 |
|||
|
|||
Alimentarni trakt i pankreas
ARCH GASTROENTEROHEPATOL 2001; 20 (No 3 – 4):
Editorial
Department of Histopathology, Institute of Digestive Diseases, Clinical Center of Serbia, Belgrade
Address correspondence to: Assistant Professor Marjan Micev, MD, M.Sci.
Institute for Medical Research, University of Belgrade
Dr Subotica 4 St.
PO BOX 721
YU-110001 Belgrade, Serbia, Yugoslavia
FAX + 381 11 643 691
E-mail: [email protected]
………………… ……………………………..
H.pylori, gastric metaplasia, duodenal ulcer Gastroenteroloska sekcija SLD-
01717, 2001.
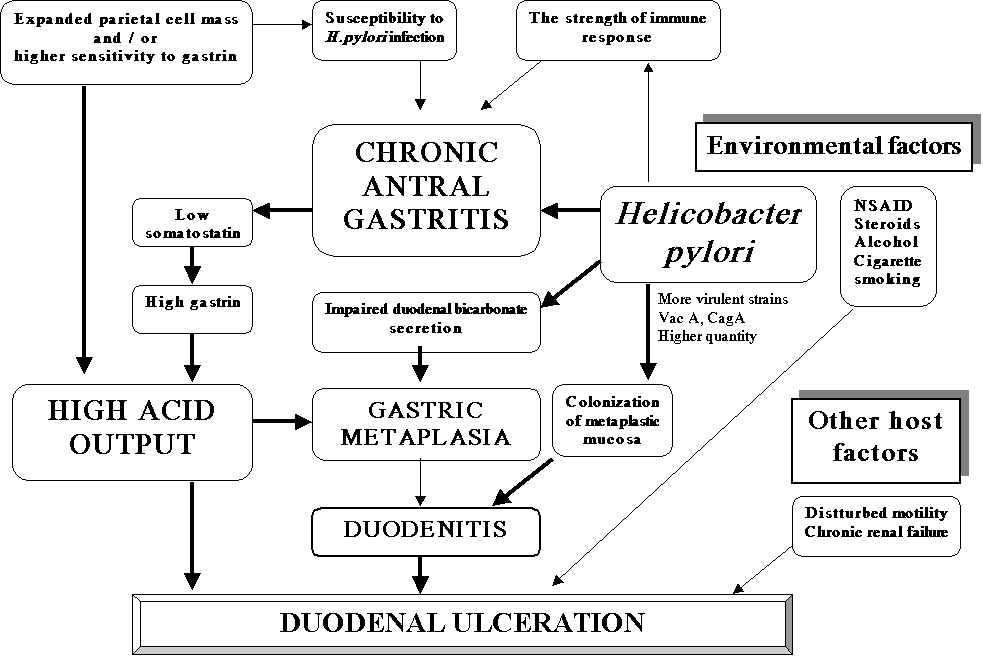
Despite the fact that proximal gastrointestinal endoscopy and mucosal biopsies have been practices for more than 45 years thus enabling the scientific understanding of peptic diseases, the introduction of a bacterial cause for ulcers in 1981 by Australian pathologist, J. Robin Warren brought the major progress in understanding the aetiology of peptic ulcer disease. Discovery that Helicobacter pylori may infect only gastric epithelium rises an interest in the role of this bacterium in the pathogenesis of duodenal ulcer (1,2). The presence of duodenal gastric metaplasia (GM) (synonymous: foveolar cell metaplasia, gastric surface epithelial metaplasia), which is common finding in peptic duodenitis and duodenal ulcer (DU) seems to be an important clue for understanding of the aetiopathogenesis of DU integrating the sequences of changes like duodenal mucosal inflammation, degenerative and regenerative changes of epithelial cells, mucosal hemorrhage and oedema as well as Brunner`s gland hyperplasia.
It is often stated that through some mechanism, e.g. rapid gastric emptying of acid into the duodenum, local mucosal neutralizing capacity may be impaired thus leading to the development of duodenal bulb GM. This sets the stage for acquisition of H. pylori infection, subsequent mucosal inflammation with ensuing epithelial cell necrosis, formation of erosions, and development of the ulceration. As a rule, DU develops in areas of GM (3). In addition, the cyclical occurrence of DU is presumably due to repeated development and ulcerative destruction of such mucosal patches of GM.
