Alimentary tract and pancreas
Alimentarni trakt i pankreas
ARCH GASTROENTEROHEPATOL 2002; 21 (
No 1 – 2 ):
The
Lynch syndrome: Report on family “S”
Lynch-ov sindrom: Prikaz porodice “S”
( accepted
June 20th, 2002 )
Aleksandar
Nagorni, Vuka Katic, Jovica Milanovic, Vesna Zivkovic, Goran Bjelakovic.
Clinic for
Gastroenterology and Hepatology and Clinic for Pathology, Clinical Centre,Ni{s
Abbreviations used in this article: CRC,colorectal cancer;
HNPCC,hereditary non-polyposis colorectal cancer; ICG HNPCC,International Collaborative Group Hereditary Non-Polyposis Colorectal Cancer; MMR,Mismatch Repair.
Address
correspondence to: Professor: Aleksandar V. Nagorni, MD, PhD,
Clinic for gastroenterology and hepatology,
Clinical Center Nis,
48 Brace Taskovica St.,
Yu-18000 Nis, Serbia, Yugoslavia.
Fax: +381 18 335 186
E-mail: [email protected]
……………………….
……………………………….
Lynch
syndrome Gastroenteroloska sekcija SLD-
01732, 2002.
ABSTRACT
Lynch
syndrome (HNPCC-hereditary non-polyposis colorectal cancer) is an autosomal
dominant inherited disorder characterized by early onset of colorectal cancer
(CRC), a preponderance of CRC in proximal colon, occurrence of multiple CRC,
mucinous and poorly differentiated adenocarcinoma. We report family “S” with 11
(25.5%) affecting members. Metachronous CRC were observed in 27.2% affecting
members. In tumor spectrum of Family “S” there were stomach, ovarian and
endometrial cancer. Genetic tests are not routinely available in our country,
so periodic screening of all family members have to be perform according to
recommendation.
Key words: Lynch syndrome,
metachronous CRC, stomach cancer
SAZETAK
Lynch-ov
sindrom (HNPKK- hereditarni ne-polipozni kolorektalni karcinom) je autozomno
dominantni nasledni poremecaj koji se karakterise ranim pocetkom kolorektalnog
karcinoma (KRK), predispozicijom za proksimalni kolon, pojavom multiplih KRK,
mucinozni i slabo diferentovani adenokarcinomi. Saopstavamo porodicu “S” sa 11
(25.5%) obolelih clanova. Metahroni KRK su zapazeni u 27.2% obolelih clanova. U
tumorskom spektru porodice “S” otkriveni su karcinomi zeluca, ovarijuma i
endometrijuma. Genetski testovi jos uvek nisu rutinski dostupni u nasoj zemlji,
pa periodicno pracenje svih clanova porodice treba izvoditi u skladu sa
preporukama.
Kljucne
reci: Lynchov sindrom, metahroni KRK, karcinom zeluca.
INTRODUCTION
Colorectal cancer (CRC) is the
fourth most common cancer and the second leading cause of cancer deaths in many
countries worldwide. In the past few years, advances in the molecular biology
and genetics of CRC have not only broadened our understanding of this disease,
but have also provided insight into the pathogenesis of both sporadic and
hereditary CRC syndromes (1).
Hereditary non-polyposis colorectal
cancer (HNPCC) firstly was recognized in 1895. Famous pathologist Aldred
Warthin then heard from his seamstress who was depressed because she was
convinced, based on her family history, that one day she would die of cancer of
the female organs or bowels. As she had predicted, she died of endometrial
carcinoma at a young age. In 1913 Warthin published Family G with endometrial,
colorectal and gastric carcinoma as a predominant malignancy in this kindred
(2).
In the fame of Henry Lynch who
studied this problem in the middle sixties International Collaborative Group
Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC) was named HNPCC as Lynch
syndrome.
Lynch
syndrome is an autosomal dominant inherited cancer syndrome characterized by
the development of (CRC) at an early age of onset, a preponderance of cancers
in the proximal segments of colon, an excess of multiple CRC, synchronous and
metachronous CRC (3,4), and poorly differentiated and mucinous CRC (5,6). Lynch
syndrome is associated with a small number of adenomas that are located
predominantly on the right side of the colon (7). ICG-HNPCC proposed the
“Amsterdam minimal criteria” in 1990 to facilitate the clinical diagnosis of
HNPCC (8). “Minimal” criteria include: 1) the presence of three or more
relatives with CRC, one who is a first degree relative of the other two, 2)
that the relatives affected by CRC span two or more generations, 3) and at
least one relative is affected by CRC before the age of 50 (8).
