Controlling the General Acid Catalyzed Reaction
Gwynne M. Osaki and James E. Jackson
Department of Chemistry and Center for Fundamental Materials Research
Michigan State University, East Lansing, Michigan 48824-1322
In a general acid catalyzed reaction the rate determining step in the
reaction is the proton transfer. Until recently the means to map
the potential energy surfaces (PES) for general acid catlyzed reactions
have not been available because the kinetics of this type of reaction occur
on a very fast time scale. The current technology makes it possible
to use computational methods to map the PES using small model compounds.
Eventually this information will be used as a guide for future experiments
using high powered, finely tuned lasers to overcome the barrier that makes
the proton transfer the rate determining step after the molecules are hydrogen
bonded. In this paper will discuss how the best proton donor/acceptor
combination was selected using semi-empirical and Hartree-Fock (HF) as
methods to model a PES in general acid catalysis. The proton donors that
were used in the calculations were NH4+ and H3O+,
while the proton acceptors included simple ortho esters and iranes.
Motivation
The control of chemical reactions has been the Holy Grail
for chemists since the time of the alchemists. Over the years much
has been learned about how to control the conditions to make a reaction
proceed more efficiently, but not until recently has there been any type
of direct manipulation of the molecules to control the reaction.
With the introduction of high-powered, short-pulsed
lasers in the 1980ís, it has become possible to control the motions
of the molecules themselves. Zewail, Zare, Crim, and many others
including Dantus here at Michigan State University have done work in this
area.
All the work in this field so far has been done in the
gas phase and in very specialized conditions, like isolating single molecules.
However, most chemistry is done in bulk and in the liquid phase.
Our work hopes to find a set of reactions that are controllable in the
liquid phase that will not need very specialized conditions. As will
be discussed in this poster, general acid catalysis is a good candidate
for a reaction that may be controllable in the liquid phase in bulk.
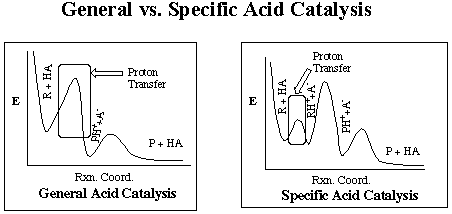
General Acid Catalysis
In acid catalysis the catalysts are oxonium ions resulting
from the dissociation of the acid in the solvent, which in most cases is
water; but can include all proton acceptors and donors in the solution.
Weak acids produce two types of acid catalysis. They are known as
general and specific. They differ in which proton donors and acceptors
are considered part of the catalyst.
In general acid catalysis, the form that is the
basis of this project, all proton acceptors and donors are included in
the catalysis, hence the name general. In other words it is dependent
on total acid concentration, not just pH. The proton transfer from
the donor to the acceptor is the rate-limiting step in this type of catalysis.
Once this energy barrier is overcome, the rest of this reaction proceeds
rapidly.
In the other type of acid catalysis, specific acid catalysis,
the rate-determining step is not the proton transfer. Overcoming
the energy barrier for this transfer does not make the reaction proceed
rapidly like in general acid catalysis. It is dependent on only the
pH, or the concentration of the specific acid, oxonium or the H+
species.
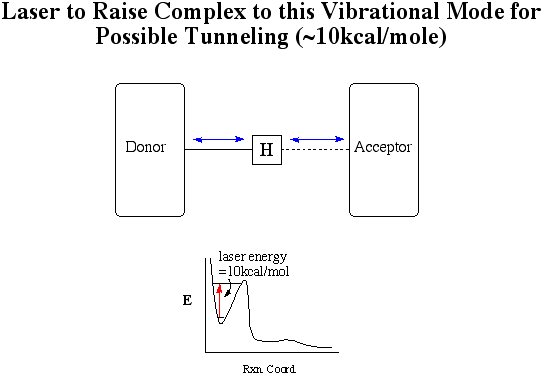
Controlling the General Acid Catalyzed Reaction
Since this reaction has proton transfer as its rate
determining step, it may be possible to control the reaction by finding
way to give the hydrogen-bonded complex just enough energy to overcome
the proton-transfer-energy barrier or arrive at an energy level where proton
can tunnel though the energy barrier. The hydrogen-bonded complex
holds the proton donor and acceptor in the right configuration to transfer
the proton. If the one can selectively break the hydrogen bond between
the proton and the donor, it will be possible to control this reaction.