Until recently, the development of peptic ulcer disease was thought to be caused only by an imbalance between aggressive factors (acid secretion, pepsinogen production) and defensive factors (mucus, bicarbonate secretion, mucosal blood flow, gastric mucosal barrier, cellular regeneration). The isolation of H. pylori from patients with chronic gastritis, DU, and gastric ulcer has revolutionized our thinking about the pathogenesis of peptic ulcer disease. Even though our understanding of vital role of H. pylori in the aetiopathogenesis of DU has changed dramatically due to worldwide studies of this bacterium over the last couple of decades, it remains unclear to what extent this infection is interrelated with complex interactions between host defense mechanism and elevated acid, pepsin levels with(out) other environmental factors (4). Current data imply that persistent infection with H. pylori may account for the high recurrence rates and the chronicity of peptic ulcer disease. Furthermore, up to 50% of the population in developed countries have an evidence of H. pylori infection by the age of 50 years. The organism is present in up to 92% of patients with active chronic gastritis, 88-100% with DU, 58-100% with gastric ulcer, and 46-94% with gastric cancer. In DU patients, eradication of this organism has been shown to markedly reduce ulcer recurrence rate and possibly change the natural history of this disease. Further evidences linking H. pylori to DU were made after several trials confirming that preexisting H. pylori infection is a risk factor for the development of DU (5,6,7). However, while the association between H. pylori and DU is strong, it is not specific. H. pylori is absent in up to 30 percent of patients with an endoscopically proven DU (8,9). A regular use of nonsteroidal anti-inflammatory drug (NSAID) and variety of other pro-ulcerogenic agents are responsible for the remaining cases. High acid output may be a cause of recurrent DUs in patients in whom H. pylori has been eradicated (10).
Multifactorial pathogenesis of DU is primarily associated
with increased acid output and active chronic duodenitis,
while gastric ulcer is primarily caused by altered mucosal
defense (10,11,12). How host factors may participate in DU
pathogenesis? There is no doubt that pre-existent increased
parietal cell mass, increased pepsinogen production, chronic renal
disease, disturbed motility, and unclear genetic predisposition,
especially the exaggerated acid response to various stimuli in
predisposed patients may promote DU development. It has been
suggested that genetic susceptibility to H. pylori infection
in patients who develop DUs could be rely on an intrinsically
higher parietal cell mass or higher sensitivity to gastrin than in
H. pylori-positive healthy adults (13,14,15). It is noticed
that patients with DU are colonised with H. pylori strains,
which are more adhesive to gastric epithelium than strains isolated
from patients with gastritis only. Furthermore, there is
genetic variation in cytokine expression, such as
IL-1bproduction, which is likely to determine the
strength of the inflammatory response to H. pylori and its
effects on acid output (16).Environmental agents such
as non-steroid anti-inflammatory drugs, steroids, alcohol,
cigarette smoking may contribute to the injury by breaking down
mucosal defenses. But H. pylori plays the most important
role in the genesis of DU, affecting some aspects of intestinal and
mucosal physiology, e.g. increased gastric acid secretion, presence
of GM, immune response, mucosal defense mechanisms (17).
The most common histopathological finding in DU patients
is antral-predominant gastritis and duodenal gastric
metaplasia. Both are attributable to H. pylori infection.
These changes define the DU diathesis (18). The virulence of
many, but not all, H. pylori strains is related to a number
of factors, some genetically determined. The most prominent one
appear to be the production of ammonium hydroxide (urease action),
phospholipases, the elaboration of cytotoxins, and inflammatory
cytokines which are putative damaging factors such as neutrophil
activating factor, and activation of the gastric
epithelium-associated immune system. Infection by more
virulent H. pylori strains, which produce the vacuolating
(vacA) toxin and the cagA protein, is accompanied by more active
inflammation. This led to more severe immune response,
aattributed to increased production of inflammatory
cytokines (19). This is in part related to stimulation of
interleukin (IL)-8 production by the epithelial cells. This is well
known potent chemotactic factor that activates neutrophils and
releases the intercellular adhesion molecule ICAM-1. Other
cytokines such as IL-1b, IL-6 and
TNFacan influence the strength of immune response,
but also can directly suppress parietal cell function (20).
Approximately 85-100% of patients with DU has CagA+ strains,
compared to 30-60% of infected patients who do not develop DU
(21).
