La electroforesis en gradiente desnaturalizante (DGGE) permite una rápida visualización de cambios de una sola base en fragmentos de DNA. La técnica se basa en la
migración de moléculas de DNA a través de geles de poliacrilamida que contiene concentraciones crecientes de agente desnaturalizante.
Cuando se combina con PCR, la DGGE es uno de los métodos elegidos para la detección de mutaciones puntuales en DNA. En este capítulo se discute tanto la teoría de la
DGGE como otros aspectos para establecer la técnica. También se presentan ejemplos de aplicaciones y varias modificaciones del protocolo original.
9.2 Consideraciones teóricas.
La electroforesis en gradiente desnaturalizante permite la separación de fragmentos de DNA que difieren en un solo nucleótido.
El método implica hacer una electroforesis de moléculas de DNA de doble cadena utilizando un incremento lineal de la concentración de agentes desnaturalizantes como formamida
y urea. También han sido empleados gradientes de temperatura de forma satisfactória. La separación de fragmentos de DNA por DGGE se basa en las propiedades de fusión
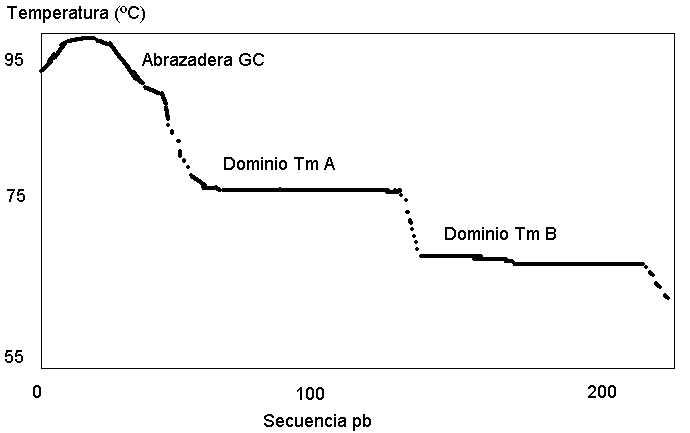
del DNA de dos cadenas: Una molécula de DNA desnaturalizará en regiones discretas (que varian en longitud desde 25 a 80 pb hasta más de 300) llamadas dominios de
fusión. Cada dominio desnaturaliza a una temperatura dada (temperatura de fusión o Tm) aquí definida como la temperatura a la que cada par de bases de DNA dúplex
está en un equilibrio 50 a 50 entre el estado helicoidal y el desnaturalizado. Debido a que interacciones amplias entre bases adyacentes tienen una influencia significativa en la
estabilidad de la doble hélice, la Tm de un DNA dado es enormemente dependiente de la secuencia de nucleótidos que se desplaza a través de un gel en gradiente
desnaturalizante, alcanzará un punto donde la concentración de agente desnaturalizante iguale a la Tm de su dominio de menortemperatura de fusión, causando una
desnaturalización parcial (ramificación) y consecuentemente un marcado retraso en su movilidad electroforética. Los fragmentos de DNA que difieren incluso en un único
nucleótido en su dominio de fusión de menor Tm experimentarán ramificaciones a diferente distancia a lo largo del gel y entonces podrán distinguirse.
La DGGE es extremadamente eficaz cuando se usa para el análisis de variantes nucleotídicas en heterocigosis. La continua desnaturalización y reanillamiento de cadenas de DNA
durante los últimos ciclos de la reacción en cadena de la polimerasa (PCR) permite tanto la formación de moléculas de DNA homodúplex como heterodúplex.
La presencia de apareamientos incorrectos en los heterodúplex reduce considerablemente su temperatura de fusión permitiendo una mejor separación y una fácil
detección visual de las mutaciones.
9.2.1 La introducción a la GC-CLAMP
Mapa de fusión del fragmento anterior
9.2.2 Predicciones computerizadas
Introducción de un dominio rico en GC.