This is because once this occurs the reaction proceeds rapidly.
Knowing the shape of the potential energy surface (PES)
for a general acid catalyzed reaction will show us what the energy barrier
looks like. This information will help us set up experiments to show
that proton transfer can be controlled in a general acid catalyzed system.
It will help to select the laser, which will be used to deliver the exact
amount of energy to overcome the barrier and will help with selecting molecules
that can be studied this way.
Why Use a Computer to Model a General Acid Catalysis Reaction?
The fast kinetics is why the potential energy surface for
general acid catalysis has not been studied experimentally. However, advances
in computer technology have made it possible to do calculations to predict
the structure of the intermediates in a timely manner, even on a personal
computer. These advances have also increased the information storage
capabilities. This is very important because the contributions of
several orbitals on each atom to the energy of the molecule(s) must be
included in each step of the calculation and saved. Calculations
improve in accuracy as the number of orbitals included in the calculation
increases. This basic structural information and information on how the
structure is changing as the calculation proceeds can take up megabytes
(MB) of storage space.
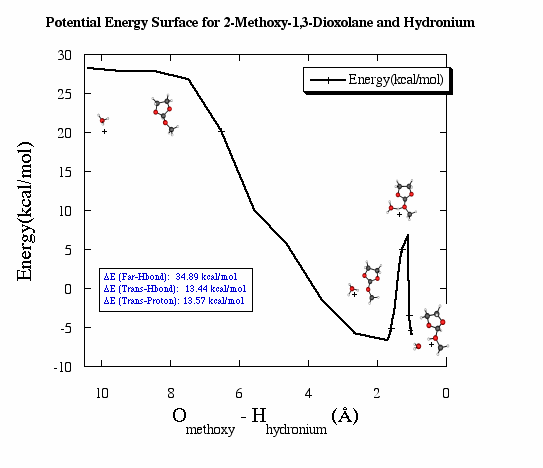
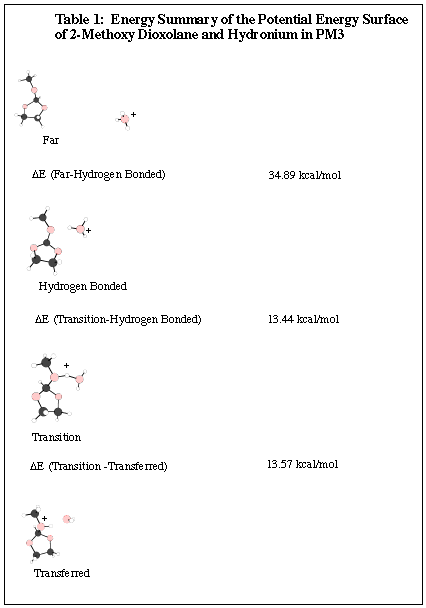
PES for 2-Methoxy-1,3-Dioxolane and Hydronium PM3 Calculation
Ortho esters, such as 2-methoxy-1,3-dioxolane, were first
used for the calculations because they are known to undergo general acid
catalysis and are available through commercial means. This is important
for the experimental work since there should be an accessible supple of
sample.
The molecules for this PES calculation were chosen because they
were smallest that could be used for ortho ester general acid catalysis.
Since every atom in has several orbitals, more computer time and space
would be needed as the molecules became larger. This also added to
the complexity of the calculation since the orbitals interact with one
another.
A semi-empirical method, PM3, was used to make this PES.
Semi-empirical methods utilize experimental values and simple basis sets,
mathematical functions describing orbitals, to calculate the energies of
the molecular configurations.
Conclusions for 2-Methoxy-1,3-Dioxolane and Hydronium
This surface showed that the proton transfer step was lower
in energy than the separated molecules. This means when the molecular
complex is excited just high enough in energy to transfer the proton, it
will not return to the separated state. In other words this reaction
will only go forward when the proton transfer is induced. Since
this PM3 calculation gave this great preliminary result, higher levels
of theory were tried. Calculations were done using ab initio methods,
specifically Hartree-Fock. In these calculations all of the orbital
information is from solutions to the Schödinger equation. This
is more accurate than using semi-empirical information since the experimental
values are from molecules have bonds not as strained as the ones in the
molecules that are being studied.
Unfortunately, just the acid proton on the methoxy
of the dioxolane, the methoxy did not break off as a methanol. This
meant we could not say conclusively from the calculations that the rate-determining
step was the proton transfer. Another molecule had to found.