Antral H.pylori infection induces gastric acid hypersecretion via interfering with mechanisms which normally inhibit gastrin release and acid secretion. On the contrary gastric acid hypersecretion itself (in subjects with intrinsically higher acid output) can develop an antral-predominant gastritis, nice “milieu” for H.pylory harbouring. This pre-infection high acid output is determined by previously mentioned genetic, nutritional and other factors. Certain serotypes of H. pylori inhibit antral D-cell function and down-regulate somatostatin secretion, which is a potent inhibitor of both gastrin synthesis and release, and gastric acid secretion (22). A number of other substances also inhibit parietal cell function thus suppressing gastric acid secretion via inhibitory G protein (G1) which supress adenylate cyclase. These substances are: prostaglandins, secretin, GIP, peptide YY etc. Somatostatin also acts by inhibiting the ECL cell, thereby suppressing histamine release, and/or may act directly on the parietal cell as well. Inflammation appears to impair this regulating function of somatostatin (23).
Another possible mechanism of gastric acid hypersecretion in DU is via increased gastrin release to normal stimuli. This GI hormone has a trophic action on parietal cells and histamine-secreting enterochromaffin-like (ECL) cells, and stimulates parietal cells largely via histamine release. In a case of H. pylori infection, basal and stimulated concentrations of serum gastrin is elevated; somatostatin concentration is decreased (23,24,25). Nevertheless, hypergastrinemia alone could hardly explain the increases in acid output in patients infected with H. pylori, although gastrin levels often return to normal within one month after its eradication. In addition, H. pylori itself does not appear to alter the sensitivity of gastric parietal cells to gastrin, although hypergastrinemia may have trophic effects over the time resulting parietal cell mass increase (26).
Several important mucosal defense factors may be
downregulated by H. pylori infection such as epidermal
growth factor (EGF) and transforming growth factor-alpha (TGF
alpha) synthesis. These peptides are potent suppressor of gastric
acid secretion, promoters of mucosal growth, proteases inductors,
and inductors of other mucous glycoprotein-degrading enzymes (27).
In addition, most patients with DUs have impaired
proximal duodenal mucosal bicarbonate secretion to intraluminal
acidification. This normalises after H. pylori eradication.
Mucosal bicarbonate secretion seems to be unrelated with associated
histopathologic abnormalities. In DU patients, impaired mucosal
bicarbonate secretion may be caused by a cellular and/or
physiological regulatory transport defect possibly related to H.
pylori (28). The presence of H. pylori -induced
asymmetric dimethyl arginin (ADMA) could interfere with NO
synthetase in the duodenal epithelial cells and contributes to an
impairment of the bicarbonate response to duodenal acidification.
The precise mechanism by which H. pylori contribute to DU formation is not known. However, current data suggest that the most probable sequence of events start with H. pylori infection and antral gastritis. This further leads to defective inhibition of gastrin release and acid hypersecretion. Gastric acid hypersecretion causes an increased duodenal acid load thus triggering GM in the duodenal bulb. Foveolar cell metaplasia, better known as gastric (surface epithelial) metaplasia (GM) appears to be an adaptive response of the mucosa to excessive acid exposure. It only occurs when the luminal pH < 2.5, what is possibly contributed by H. pylori-induced impaired duodenal mucosal bicarbonate secretion (28,29). The strong association of GM and active duodenitis suggests that the same factors give rise to islands of GM and probably play a role in the development of mucosal inflammation (peptic duodenitis), including metaplasia itself. This phenomenon allows migration of the bacterium from the stomach into the duodenum (22,30). Colonization of H. pylori further weakens duodenal mucosal defenses making it more susceptible to acid injury and allows progresses to erosive peptic duodenitis and ultimately to DU Table 1.
In this issue of Archives Grgov et al. discuss how
difficult is to link a singular causative factor with the aetiology
of duodenal GM. The results of this prospective study are that
there is no clear-cut association between H. pylori
infection and GM in patients with DU and non-ulcer dyspepsia
(NUD). This observation may be further supported by the fact
that Koch’s postulates are not proven for H. pylori in
peptic ulcer disease, but there is strong evidence that eradication
of bacterium significantly lower ulcer relapse rate
(31,32).
There is plenty of evidence that GM regress in patients with
eradicated H.pylori, while in non-eradicated persons there
was no significant change (33). Also, in over 50% of children DU
disease occur with both H pylori infection and GM. In
contrast, DU does not occur in children when both were absent.
Thus, the presence of GM colonised by H pylori appears as
the major risk factor for DU disease in children (34).
As it was previously stated, a critical factor for the
development of DU seems to be the quantity of virulent H.
pylori strains in the duodenal bulb. High density of
cagA-positive strains in the duodenum accompanied with severe
duodenitis is an important determinant of DU disease (35). But, the
same was not proved to be a case in NUD patients with duodenal GM
(36). Differences in the gastric topography of H.
pylori density and inflammatory scores between DU and gastric
ulcer may contribute to differences in development and presentation
of both peptic ulcer conditions. But no evidence this was not a
case in duodenal GM (37). However, some additional environmental
and host factors may be involved. The allele frequency and the
genotypes that possessed the DQA1*0102 allele were significantly
more common in H. pylori-negative than in H.
pylori-positive DU patients (38,39).