9.3 Implementación
9.3.1 Simulación por ordenador y elección de cebadores.
Si está disponible la secuencia completa de los fragmentos de DNA, el comportamiento de fusión de la molécula de DNA puede ser predicho con los programas antes
mencionados: MELT87 y SQHTX. Los Laboratorios BioRad venden un paquete de software para Macintosh Ó (MacMelt). Este programa permite la
identificación de los diferentes dominios de fusión en la molécula de DNA y su Tm específica. Los cebadores de PCR pueden ser diseñados basándose en la
información proporcionada por el ordenador. De esta forma podemos maximizar la efectividad del análisis y evitar los problemas previsibles. Preferiblemente, suelen elegirse
fragmentos de unas 50 a 100 pb comparando uno ó 2 dominios de fusión; 3 ó más dominios de fusión se evitarán puesto que esto normalmente produce un
descenso en la sensibilidad de detección, especialmente dentro de los dominios de fusión con mayor Tm. Deben evitarse diferencias significativas entre 2 dominios de fusión,
puesto que la ramificación del dominio de fusión de menor temperatura debería causar un retraso electroforético de la molécula de tal magnitud que no
alcanzará el punto donde el segundo dominio de fusión se desnaturalize también.
Si esto se consigue evitar, se puede analizar el fragmento en 2 gradientes diferentes o los productos de PCR digerir con una enzima de restricción y ser analizados separadamente.
La presencia de la abrazadera GC en cualquier lado del fragmento de DNA también puede tener profundos efectos en el mapa de fusión y el
porcentaje de cambios detectables. La simulación por ordenador ayudará en el proceso de toma de decisiones y prevendrá localizaciones erróneas del cebador de la
abrazadera GC. La secuencia de la abrazadera CG descrita por Sheffield et al. serviría como un dominio de alta Tm adecuado en muchos de los casos. Sin embargo, si están siendo
analizadas las secuencias ricas en GC(inusuales), se pueden diseñar y evaluar por ordenador secuencias abrazadera largas y/o ligeramente modificadas.
9.3.2 DGGE perpendicular
Si no está disponible la secuencia nucleotídica completa del fragmento bajo análisis (solamente son necesarias las secuencias completas de 5’ y 3’ para
diseñar los cebadores de PCR), el comportamiento de fusión puede ser determinado experimentalmente por DGGE perpendicular. En este caso, el gradiente desnaturalizante es
perpendicular a la dirección de la electroforesis y la muestra se aplica a lo largo de toda la anchura del gel. La molécula de DNA migrará a través de una
concentración constante de desnaturalizante con un grado de electroforesis constante. La curva resultante indica el número de dominios de fusión y el porcentaje de agentes
desnaturalizantes a los que ocurre la desnaturalización de cada dominio. La preparación de geles perpendiculares ha sido ampliamente descrita por Myers et al.
Sin embargo, el análisis por DGGE perpendicular de cualquier fragmento de DNA requiere cantidades relativamente grandes de DNA en forma de material clonado o como producto de PCR.
Además, esto no proporciona información sobre la posición relativa de cada dominio dentro del fragmento y del efecto de la presencia de la abrazadera GC a cada lado de la
molécula lineal, a menos que se sinteticen cebadores diferentes (costosos) y se prueben diferentes combinaciones de cebadores.
9.3.3 Geles paralelos
El DGGE paralelo es una electroforesis en gel de poliacrilamida convencional donde las moléculas de DNA migran a través de un incremento lineal de concentracion de agente
desnaturalizante (formamida y urea), que son vertidas con marcadores estándar de gradientes. Con objeto de permitir una reproducibilidad, la carrera de la electroforesis se produce a la
temperatura constante de 60ºC. Esta temperatura fue elegida empíricamente para exceder la Tm de un fragmento de DNA rico en A-T en ausencia de agentes desnaturalizantes. Hay
aplicaciones específicas, sin embargo, que pueden requerir otras temperaturas diferentes de 60ºC. Por ejemplo, para secuencias extremadamente ricas en G-C, pueden emplearse
temperaturas por encima de 75ºC. Para asegurar, el mantenimiento uniforme de la temperatura elegida durante la electroforesis del gel, se sitúa en un tanque de agua y se sumerge en
tampón de electroforesis el cual se mantiene a la temperatura deseada mediante una combinación de termostato calentador/agitador.
