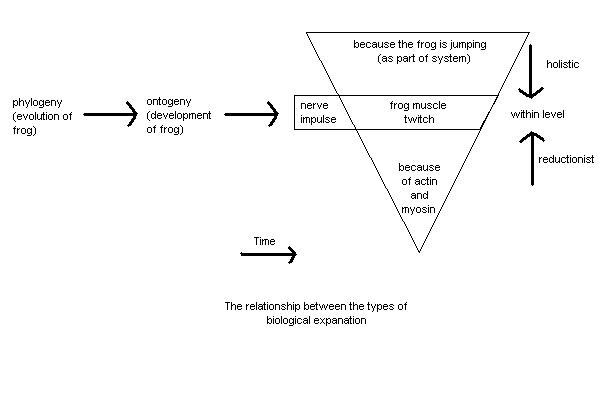
Disordered Molecules and Diseased Minds - Professor Steven P.R. RoseBrain Research Group, The Open University, Milton Keynes, MK7 6AA This paper, by the Professor of Biology at the Open University and an internationally respected neurobiologist, questions whether psychiatric research is in any way scientific. Although written in the deceptively polite language of science, he is actually scathing of the pretensions of psychiatry to be scientific, which coming from someone of Professor Rose�s reputation is extremely serious. It is obviously written for people with some knowledge of neurobiology, but it is worth skipping stuff you don�t understand to see just how damning his criticism is. Summary - This paper reviews problems associated with the concept of causation in interpreting relationships between altered biochemical measures and psychiatric states, and critically reanalyses claims for biochemical and genetic �markers� in depression and schizophrenia. To begin with the disclaimers: I am not a psychiatrist, nor yet an M.D. I have never sought to study the mode of action of a drug on an animal model of psychiatric disturbance. My claims to legitimacy among the contributors to this symposium can only be those of a �basic� neurobiologist whose research concern is the interpretation of the relationships between brain structures and biochemistry on the one hand and changes in the behaviour of the laboratory animals I study on the other. As such, I offer a series of questions to which the other contributors to this symposium may well supply answers. Our theme is �biological markers in mental disorders� - a fertile enough field over the past decades for the generation of research programmes, grant requests, hypotheses as to the �causes� of mental distress and interpretations as to how agents which alleviate that distress may work. Yet despite the wealth of published papers, the increasing sophistication of biochemical, metabolic and genetic methodology, the synthetic skills of the pharmaceutical industry (and the vast tonnage of prescribed psychotropic drugs worldwide) it is hard to avoid a sense of unease, of uncertainty as to the questions which the whole endavour is addressing. Let me emphasise two things at the outset. First, I am not going to go over for the umpteenth time the long-drawn out battle between medical and social models of mental distress. Second - and this, I am sure is common ground between all contributors - I am not a dualist in the Jack Eccles mould, but have a materialist belief in the unity of mind and brain. What does such a glib statement mean? Surely that any event or activity which can be described in �mind language� must also have a description in biological, brain language. Thus, the acts of speaking and listening could be redescribed in terms of nerve impulses, activities in particular sets of cells; so many synapses activated, so much transmitter released, in the brains of speaker and listeners alike. We will all agree that my brain is in a different state when I am talking than when I am not. That different state may be described as a �marker� for my speaking rather than being silent. With appropriate of CAT scans, EEGs and acetylcholine electrodes I might be able to define that marker more precisely - indeed a good part of my current research is devoted to trying to achieve such definition for one type of behavioural process - that of memory formation in the young chick. But come back to the markers for my speaking. I suspect that we will agree on at least one fundamental limitation to the identification of such markers. However good they are, and however good materialists we are, we do not expect that it will prove possible to translate the content of my speech into changing molecular structures or cellular acitivities. Whilst all mind process translate into brain processes and vice versa, there is an important way in which the languages of mind and brain are incommensurable. The information content of my speech does not translate into statements about molecules or cells, even though both speech and molecules are aspects of the same unitary phenomenon. Saying �I love you�; �that man makes me angry� or �I feel miserable or suicidal� carries sets of meanings which are given by personal history, culture, social and economic circumstances and are not reducible to the mere motion of molecules. I am labouring this point because, presumably, our search for biological markers for mental disorder is motivated by the belief that to find such markers will help us help us either to explain, or cure, or at least intervene therapeutically in that disorder. Why else should the World Health Organsiation be interested in such an endeavour? The core of the problem can be stated quite simply; a few years ago I shared a platform with a colleague discussing the problem of learning disability before an audience of parents of �learning disabled� children. We need biochemical research, my colleague stated, to discover �the disordered molecules which cause diseased minds�. It is just this question of cause which needs to be explored in some detail, because therein, I believe, some of our conceptual problems lie. Consider a simple biological observation, the firing of an axon in the motor nerve leading to the leg of a frog. There are at least five ways in which biologists might answer the question: what is the cause of the firing of the axon? (1) The first is the most straightforward; the axon fires because of a potential difference change at the axon hillock; this in turn is caused by the sum of depolarisations occurring at synapses on the cell body or dendrites; these