In addition to CRC patients with
Lynch syndrome are at risk of developing extracolonic tumors including
endometrial, ovarian, ureteral, stomach, renal pelvis, small bowel,
hepatobiliary and skin, which are included in the modified Amsterdam criteria
(9).
The genetic defect underlying Lynch
syndrome is a defect in DNA mismatch repair genes. In the presence of normal
DNA mismatch repair, abnormally paired nucleotides are excised and replaced
with the proper nucleotide base pair. However, if inactivation of mismatch
repair system, characteristic of Lynch syndrome, is present, errors in base
pairing can accumulate and predispose the cell to malignant progression. This
persistent error in base pairing in the DNA from patients with Lynch syndrome
has been referred to as the “mutator” phenotype, the “replication error repair”
phenotype, or microsatellite instability (1).
Six human genes have been
identified as participating in the function of mismatch repair. These genes
include hMSH2, hMLH1, PMS1, PMS2, hMSH6 and hMSH3 (10-15), although germline
mutations in MSH2 and MLH1 account for the majority of Lynch syndrome families
(16,17).
REPORT OF A FAMILY
Our
interesting for hereditary CRC began in 1989 when a 58-year old man (proband)
was admitted to the Clinic for gastroenterology and hepatology, Clinical Center
of Nis, because of intestinal bleeding and changes in the large bowel
movements. First episode was recorded six months before admission. Twenty-three
years ago adenocarcinoma of transverse colon was diagnosed and right
hemicolectomy was done. In the same time his son was admitted to the Clinic
because of liver metastasis of colorectal cancer, two years after surgery for
cecal adenocarcinoma. Colonoscopy revealed adenocarcinoma of descending colon
at the splenic flexure. Additional analyses: laboratory, abdominal ultrasound
and CT are excluded the spreading of disease and left hemicolectomy was done.
The proband was followed-up by colonoscopy and ultrasound, but four years after the second operation of metachronous CRC, stomach cancer was diagnosed (adenocarcinoma), and patient was operated.
We began to collect family data and pedigree of family “S” was made (Fig. 1).
We report the family “S”more than a 10 years after collecting data.
In family “S” there are 43 members, but 11 (25.5%) members are involved with a total of 16 malignant tumors. In nine patients there were one or more CRC (metachronous), but in two family members, single stomach and ovarian cancer were detected. Most (12) malignant tumors are CRC, but two stomach cancer and endometrial and ovarian cancer were diagnosed, too. In 3 (27.2%) of involved family members, metachronous CRC were observed 4-23 years after diagnostics of initial CRC. The earliest onset of the disease was diagnosed in the proband (35 years), and in more than a half of involved patients, malignancy was detected before 50 years of age.
There were 9 (75%) of right sided, but 3 (25%) of left-sided CRC. In one family member, metachronous sigmoid CRC was small (6 mm) flat lesion revealed four years after surgery for cecal adenocarcinoma. Histology revealed in most of cases mucinous and poorly differentiated adenocarcinoma.
The proband is under regular control, without any sign for metachronous growth. The proband is responsible not only for our interest in hereditary cancers, but for the other healthy family members, who follow him and come for regular, determinated gastroenterological and gynecological examinations.
DISCUSSION
Lynch syndrome is an autosomal dominantly inherited disorder of cancer susceptibility with very high penetrance rate of 80-85% (18-21). This syndrome accounts for about 1-2% of all CRC occurrences (22). Mutations of the major mismatch repair (MMR) genes MLH1 and MSH2 are detected in 30-80% of HNPCC kindreds from different ethnic backgrounds (23-26). To date the only known cause is an inherited mutation in one of the following (MMR) genes: hMSH2, hMLH1, PMS1, PMS2, hMSH6, hMSH3 (10-15).
ICG-HNPCC at its meeting held in Amsterdam in 1990 was established a set of selection criteria for families with Lynch syndrome to provide a basis for uniformity in collaborative studies. By the time the Amsterdam criteria have also been criticized. Some investigators feel that the criteria exclude some classic Lynch families because they do not take into account the extracolonic cancers that are tumor spectrum of the syndrome (8). It is now concluded that many true HNPCC families would be missed if the criteria are applied to clinical diagnosis, and that families not meeting the criteria might be falsely reassured and excluded from genetic counseling, DNA testing, or surveillance (27). To resolve some of these problems Rodriguez-Bigas et al. (28) developed additional taking into account the molecular and pathologic features of CRC and adenomas in addition to family history.