La elección del rango de desnaturalizante se basa en la Tm de los dominios a analizar: el gradiente inicialmente será elegido con un 25 a 30%
de diferencia entre los valores extremos del gradiente desnaturalizante centrada alrededor de la Tm del dominio. El factor de conversión entre la Tm y el porcentaje de desnaturalizantes
para geles corridos a 60ºC viene dado por esta fórmula empírica:
![]()
Por ejemplo, un fragmento de DNA con un único dominio de fusión con una Tm de 72ºC (=48% desnaturalización), será analizado en un rango de gradiente de desnaturalización desde 33 a 63%. Gradientes de desnaturalización de pequeño rango (10-15% de diferencia) también pueden permitir alta resolución.
9.4 Materiales
9.4.1 Instrumental de fabricación de geles.
El aparato para fabricación de geles usado normalmente en el laboratorio está hecho a mano, basado en el descrito originalmente por Myers et al. El equipamiento puede obtenerse de forma comercial. Alternativamente, pueden adaptarse equipos para electroforesis vertical preexistentes. Para verter el gel en gradiente desnaturalizante se usa un equipo convencional.
9.4.2 Reactivos químicos y soluciones
-
Solución stock de acrilamida al 40%* (acrilamida:bis-acrilamida 37,5:1): disolver 100g de acrilamida y 2,7g de bisacrilamida en H 2 O hasta un volumen final de 250 ml.
-
Tampón de electroforesis TAE 20x (0,8M Tris base, 0,4M acetato sódico, 0,02M EDTA, pH 8,0): disolver 96,912g de Tris base, 7,445g de Na 2 EDTA, y 54,432g de acetato sódico en H 2 O hasta un volumen final de 1l. Ajustar el pH con ácido acético glacial (unos 36ml).
-
Solución stock de acrilamida al 6%* (solución stock 0% desnaturalizante) en tampón TAE. Para 500ml: 75 m l de acrilamida (stock al 40%), 25ml de TAE (stock 20x), y H 2 O hasta 500ml.
-
Stock de solución desnaturalizante al 80%* (acrilamida 6%**, formamida 32%**, urea 5,6M en tampón TAE 1x): Para 500ml: 75ml de acrilamida (Stock 40%), 160ml de formamida desionizada*** (stock 100%), 170g de urea de grado de electroforesis, 25ml de TAE (stock 20x), añadir hasta 500ml de H 2 O.
-
Stock de persulfato amónico (APS) al 10% : Disolver 10g de persulfato amónico en un volumen final de 100 ml de H 2 O. Esta solución normalmente se prepara en el momemto, pero también puede almacenarse en pequeñas alícuotas (1 ml) a –20ºC.
-
TEMED (N,N,N’,N’-tetrametiletilendiamina).
-
Solución de carga de gel 5x (0,25% bromofenol, 0,25% xilencianol, 20% Ficoll): disolver 20g de Ficol, 250mg de bromofenol azul, y 250 mg de xilencianol en un volumen final de 100 ml de H 2 O.
-
10mg/ml de bromuro de etidio disolver 1 g de bromuro de etidio en 100 ml de H 2 O.
Notas:
*Estas soluciones deben ser almacenadas en recipientes de cristal oscuro a 4ºC.
**Pueden emplearse diferentes porcentajes de acrilamida, dependiendo del tamaño de los fragmentos de DNA.
***Para desionizar la formamida: añadir 2g de mezcla resina cama (Baker) a 100 ml de formamida y agitar durante 20 minutos a temperatura ambiente.
Filtrar para eliminar la resina y almacenar a –70ºC en recipientes de cristal oscuro.
9.5 Protocolos
9.5.1 Preparación del gel
Los geles paralelos contienen un gradiente de concentración de formamida y urea que se incremento linealmente desde la parte alta a la parte baja gel. Los geles se usan para analizar un número de muestras cargadas en pocillos a lo largo del gel.