are caused by prior firing of axons of nerves which synapse on the cell in question... and so forth. There is a simple linear chain of events; sensory nerve firing > interneuronal events > synaptic depolarisation > motor nerve firing. This chain is linear as set out on the page: it is also linear and irreversable in time. It is causal in the physiologist�s sense. (2) But we may also say: the axon fires because the frog is jumping to escape a predator. Here the phenomenon of the axon firing is described in terms of the part it plays in the system which is the frog: this is a holistic or engineer�s type of explanation. (3) And we may say: the axon fires because of the passage of ions across the cell membrane. The axon�s firing is now described in terms of its constituent molecular processes: this is a reductionist, or biochemist�s explanation. (4) Further, we may say: the axon fires because of the way in which, during neural development in the egg and the tadpole, brain and muscle become wired up and neuromuscular connections formed: this is an ontogenetic, or developmental explanation. (5) And finally, we may say: the axon fires because, during evolution, natural selection has favoured those frogs which are able to jump swiftly to avoid predators: this is an evolutionary, or in the ethologist�s sense, a functional explanation. The relationship between these different types of explanation is summarised in Fig. 1, and discussed more fully elsewhere (Rose, 1982; Lewontin et al., 1984) Note however that explanations (2) and (3) are quite different in kind from the others. In explanation (1) the word cause is used to describe a linear sequence of events: first (a) occurs, then (b) and so forth. But we cannot say first, the frog jumps to escape the predator, then the axon fires. The axon firing is part of the jump. And we cannot say first ions cross the membrane, then the axon fires: the ions crossing the membrane are the axon firing - merely another way of describing the phenomenon. |
|
I know this diagram isn't very clear - until I fix it, you can see it much more clearly in Windows by right-clicking, choosing copy, and pasting it into Windows Paint or a similar program. Similar procedures apply for Linux or Mac users.
The way in which biologists use the word �cause� often confounds statements about linear sequences of events - within-levels explanations of a phenomenon - and statements of identity or partial identity - between-levels descriptions of a phenomenon. Contrary to the view of my biochemist colleague, disordered molecules do not cause diseased minds in the same way as dendritic summation causes axonal firing. At best such disordered molecules may be considered to be identical with - alternative descriptions for - the diseased mind. If we want our question about the causes of mental disorder to be answered along the lines of my first type of explanation - that is, what happened such that individual such-and-such showed mental disorder or distress - then offering statements about changed catecholomine metabolism is no sort of an explanation at all: it is merely, at best, a translation of the problem from one language to another. Nonetheless, the power of the medical model, the power of reductionist thinking, is such that I would lay odds that most psychiatrists, and virtually all pharmacologists and biochemists, really do believe that the description of the phenomenon of mental distress in terms of the properties of particular molecules or ensembles of cells is more fundamental, more scientific and more rigorous than the description of the same phenomenon by a clinicician, a sociologist or a novelist. The reasons why reductionism is so seductive lie deep in the history of the origins of scientific thought, and indeed of the social order which sustains this thought, and I do not intend to explore them further here. (Lewontin et al., 1984) Once one concedes the reductionist model, the battery of techniques available in the search for markers is well known to us all. One may analyse accessible body fluids of patients and �normal� controls and attempt to detect statistically reliable differences in likely metabolites - a procedure roughly analogous to attempting to find out which way the inhabitants of a house vote at elections by examining their grocery purchases and garbage disposal. One may analogise other body tissues ot the brain - in recent years psychopharmacology has shown an extraordinary obsession for blood platelets. One may endeavour to develop animal models, a procedure fraught with its own two-step reductionist logical problems of trying to explore the mind events of one highly complex and social species in terms of the blood biochemistry of another. And above all, one may argue backwards from the effect of drugs. This procedure, described by Bignami (1982) as ex juvantibus logic (see also Sedgewick, 1983) is, as a rough check on the literature of the last decade on the biology of schizophrenia and the affective disorders reveals, by far the most commonly adopted. The argument goes roughly like this. Drug X is shown - by double blind statistically controlled properly authenticated clinical trials - reliably to improve the mood of depressed patients, or decrease the existential communication disorder of schizophrenic patients. If we can then show that drug X interacts with system Y - perhaps neurotransmitter uptake or binding in blood platelets or slices of rat brain, then it is reasonable to assume that system Y is in some way faulty in the depressed or schizophrenic patient, and therefore this fault is the �cause� of the disorder. Let me draw examples almost at random from the literature: first a review of Langer and colleagues entitled �High affinity binding of 3H-imipramine in brain and platelets and its relevance to the biochemistry of affective disorders� (Langer et al., 1981) The authors start by stating the �catecholamine� and �indoleamine� hypotheses of affective disorders�. The fact that tricyclic depressants inhibit neuronal uptake of monoamines is consistant with these hypotheses, they say, but not all drugs which serve as antidepressants inhibit such uptake. So one should investigate other sites of biochemical action of the drugs. They proceed to show that 3H-imipramine binds to many brain areas, including hypothalamus and amygdala, and that antidepressant drugs do inhibit imipramine binding. The authors then turn their attention to platelets. Platelet imipramine binding is affected by age but not sex in healthy volunteers. But it is found to be lowered in �untreated severely depressed patients� �suggesting that this binding site might be involved in the biochemical changes related to affective disorders�. A massive jump to a conclusion. Yet within the next paragraph we learn that the patients included both those suffering from reactive and uni-or bi-polar endogenous types of depression, even though there is no correlation between degree of depression and degree of lowering of binding. Our ingenious authors are not easily tied down by such a doubt however. �This might be explained by a cancelling out of two concurrent changes in 3H imipramine binding.� There is an effect of the drug, and simultaneously an effect due to improvement caused by the drug. And if this won�t do, then the decrease in binding sites may be �a genetic (sic) marker of a susceptibility to depression� - apparently to be studied by looking at platelet 3H-imipramaine binding in close relations of depressed patients. What a thicket of non sequiturs this experimental logic offers us! If a drug diminishes a symptom, it cannot be inferred that the biochemical system on which the drug acts is the cause of that symptom, or even its brain correlate. If aspirin reduces toothache, it does not follow that investigating the biochemical mode of action of aspirin will also diminish the pain resulting from a broken leg allow us to deduce that the �causes� of toothache and of the broken leg are identical. In the Langer case, we see how failure to find a biochemical difference - in binding sites for 3H-imipramine between different categories of depressed patients - is explained in terms of a common genetic propensity for the different types of depression. What would have happened had a difference been found? A second paper, by Anath (1978), �Clinical prediction of antidepressant response� tells us. Anath uses differences in individual responses to drugs to define the clinical diagnosis of the disorder. Thus he cites an earlier study of Kupfer et al (1975) to show that the �unipolar-T (tricyclic responder) group showed premorbid personality traits of chronic anxiety and obsessiveness, while the unipolar-L (lithium responder) group manifested characteristics of cyclothymic personality including mood swings, elation and exitation.� That is patients who show one type of response to a drug are one type of depressive; those who show another are a different clinical class. Ex juvantibus logic is thus no loose logic. If some groups of patients respond to a drug and others do not, this difference becomes the motor for clinical diagnosis - for instance depressed patients who respond to tricyclics are regarded as endogenous, those who do not are reactive. If groups clinically described as different both respond to a treatment or show a common marker this shows a common genetic prediposition to disorder. In either case the molecular explanation rules supreme. I should emphasise that I have not chosen poor papers of their type to criticise. The authors are respected workers in their field. That is why I worry about the logical and scientific standing of the field itself. Only real conceptual problems could explain the extraordinary range of biochemical markers which have been offered as providing �the� unitary cause of schizophrenia they have been will catalogued by Andreoli (1980). They include anomalus substances in schizophrenic blood (1960s) ; the dopamine hypothesis (1972 on); the noradrenaline hypothesis (1977 on); the endorphine hypothesis (1976 on); the amino acid hypothesis (1972 on); the acetylcholine hypothesis (1973 on); the histamine hypothesis (1937 on); the transmethylation hypothesis (1952 on); and several viral hypotheses (1973 on). At best, such biochemical marker studies offer correlates of behavioural manifestations. Logically, the biochemical change being studied may precede in time the behavioural state - in which case it may be the correspondent at the biological level of some behaviour/mind state antecedent to the �disorder�; or it may coincide in time with the behaviour; or it may follow it. Early studies on biochemical markers were led astray by identifying as �causes� of the disorder substances which were the degradation products in the urine of drugs used to treat the disorder, and we know better than to accept such iatrogenic �causes� today. But supposing depression �causes�decreased 3H-imipramine binding in the blood just as influenza virus causes decreased nasal mucus secretion? Or, it that appears too trival a criticism, take a more substantive one. Suppose that a depressed patient has a decreased level of synthesis of a neurotransmitter in a particular brain region. Synthesis of that neurotransmitter will be an energy-demanding process. If less is synthesised, there will be less glucose utilisation in