The role of GM in patients with DU or NUD is far from be
elucidated, but this phenomenon is most oftenly attributed to the
gastric acid hypersecretion and impaired duodenal mucosal
bicarbonate secretion induced by H. pylori. These factors
contribute to the low duodenal luminal pH. There are authors who
reported that the extent of duodenal GM is unrelated to the
presence or absence of ulceration but is partly due to H.
pylori and acid hypersecretion. Other group noticed that
prevalence and extent of GM are not related to H pylori
neither in patients with DU, nor in controls (40,41,42,43,44). High
acid response to gastrin may be more important
(45).It is concluded that the unchanged gastric acid
output after eradication of H. pylori is a more important
factor in the development of GM than the H. pylori related
inflammatory process (46). On the contrary, the lower gastric
acidity in patients with NUD than in patients with DU and the lack
of correlation between gastric pH and the various GM degrees in the
two H. pylori-positive populations suggest that gastric
hyperacidity may not be associated with duodenal GM and the
disappearance of H. pylori infection does not determine any
increase in gastric pH and any reversal of gastric-type epithelium
in the duodenum (47,48). Among conflicting reports on this matter,
some authors demonstrated a low incidence of GM and H.
pylori in the duodenum in patients with DU or that the
prevalence and the extent of GM were not related to H.
pylori in patients with DU or NUD. Also, the prevalence and the
extent of GM did not change until 1 year after H. pylori
eradication (49,50).
Despite the fact that whether GM cells originate from pluripotent
stem cells, possibly under the control of homeobox genes or from
Brunner`s gland duct cells, this phenomenon is a consequence of
disturbed mucosal renewal in altered local conditions and/or
stimuli (51). The process of proliferation and cellular
differentiation are topologically well organized with gradients in
gene expression established and maintained within duodenal
epithelium. The present findings indicate that GM is a reversible,
although this process is not dynamic and requires more time. The
presence of GM in the duodenum after H. pylori eradication
indicates an increased risk for the recurrence of DUs, thus
providing a useful information for a clinician (52). Increased IL-8
activity in the duodenal mucosa with GM and the impaired PGE2
generation in DU disease may be important for ulcerogenesis in
H. pylori-positive DU patients (53, 54). Some other findings
support the putative role of secretory peptides expressed in
gastric epithelial cells and in GM at the margin of DU ( trefoil
peptide pS2, possible human spasmolytic polypeptide -hSP) in
mucosal healing. This provides further evidence for an autocrine
'ulcer-GM-repair' loop involves this trefoil peptide (55).
Furthermore, an alteration in the catecholaminergic system (reduced
mucosal norepinephrine concentration in the outer edge of DU) may
be associated with one of the pathogenic factors of DU (56). The
prevalence of parietal cells in the duodenal bulb is notably higher
than previously reported in endoscopic studies, but does not
contribute to the pathogenesis of DU in the duodenal bulb (57).
Methylene blue mucosal staining helps to investigate the
extent of GM of the duodenal bulb. This method demonstrates higher
incidence of GM in the duodenal bulb in patients with healed DU
than in non-ulcer patients (58,59).
The extent of GM is often classified as focal (grade 1),
multifocal (grade 2), and diffuse type (grade 4). According to the
amount of mucus in the metaplastic cells GM can be divided into
three types: complete, intermediate and incomplete. It was recently
reported that the complete GM was frequently detected in the H.
pylori-negative group, whereas the incomplete type was
frequently observed in the H. pylori-positive group. After
eradication, the incomplete type changed to the complete type with
a decrease of histological inflammation (60). Using
the methylene blue test and competitive polymerase chain reaction,
Futarni concluded that the amount of H. pylori in the
duodenal bulb might be related to the amount of H. pylori in
the gastric antrum and to the extent of GM in the duodenal bulb
(61). There are studies that showed a healed DU with a
normal-shaped bulb, which is not frequently accompanied by GM. On
the contrary, a healed ulcer with a markedly deformed bulb has a
high incidence and degree of GM (62, 63). At this point it is
questionable whether GM represent causal factor or a consequence of
a series of chronic local disorders in the duodenal bulb. It is
hard to believe that H. pylori is an innocent bystander in
cases of duodenal peptic disease with developed GM, although it is
quite possible that it is not always mandatory and/or solely
sufficient.
