001;(1):219-220.
10. Park WS, Miyano-Kurosaki
N, Hayafune M, Nakajima E, Matsuzaki T, Shimada F, Takaku H. Prevention of
HIV-1 infection in human peripheral blood mononuclear cells by specific RNA
interference. Nucleic Acids Res. 2002
Nov 15;30(22):4830-5.
11. Shirane D, Sugao K,
Namiki S, Tanabe M, Iino M, Hirose K. Enzymatic production of RNAi libraries
from cDNAs. Nat Genet. 2004 Feb;36(2):190-6. Epub 2004 Jan 04.
12. Novina, C. D. et al. siRNA-directed inhibition of HIV-1 infection. Nature Med. 8, 681–686 (2002).
13. Lee, N. S. et al.
Expression of small interfering RNAs targeted against HIV-1 rev transcripts
in human cells. Nature Biotechnol. 20, 500–505 (2002).
14.
Park WS, Hayafune M, Miyano-Kurosaki N,
Takaku H. Specific HIV-1 env gene silencing by small interfering RNAs in human
peripheral blood mononuclear cells.
15. Coburn, G. A. &
Cullen, B. R. Potent and specific inhibition of HIV-1 replication using RNA
interference. J. Virol. 76,9225–9231 (2002).
16. He ML, Zheng
B, Peng Y, Peiris JS, Poon LL, Yuen KY, Lin MC, Kung HF, Guan Y. Inhibition of
SARS-associated coronavirus infection and replication by RNA interference.
JAMA. 2003 Nov 26;290(20):2665-6.
17. Boden D, Pusch O, Lee F, Tucker L, Ramratnam B. Human immunodeficiency virus type 1 escape from RNA interference. J Virol. 2003 Nov;77(21):11531-5. Viruses escape RNAi by harboring point mutations.
18. Schwarz DS, Hutvagner G,
Haley B, Zamore PD. Evidence that siRNAs function as guides, not primers, in
the Drosophila and human RNAi pathways.
Mol Cell. 2002 Sep;10(3):537-48.
Describes that siRNAs act as guides and do not prime
dsRNA synthesis.
19.
Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schäffer, Jinghui Zhang,
Zheng Zhang, Webb Miller, and David J. Lipman (1997), Gapped BLAST and
PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402.
Article that describes the Protein blast program.
20. Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Research, submitted, June 1994.
Article that describes the multiple sequence alignment program ClustalW
Database:
www.ncbi.nlm.nih.gov

Fig4. MSA profile of the
protease enzyme.
NOTE: This paper won the prize
at the IIT