the brain region concerned - and this will show up in 2-deoxyglucose or related CAT-scan approaches. The lowered glucose metabolism is a necessary correlate of the changed neurotransmitter synthesis, but it would be hard to argue that even in the reductionist sense it was the cause of the depression. All investigations of the relationship of brain biological events to mind/behaviour phenomena, if they are to cast meaningful light on the translation between the two types of description of events, need to show rigorously, for each biological process or marker under consideration, that the change in the marker is necessarily, sufficiently and exclusively related to the behaviour being studied. In another context, I have tried to identify the criteria that would need to be fulfilled to show this correspondence between biochemistry and behaviour in the case of animal models (Rose, 1981). The six criteria offered there, which are minimal demands, can scarcely be fulfilled even for very simple models of animal learning, although we (e.g. Rose and Harding, 1984) and others (e.g. Kandle and Schwartz, 1982) have gone some way towards doing so. Frnakly I find it hard to see how such rigorous criteria can be met in the analysis of mental disorder, granted the problem of animal models and the limitations of molecualr roulette and ex juvantibus logic. Even if they could be met, if we could have a complete description of the differences between the brain of a mentally distressed person and that of a normal person, we would still have a statement of correspondents only; it would not be causal in the sense which reductionist thinking implies. The biological description would only become causal in the event that we could show a relationship, not between the biological markers of the mental distress but between the biological markers and the mind events leading to that distress. One could then map a correspondent of the type: predisposing mind event > mental distress biological state (a) > biological state (b) (There are vertical arrows that I can�t put in this computer programme) This is the logic, presumably, of the search for genetic markers in mental disorder. It must be granted that, even if today�s brain state does not predate today�s mind distress, today�s genotype must so do. Yet despite the conventional wisdom to the contrary, the evidence for a genetic predisposition to the most frequent forms of diagnosed mental disorder - schizophrenia and the affective disorders - is at best weak. The re-evaluation of the Danish and adoption studies by Kamin (Lewontin et al., 1984), Schiff (Casson et al., 1980) and Lidz (Lidz et al, 1981, Lidz and Blatt, 1983) cast doubt on what has widely been regarded as some of the strongest data in the field. In any event, the statement that individuals with a particular genotype are more at risk at being diagnosed as schizophrenic or depressed, if verifiable, should not be surprising. Unless all individuals of that genotype show the disturbance, the significance of a �genetic predisposition� only becomes meaningful in an ontogenetic, developmental context, and it is here that the real difficulty of unpicking the meaning of causation statements becomes profound. The problem lies in the relationship of genotype to phenotype. First, the norm of reaction of gene to environment during development means that the phenotype generated by any particular gene differs at different stages of development and in differing body tissues, to say nothing of different genotypes. (This is not quite what geneticists mean by the terms pleiotropy and phenocopy, but it is not so very different.) The obvious example is, of course, phenylketonuria, where the same genotype can generate an irreversably mentally retarded or �normal� child depending on the prescence or absence of phenylalanine in the diet. More to our immediate point, however, the fact that the site of the genetic lesion in phenylalanine metabolism is known does not give us any way of understanding which of the multitude of diffuse effects of this deficit on the physiology of the child is a �marker for� or �causative of� the mental retardation. Second, the metaphore of �environmental effects� on �genotypic expression� is inadequate, because it defines the organism - and the genes within it - essentially as a passive responder to environmental challenge. Yet organisms do not passively receive their environments, but actively seek them out and transform them (Lewontin et al., 1984). In a paradoxical way this truth has been long understood in the literature of mental disorder. When in the 1930s, Faris and Dunham accumulated evidence to show that incidence of schizophrenia was highest in the derelict inner city of Chicago and lowest in the affluent suburbs (see Dunham, 1963); the at first sight obvious environmentalist interpretation was rejected on the grounds that it was precisely the schizophrenic personalities - read �schizophrenic genotypes� - who �chose� to live in the inner city. There is yet a further problem in understanding the significance of assumed genetic markers for mental disorder. If a disorder is extremely rare in the population, a discussion of genetic predispositions may prove helpful to understanding its aetiology. If it is very common, the genetics seems to be less and less relevant. Recall that in Brown and Harris� (1978) major study of Camberwell, they found that some quarter of working class women with children living in the area were suffering from a definite neurosis - mainly depression - whose incidence could be related to significant life events, whereas the incidence amongst comparable middle class women was only some 25% of this