-
Limpiar concienzudamente las placas de cristal, espaciadores, peine y aparato para hacer gradientes con un detergente fuerte. Enjuagar los cristales con etanol y secar cuidadosamente.
-
Alinear los espaciadores a lo largo de los bordes del cristal más largo (sin salientes). Colocar el cristal pequeño en posición y sujetarlos juntos con pinzas de encuadernación. Sellar los bordes y la parte baja con cinta de sellar geles.
-
Poner el peine en posición y localizar las placas de cristal en el marco para DGGE de forma que el cristal con salientes está situado al lado opuesto de la junta de goma, formando la cámara de electroforesis superior. Cerrar las tuercas. Dejar espacio para que salga el aire.
-
Situar el aparato de fabricación de gradientes encima de un agitador magnético, unos 25cm sobre el marco de DGGE. Conectar el tubo de salida en lo alto del cristal cerca del peine y cerrar este tubo de forma que conecte los 2 compartimentos del aparato para fabricar gradientes. Preparar 2 soluciones de igual volumen (15 ml: en nuestro set, con un volumen de 30 ml se llenarán los cristales) el cual dará el rango de concentración deseado. Añadir 16 m l de TEMED y 160 m l de APS a cada solución. Mezclar bien.
-
Verter 15 ml de solución con la mayor concentración de desnaturalizante en la cámara del aparato para fabricar gradientes que está conectada a la cavidad de los cristales. Abrir y cerrar brevemente la llave entre los 2 compartimentos para permitir que la solución llene el tubo de conexión. Asegúrese de que ninguna burbuja de aire bloquea la vía de paso entre los 2 compartimentos.
-
Verter 15 ml de solución con el menor porcentaje de desnaturalizante en el otro compartimento.
-
Mientras agitas la solución en el compartimento con la mayor concentración de desnaturalizante, abre la llave entre los 2 compartimentos y la salida de los cristales. Evita las burbujas de aire.
-
El líquido pasa por gravedad a través del tubo de plástico en la cavidad entre las 2 placas de cristal. El gel será vertido en unos 5 minutos.
-
Dejar el gel para que polimerize unos 30 a 60 minutos.
-
Quitar el peine del gel y la tapa de la parte inferior de las placas de cristal.
9.5.2 Electroforesis del DNA
-
Sitúar el marco con el gel en el contenedor del baño TAE 1x calentado a 60ºC. Ajusta el tampón para que suba justo sobre el nivel de los pocillos. Evitar el contacto del tampón con la cámara de electroforesis superior. Conecta el termostato combinado para que el tampón circule desde la cámara superior de tampón (que contiene el cátodo), mientras se derrama a través de un hueco en la parte posterior del marco en el acuario.
-
Precorrer el gel durante unos 30 minutos a 60 voltios (unos 50 mA).
-
Añadir solución de carga del gel a las muestras (0,1 a 0,5 volúmenes de producto total de PC). Un pequeño volumen final (<10 m l) puede originar bandas muy finas.
-
Enjuagar las ranuras con una jeringuilla provista de una aguja inmediatamente antes de cargar las muestras.
-
Nosotros solemos desarrollar nuestras carreras de DGGE durante unas 16 horas (durante la noche) a 60 voltios (50 mA). Sin embargo se pueden usar diferentes tiempos y condiciones de carrera.
9.5.3 Tinción del gel
-
Parar la electroforesis y sacar el marco del baño. Quitar las placas de cristal del marco y levantar suavemente el cristal con salientes: usar la otra placa para apoyar el gel durante el transporte y el teñido.
-
Teñir el gel durante 20 a 30 minutos en 250 ml de TAE 1x, conteniendo 0,5 m g/ml de bromuro de etidio.
-
Si se observa un excesivo color de fondo puede introducirse un paso de desteñido de unos 15 a 20 minutos en 250 ml de TAE 1x o agua.
-
Examinar el gel bajo luz ultravioleta (254 nm).