Taking into account some new reported data (17-21) ICG proposed definition of Lynch syndrome:
· Familial clustering of colorectal and/or endometrial cancer
· Associated cancers: cancer of the stomach, ovary, ureter/renal pelvis, brain, small bowel, hepatobiliary tract, and skin (sebaceous tumors)
· Development of cancer at early age
· Development of multiple cancers
· Features of CRC: 1) predilection for proximal colon; 2) improved survival; 3) multiple CRC; 4) increased proportion of mucinous tumors, poorly differentiated tumors, and tumors with marked host-lymphocytic infiltration and lymphoid aggregation at the tumor margin
· Features of colorectal adenoma: 1) the numbers vary from one to a few; 2) increased proportion of adenomas with a villous growth pattern; 3) a high degree of dysplasia, and 4) probably rapid progression from adenoma to carcinoma
· High frequency of MSI (MSI-H)
· Immunohistochemistry: loss of MLH1, MSH2, or MSH6 protein expression
· Germline mutation in MMR genes (MSH2, MLH1, MSH6, PMS1, PMS2).
The new set of clinical criteria (Amsterdam criteria II-Revised ICG-HNPCC criteria) was proposed and accepted by the majority of ICG-HNPCC group (27):
· There should be at least 3 relatives with and HNPCC-associated cancer (CRC, cancer of the endometrium, small bowel ureter, or renal pelvis)
· One should be a first-degree relative of the other two
· At least two successive generations should be affected
· At least one should be diagnosed before age 50
· Familial adenomatous polyposis should be excluded in the CRC(s) if any
· Tumors should be verified by pathological examination.
It is important in the discussion of criteria, that criteria aim to provide a common nomenclature for the selection of families for studies and for the comparison of the results of these studies. Criteria are not permanent, they will change as our knowledge of Lynch syndrome advances (27).
There are a few forms or variants of HNPCC. Muir-Torre syndrome (6,29-31) is characterized by tumor spectrum of Lynch syndrome and skin tumors.
“Café-au-lait spots”(macular, sharply demarcated, evenly oval pigmented spots, present at birth) is another variant of the Lynch syndrome (32), characterized with early onset of CRC, oligopolyposis, glioblastoma and lymphoma.
To the best of my known this report is the first in our literature. Reported family “S” fulfills “minimal Amsterdam”(Amsterdam Criteria I) and Revised ICG criteria (Amsterdam Criteria II). More than 25% (11) of family members were affected with malignancy; three successive generations were involved. In 75% of affected members CRC was detected proximal to the splenic flexure and correlate with literature data (33-35). Metachronous CRC were detected 4-23 years after initial CRC in 27.2% of affected family members and this finding correlate with literature data (36,37).
In 2 (18.3%) patients stomach cancer was detected. There are conflicting data in literature (27,38) about inclusion of the stomach cancer in the tumor spectrum of the Lynch syndrome. Some authors considered that gastric carcinoma is the second most common extra-colonic malignancy associated with Lynch syndrome (39,40). The cumulative risk of carcinoma of the stomach in putative HNPCC gene carriers has been estimated at 19 % (41). The predominance of intestinal type of gastric cancer in Lynch syndrome families (as in our report) was unexpected because the diffuse type usually is associated with familial occurrence (42). It is considered that high prevalence of stomach cancer in some Asian countries may occasionally result in the chance of familial aggregation of CRC and stomach cancer; the inclusion of stomach cancer in the criteria might therefore decrease the certainty that we were dealing with HNPCC (27).
Periodic screening of patients with Lynch syndrome and healthy family members of HNPCC families is a secondary prevention of carcinoma and may lead to reduction of morbidity and mortality. Patients should be screened not only for CRC, but also for other malignancy of HNPCC tumor spectrum.
The American College of Gastroenterology recommend that a complete colonoscopy be performed in members of a family who meet any of the revised Amsterdam criteria for Lynch syndrome every 1-3 years beginning at the age of 20-25 years when the earliest cases of CRC can occur, until the of 40 years (40,43,44). From 40 years, yearly colonoscopy should then be performed (43,45).
Genetic tests are available for the diagnosis of Lynch syndrome in families suspected of having the disease based on the revised criteria (27). Unfortunately these tests are not available routinely in our country. The first test should be perform in the youngest family member with CRC (46). If the mutation is detected in the index patient, then the accuracy of the test in family members approaches 100% since all family members inherit the same mutation. These results are important to decide who needs colonoscopic screening. Genetic tests for HNPCC are far from ideal, so periodical screening of all family members of Lynch syndrome families have to be performed until genetic tests become routinely available.