PES for 2-Methoxy-2-Methyl-Oxirane and Ammonium HF 6-3lG* Calculation
Oxiranes were tried next. The simplest one, the plain oxirane,
was no better than the ortho esters. It did not go on to do the rest
of the chemistry after the proton transfer. In this case it was to open
the C-C-O ring. However, after adding the methyl and the methoxy
groups the ring, the ring opened up with no problem once the proton was
placed on the oxygen of the ring. However, hydronium was too strong
of an acid for this calculation. This complex did not optimize at the hydrogen
bond, but at the open ring. Another simple acid, ammonium, was used
instead to build the PES.
The PM3 PES was not very promising. The energy of
the transition state was higher than the energy of the separated molecules.
Since oxiranes are high strained molecules because of the three-membered
ring, the ab initio calculations were then done. The Hartree-Fock
method and the 6-31G* basis set were used. This basis set was chosen
because it was the smallest one that gave reliable data. STO-3G did
not allow the ring to be broken up with just the acid proton and the oxirane.
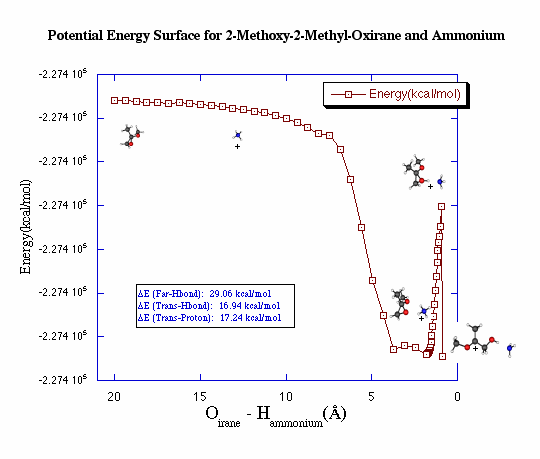
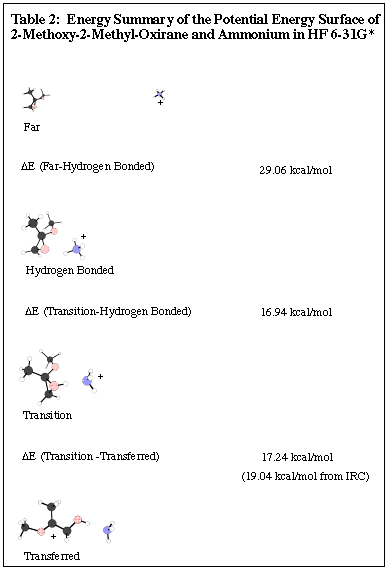
Conclusions for 2-Methoxy-2-Methyl-Oxirane and Ammonium from HF 6-3lG*
The PES calculated for 2-methoxy-2-methyl-oxirane and ammonium
shows that the separated compounds are higher in energy than the transition
state of the proton transfer. In other words, reaction will only
go forward if the molecule is excited just enough to transfer the proton.
When the proton is transferred to the oxirane, the ring opens up.
This shows that the reaction continues on after the proton transfer, which
is what is expected and observed experimentally.
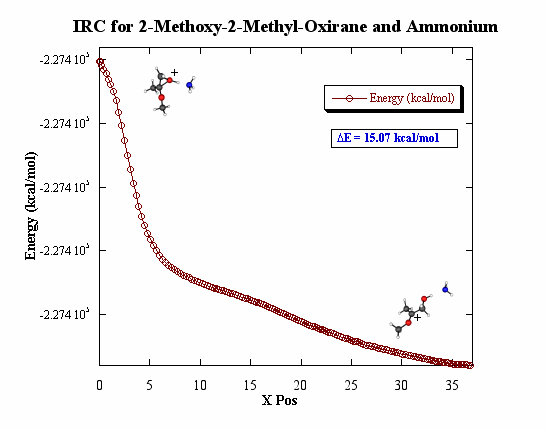
The IRC, intrinsic reaction coordinate, calculations gives a
better picture of what the real PES should look like because all of the
configurations of the molecules will be calculated as the proton is moved
toward the oxirane. This PES shows calculations that were made by only
moving the proton toward the oxygen on the oxirane ring along one path.
The IRC showed that minimum for the hydrogen bonded complex in these first
set of calculations is not a local minimum.
Future Work