9.6 Aplicaciones
El protocolo PCR-DGGE ha sido aplicado al análisis molecular de un gran número de loci genotípicos. En genética molecular humana, la DGGE ha resultado ser
particularmente útil en el análisis de las condiciones heredadas causadas por un espectro de mutaciones heterogéneas o por mutaciones frecuentes de novo , Ej:
b -talasemia, hemofilia A y B, y fibrosis quística.
En genética del cáncer, la identificación de mutaciones germinales y somáticas que dirigen a través de procesos de múltiples pasos a fenotipos malignos
ha permitido la definición de importantes modelos biológicos como el de la secuencia adenoma-carcinoma en tumorigénesis colorectal. En estos casos la DGGE puede ser empleada
para detectar mutaciones en genes susceptibles de degenerar en tumores como k-ras, P53, Hmsh2, y APC, y monitorizar su acumulación en la progresión del tumor.
Aplicaciones más orientadas a la investigación incluyen el examen de la fidelidad de varias polimerasas usadas en PCR, el análisis de
un espectro de mutaciones in vitro de varios mutágenos, estudios genéticos de evolución y poblaciones, y la detección de transiciones conformacionales en los
ácidos nucleicos.
9.7 Modificaciones
Desde su primera descripción, DGGE ha sido objeto de muchos esfuerzos para mejorar algunas de sus características. En su forma básica e ideal, la DGGE con abrazadera GC
detectará la gran mayoría de cambios de base en un fragmento de unos 500 pb que contenga un único dominio de fusión. Sin embargo, cuando hay grandes fragmentos para
analizar, la presencia de 2 ó más dominios de Tm harán más difícil la resolución de mutaciones localizadas en los más termoestables. Varios
investigadores han intentado hacer PCR a blancos de DNA relativamente grandes (2-3kb) y digerir el producto de PCR en fragmentos de unas 500pb antes de hacer DGGE. Esta aproximación
también puede ser implementada usando cebadores con abrazaderas GC en ambos lados de la secuencia diana: de esta forma, aunque el análisis por DGGE de los fragmentos de
restricción internos será menos efectivo, la sensibilidad de detección de mutaciones teórica para todo el conjunto estará próxima al 70 ó 75% (50%
para fragmentos internos y 100% para los 2 fragmentos externos unidos a la abrazadera GC).
También ha sido aplicado satisfactoriamente el uso directo de enzimas de restricción digiriendo muestras de DNA genómico para
análisis por DGGE. El DNA genómico es digerido, hibridado en solución para marcar un DNA de cadena simple y analizado por electroforesis en gradiente desnaturalizante. Abrams
et al. han desarrollado un protocolo modificado para generar moléculas heterodúplex entre una muestra de DNA con abrazadera GC marcada radioactivamente y fragmentos de
restricción de DNA genómico que se analizan entonces por DGGE. También pueden usarse sondas de RNA marcadas radiactivamente.
En DGGE genómico (gDGGE), el DNA
genómico es digerido con una enzima de restricción, sometiendo a electroforesis a través de un gel en gradiente desnaturalizante, transferido a filtros de nylon e hibridados
a una sonda única de DNA. Las ventajas de la DGGE sobre sus protocolos parentales son que 1) no está limitada a una secuencia blanco específica ni a su longitud (puede usarse
cualquier sonda única disponible para cualquier longitud), 2) no requiere información de la secuencia, y 3) son detectables modificaciones covalentes en el DNA genómico de
otra manera perdidas por amplificación enzimática, como la metilación. Por otro lado, como se confía en la presencia de “abrazaderas naturales” (un dominio
de fusión con una Tm tan alta que aquellos de los dominios donde la variación putativa se localiza) en los fragmentos de restricción para ser analizado, sólo una
subsección (20%-60%) de todos los cambios únicos de base serán detectados por DGGE. Por otro lado, el uso de 2 o más enzimas de restricción o una
combinación de ellas podrían mejorar la sensibilidad de la detección. Mas aún, puesto que la gDGGE no está basada en la PCR, la formación de
heterodúplex no es factible y uno tiene que confirmar en la resolución de 2 moléculas de DNA de doble cadena que difieren en un cambio de base. No obstante, gDGGE ha sido
aplicado satisfactoriamente a la identificación de variaciones de secuencias polimórficas en el cromosoma 21 humano y a visualizar mutaciones en Drosophila.