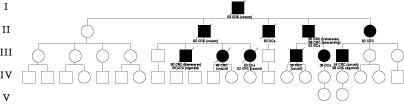
Figure 1. Pedigree of family “S”. (CRC - colorectal cancer, GCa -
gastric cancer; ECa - endometrial cancer; OCa - ovarian cancer)
REFERENCES:
1. Souza RF. Review article: a molecular rationale for the how, when and why colorectal cancer screening. Aliment Pharmacol Ther 2001; 15: 451-62.
2. Warthin
AS. Heredity with reference to carcinoma. Arch Intern Med 1913; 12: 546-55.
3. Lynch HT, Watson P,
Lanspa SJ, et al. Natural history of colorectal cancer in hereditary
nonpolyposis colorectal cancer (Lynch syndromes I and II). Dis Colon Rectum
1988; 31: 439-44.
4. Vasen HFA, den Hartog Jager FCA, Menko FH, Nagengast FM. Screening
for hereditary non-polyposis colorectal cancer. A study of 22 kindreds in the
Netherlands. Am J Med 1989; 86: 278-81.
5. Mecklin JP, Sipponen P, Järvinen HJ. Histopathology of colorectal
carcinoma and adenomas in cancer family syndrome. Dis Colon Rectum 1986; 29:
849-53.
6. Rustgi AK. Hereditary gastrointesstinal polyposis and nonpolyposis
syndromes. N Engl J Med 1994; 331: 1694-702.
7. Lynch HT, Schuelke GS, Kimberlingg WJ, et al. Hereditary nonpolyposis
colorectal cancer (Lynch syndromes I and II). II. Biomarker studies. Cancer
1985; 56: 939-51.
8. Vasen HF, Mecklin JP,
Khan PM, Lynch HT. The International Collaborative Group on Hereditary
Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991; 34: 424-5.
9. Watson P, Lynch HT.
Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer 1993;
71: 677-85.
10. Fishel R, Lescoe
MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with
hereditary non-polyposis colon cancer. Cell 1994; 77: 167.
11. Leach FS,
Nicolaides NC, Papadopoulos N, et al. Mutatuons of a mutS homolog in hereditary
nonpolyposis colorectal cancer. Cell 1993; 75: 1215-25.
12. Bronner CE, Baker
SM, Morrison PT, et al. Mutation in the DNA mismatch repair gene homologue
hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994;
368: 258-61.
13. Nicolaides NC,
Papadopoulos N, Liu B, et al. Mutations of two PMS homologues in hereditary
nonpolyposis colon cancer. Nature 1994; 371: 75-80.
14. Malkhosyan S,
Rampino N, Yamamoto H, Perucho M. Frameshift mutator mutations. Nature 1996;
382: 499-500 (Letter).
15. Yin J, Kong D, Wang
S, et al. Mutation of hMSH3 and hMSH6 mismatch repair genes in genetically
unstable human colorectal and gastric carcinomas. Hum Mutat 1997; 10: 474-8.
16. Liu B, Parsons RE,
Hamilton SR, et al. hMSH2 mutations in hereditary nonpolyposis colorectal
cancer kindreds. Cancer Res 1994; 54: 4590-4.
17. Han HJ, Maruyama M,
Baba S, Park JG, Nakamura Y. Genomic structure of human mismatch repair gene,
hMLH1, and its mutation analysis in patients with hereditary non-polyposis
colorectal cancer (HNPCC). Hum Mol Genet 1995; 4: 237-42.
18. Lynch HT, Smyrk TC,
Watson P, et al. Genetics, natural history, tumor spectrum, and pathology of
hereditary nonpolyposis colorectal cancer: an updated review. Gastroenterology
1993; 104: 1535-49.
19. Marra G, Boland CR.
Hereditary nonpolyposis colorectal cancer: the syndrome, the genes, and
historical perspectives. J Natl Cancer Inst 1995; 87: 1114-25.
20. Lynch HT, Smyrk T,
Lynch J. An update of HNPCC (Lynch syndrome). Cancer Genet Cytogenet 1997; 93:
84-99.
21. Jass JR. Diagnosis
of hereditary nonpolyposis colorectal cancer. Histopathology 1998; 32: 491-7.
22. Aaltonen LA,
Salovaara R, Kristo P, et al. Incidence of hereditary nonpolyposis coloectal
cancer and the feasibility of molecular screening for the disease. N Engl J Med
1998; 338: 1481-7.
23. Liu B, Parsons R,
Papadopoulos N, et al. Analysis of mismatch repair gene in hereditary non
polyposis colorectal cancer patients. Nat Med 1996; 2: 169-74.
24. Nystrom-Lahti M, Wu
Y, Moisio M, et al. DNA mismatch repair gene mutations in 55 kindreds with
verified or putative hereditary non-polyposis colorectal cancer. Hum Mol Genet
1996; 38: 763-76.