rate. Are we to assume that all of these working class women shared in addition a particular genetic predisposition? Clearly the life and environmental circumstances of this group are much better predictors of their likehood of suffering depression than any amount of gene-library-making or biochemical analysis. In common parlance, environmental �causes� are those to be explored if explanations of maximal utility are to be sought. And whilst the prevalence of the depression diagnosis in women compared with men is enough to make a simple genetic argument unlikely anyhow, we should also note that Roy (1981) showed that in depressed men, parental loss before 17 years, poor marraige and unemployment were major vulnerability factors. The question then for those who maintain that there is a utility to the search for biological markers in depression is this: are we being asked to believe that in all of these causes of depression there are going to be found common biochemical abnormalities? That is, there is a unitary phenomenon, depression (or a simple two-or-three subset classification thereof) to which we will ultimately find that there is a matching unitary of two-or three subset range of bichemical abnormalities? Do all depressed women in Camberwell show, if not common genes, then common levels of catecholamine, indoleamine or imipramine binding to their platelets? And if so, is this not much more likely to be related to depression as the nasal mucus to the influenza rather than as the PKU to mental retardation? There is one further question which the emphasis by psychopharmacology on ex juvantibus logic raises insistently in my mind. Let us accept that antideprassant drugs do act by changing the absolute levels, ratios or specific binding sites of one or more neurotransmitters in particular brain regions. Yet rigorously conducted clinical trials of such agents consistently show a placebo effect of around 30% - sometimes more, somtimes less, depending on the exact experimental design, the severity of the patient�s depression, the duration of the trial and so forth. I may have missed it in the literature, but I am not aware of any biological psychiatrist endeavouring to answer the question: how does the placebo exert its effect? Social psychiatry can of course refer to such explanations as patients� and doctors� expectations of success, the subtle dialectical relationship of patient to healer. But what do we materialists who hold that to every mind state there corresponds a brain state believe? Does the placebo also alter neurotransmitter metabolism, and if so, how? If it does not, there can be no specific one-for-one relationship between the depression diagnosis and altered biochemistry. If it does, alas for the specificity of drug binding studies, S.A.R. and the gamut of molecular roulette! My paper has been critical; it is not intended to be negative. I said at the outset that in pointing to the logical problems involved in biological marker studies I was not trying to rewage a battle between social and medical models of mental disorder. A unitary materialist understanding of mind/brain relationships, of the sort which I have assumed we all share, must reject such a dichotomy. Just as biological statements about the brain must translate into mind statements, so social, behavioural and mental statements must translate into biological ones. What this means is that we cannot accept the easy. pluralisic options which divide the world into two sorts of phenomena: either X is depressed because of an endogenous neurotransmitter disorder or because she has just lost her job or her husband. Psychiatry adopts this eclecticism too easily; think of the distinction between organic and functional disorders that the standard text books offer. Or look at the way in which critics of institutional psychiatry like Szasz discriminate between states of mind which he believes are organically caused by observable brain lesions, and therefore the province of medicine, and those for which he claims no brain cause can be found - schizophrenia and the affective disorders - and which are therefore part of an individual�s personal responsibility and not the policies of institutionalised medicine. The logic of Szasz�s approach is that as more and more biological �markers� for such disorders are found - as they will be - the province of institutionalised psychiatry will expand and the area of personal responsibility decline. Far from it, we have to understand that matters of mind are simultaneously amenable to social and personal explanation and biological description, just as the firing of the axon in the frog�s motor nerve is simultaneously amenable to holistic, reductionist, physiological, ontogenetic and evolutionary explanations. Where does this leave the search for biological markers? First, in need of more rigorous criteria for tests which marker-candiadates must fulfill. Second, an acceptance that there are likely to be many-for-many correspondences between brain biological events and mental states, rather than the one-to-one or one-to-many which is all that most present psychopharmacological hypotheses allow. This means we should expect a multitude of biochemical systems to show very subtle changes in individuals diagnosed as depressed or schizophrenic. The very magnitude and range of the found or hypothesised changes in neurotransmitters in allevating existential distress in mental distress in mental disorders is more on a par with aspirin in toothache than with any more direct, mechanistic one-drug-one-disorder mode of thinking. It is up to the other contributors to this symposium to prove me wrong. Intrepid Carpets Home Page |