Una huella
dactilar bidimensional de genomas complejos (tipificación 2-D DNA) combina el fraccionamiento por tamaños de los fragmentos de restricción genómicos en la primera
dimensión con su separación dependiente de secuencia a través del gel en gradiente desnaturalizante en la segunda dimensión. Transferido a membranas de nylon e
hibridado con micro- y minisatélites o sus secuencias respectivas, origina patrones complejos de puntos (más de 600 dependiendo de la sonda), una porción de los cuales son
polimórficos entre individuos no relacionados. Esta tecnología tiene potencialidad para el análisis de inestabilidad genómica con relación al cáncer,
envejecimiento y exposición a agentes mutagénicos.
Otra modificación del protocolo DGGE original es la electroforesis en gel con desnaturalización constante (CDGE). Geles con
concentraciones fijas de agente desnaturalizante permiten incrementar la resolución de fragmentos mutantes puesto ellos migrarán de forma constante con una diferencia de movilidad
electroforética a través de toda la longitud del gel. Aunque el CDGE se ha propuesto como muy útil para la visualización de mutaciones desconocidas en genes p53 y
HPRT, el método no representa una alternativa válida a DGGE para buscar cambios de base que no han sido previamente caracterizados en fragmentos de DNA relativamente largos puesto
que cada variante requiere condiciones electroforéticas específicas para una resolución óptima.
9.8 Comparación con otros métodos
Cuando la comparamos con otros protocolos de detección de mutaciones tales como la protección por Rnasa y rotura química con hidroxilamina y tetróxido de osmio (HOT) , la DGGE muestra una gran sensibilidad de detección de mutaciones y un protocolo relativamente menos laborioso. Una comparación directa entre polimorfismos conformacionales de cadena sencilla (SSCP) y DGGE ha indicado que esta última es más sensible puesto que el 100% de las variaciones probadas pudieron ser detectadas vs el 90% en el caso de SSCP. Sin embargo, hay que indicar que el descenso en eficiencia del SSCP podría ser abordado optimizando los protocolos empleados.
9.9 Conclusiones
En conclusión, DGGE ciertamente representa uno de los métodos de elección para el análisis de mutaciones de DNA de doble hebra y ha sido empleado en un amplio
espectro de aplicaciones tanto en investigación como diagnóstico.
Entre la ventajas del DGGE están: 1) la gran sensibilidad de detección de mutaciones ( ~ 90%), 2) mejora en la detección de heterocigotos
(patrón de 4 bandas), 3) La posibilidad de optimizar el análisis mediante simulación por ordenador, 4) un protocolo no radiactivo sin agentes químicos extremadamente
tóxicos o mutagénicos, y 5) fácil aislamiento de alelos mutantes para la determinación de su secuencia. Las desventajas del DGGE pueden ser representadas por: 1)
trabajo preliminar laborioso (simulaciones experimentales o computerizadas) antes de que pueda realizarse el análisis de los fragmentos, 2) uso de cebadores de PCR relativamente largos (
~ 60 pb) y costosos, y 3) el limitado tamaño de los fragmentos de DNA grandes ( ~ 500 pb) que pueden ser analizados
eficientemente por DGGE. Han sido aplicadas satisfactoriamente varias alternativas de uso de cebadores con abrazadera GC, el análisis por DGGE de grandes regiones genómicas requiere
innegablemente un trabajo preliminar sustancial para maximizar su eficiencia. No obstante, la introducción de programas de ordenador que permiten la simulación del comportamiento de
fusión del DNA, ha reducido considerablemente la cantidad de trabajo experimental preliminar necesario.
La elección de un protocolo de detección de mutaciones siempre estará basado en si la especificidad de la aplicación está conforme con que: el DGGE es una
opción válida cuando se requiere un análisis de mutación muy exacto y completo y cuando el tiempo necesario para establecer la técnica y para definir una
estrategia óptima no es un factor limitante.