25. Viel A, Genuardi M,
Capozzi E, et al. Characterization of MSH2 and MLH1 mutations in Italian
families with hereditary non-polyposis colorectal cancer. Genes Chromosom
Cancer 1997; 18: 8-18.
26. Weber TK, Conlon W,
Petrelli NJ, et al. Genomic DNA-based hMSH2 and hMLH1 mutation screening in 32
eastern United States hereditary nonpolyposis colorectal cancer pedigrees. Cancer
Res 1997; 57: 3798-803.
27. Vasen HFA, Watson
P, Mecklin J-P, Lynch HT, and the ICG-HNPCC. New clinical criteria for
hereditary Nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by
the International Collaborative Group on HNPCC. Gastroenterology 1999; 116:
1453-6.
28. Rodriguez-Bigas MA,
Boland CR, Hamilton SR, et al. A National Cancer Institute workshop on HNPCC
syndrome: meeting hihglights and Bethesda guidelines. J Natl Cancer Inst 1997;
89: 1785-1792.
29. Anderson DE. An
inherited form of large bowel cancer: Muir’s syndrome. Cancer 1980; 45: 1103-7.
30. Lynch HT, Lynch PM,
Restep J, Fusaro KM. The cancer family syndrome: Rare cutaneous phenotypic
linkage of Torre’s syndrome. Arch Intern Med 1981; 141: 607-11.
31. Godard V, Coulet F,
Bernaudin J-F, Housset M, Soubrier F. Mutation oh MSH2 gene in Muit-Torre
syndrome. Ann Dermatol Venereol 1999; 126: 1-4.
32. Trimbath JD,
Petersen GM, Erdman SH, Ferre M, Luce MC, Giardiello FM. Café-au=lait spots and
early onset colorectal neoplasia; a variant of HNPCC? Familial Cancer 2001; 1:
101-5.
33. Nagorni A.
Hereditary non-polyposis colorectal cancer. Arch Gastroenterohepatol 1996; 15:
116-22.
34. Lovett E. Family
studies in cancer of the colon and rectum. Br J Surg 1976; 63: 13-8.
35. Lynch PM, Lynch HT,
Harris RE. Hereditary proximal colonic cancer. Dis Colon Rectum 1977; 20:
661-8.
36. Mecklin JP,
Järvinen HJ. Clinical features of colorectal carcinoma in cancer family
syndrome. Dis Colon Rectum 1986; 29: 160-4.
37. Lynch HT, Watson P,
Lanspa SJ, et al. Natural history of colorectal cancer in hereditary
nonpolyposis colorectal cancer (Lynch syndrome I and II). Dis Colon Rectum
1988; 31: 439-44.
38. Aarnio M, Salovaara
R, Aaltonen LA, Mecklin J-P, Järvinen HJ. Features of gastric cancer in
hereditary non-polyposis colorectal cancer syndrome. Int J Cancer 1997; 74:
551-5.
39. Vasen HFA,
Offerhaus GJA, Den Hartog Jager FCA, et al. The tumor spectrum in hereditary
non-polyposis colorectal cancer: a study of 24 kindreds in the Netherlands. Int
J Cancer 1990; 46: 31-4.
40. Lynch HT, Smyrk TC,
Watson P, et al. Genetics, natural history, tumor spectrum, and pathology of
hereditary non-polyposis colorectal cancer: an updated review. Gastroenterology
1993; 104: 1535-49.
41. Aarnio M, Mecklin
J-P, Aaltonen LA, Nystrom-Lahti M, Järvinen HJ. Life-time risk of different
cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J
Cancer 1995; 64: 430-433.
42. Lehtola J. Family
study of gastric carcinoma: with special reference to histological types. Scand
J Gastroenterol 1978; 13 (suppl. 50): 1-54.
43. Rex DK, Johnson DA,
Lieberman DA, Burt RW, Sonnenberg A. Colorectal cancer prevention 2000:
screening recommendations of the American College of Gastroenterology. Am J
Gastroenterol 2000; 95: 868-77.
44. Terdiman JP, Conrad
PG, Sleisenger MH. Genetic testing in hereditary colorectal cancer: indications
and procedures. Am J Gastroenterol 1999; 94: 2344-56.
45. Järvinen HJ,
Mecklin JP, Sistonen P. Screening reduces colorectal cancer rate in families
with hereditary nonpolyposis colorectal cancer (see comments). Gastroenterology
1995; 108: 1405-11.
46. Burt RW. Colon
cancer screening. Gastroenterology 2000; 119: 837-53.