Marshall BJ. The Campylobacter pylori story. Scand J
Gastroenterol Suppl
1988;146:58-66.
McGowan CC, Cover TL, Blaser MJ. Helicobacter pylori and gastric acid: biological and therapeutic implications. Gastroenterology 1996;110:926-8.
Fenoglio-Preiser CM, Noffsinger AE, Stemmermann GN, Lanty PE, Listrom MB, Rilke FO, eds. Gastrointestinal pathology, 2nd ed. Philadelphia-New York: Lippincott-Raven Publishers, 1999.
Dixon MF: Acid, ulcers and H pylori. Lancet 1993, 342:384-5.
Sipponen, P, Varis, K, Fraki, O, et al. Cumulative 10-year risk of symptomatic duodenal and gastric ulcer in patients with or without chronic gastritis: A clinical follow-up study of 454 patients. Scand J Gastroenterol 1990; 25:966.
Hopkins, RJ, Girardi, LS, Turney, EA. Relationship between H. pylori eradication and reduced duodenal and gastric ulcer recurrence: A review. Gastroenterology 1996; 110:1244.
Leoci, C, Ierardi, E, Chiloiro, M, et al. Incidence and risk
factors of duodenal ulcer. J Clin Gastroenterol 1995; 20:104.
Ciociola, AA, McSorley, DJ, Turner, K, et al. Helicobacter pylori infection rates in duodenal ulcer patients in the United States may be lower than previously estimated. Am J Gastroenterol 1999; 94:1834.
Laine, L, Hopkins, RJ, Girardi, LS, et al. Has the impact of Helicobacter pylori therapy on ulcer recurrence in the United States been overstated? A meta-analysis of rigorously designed trials. Am J Gastroenterol 1998; 93:1409.
Harris, AW, Gummett, PA, Phull, PS, et al. Recurrence of duodenal ulcer after Helicobacter pylori eradication is related to high acid output. Aliment Pharmacol Ther 1997; 11:331
El-Omar, EM, Penman, ID, Ardill, JE, et al. Helicobacter pylori infection and abnormalities of acid secretion in patients with duodenal ulcer disease. Gastroenterology 1995; 109:681-1.
Collen MJ, Sheridan MJ. Gastric ulcers differ from duodenal ulcers. Evaluation of basal acid output. Dig Dis Sci 1993; 38:2281-6
Graham, DV. Helicobacter pylori and perturbations in acid
secretion: The end of the beginning. Gastroenterology 1996;
110:1647.
Peura, DA. Ulcerogenesis: Integrating the roles of Helicobacter pylori and acid secretion in duodenal ulcers. Am J Gastroenterol 1997; 92:8S.
Gillen, D, El-Omar, E, McColl, K. Parietal cell sensitivity to
gastrin distinguishes helicobacter pylori infected patients from
infected healthy volunteers. Gut 1995; 37 Suppl 1:A8.
El-Omar EM. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000; 404: 398-402.
McColl KE, el-Nujumi AM, Chittajallu RS, Dahill SW, Dorrian CA, el-Omar E, et al. A study of the pathogenesis of Helicobacter pylori negative chronic duodenal ulceration. Gut 1993;34:762-8
Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis: The updated Sydney System. American Journal of Surgical Pathology 1996; 20: 1161-81.
Warburton VJ, Everett S, Mapstone NP, Axon ATR, Hawkey P, Dixon MF. Clinical and histological associations of cagA and vacA genotypes in Helicobacter pylori gastritis. J Clin Pathol 1998; 51: 55-61
Beales I, Calam J. Interleukin 1b and tumour necrosis factor-a inhibit acid secretion in cultured rabbit parietal cells by multiple pathways. Gut 1998; 42: 227-34.
Weel, JF, van der Hulst, RW, Gerrits, Y, et al. The interrelationship between cytotoxin-associated gene A, vacuolating cytotoxin, and helicobacter pylori-related diseases. J Infect Dis 1996; 173:1171
Blaser MJ. Hypotheses on the pathogenesis and natural history of Helicobacter pylori-induced inflammation. Gastroenterology 1992;102:720-7
Calam, J. The somatostatin-gastrin link of helicobacter pylori infection. Ann Med 1995; 27:569
Peterson, WL, Barnett, CC, Evans, DJ, et al. Acid secretion and serum gastrin in normal subjects and patients with duodenal ulcer: The role of H. pylori. Am J Gastroenterol 1993; 88:2038.
El-Omar, E, Penman, I, Dorrian, CA, et al. Eradicating Helicobacter pylori infection lowers gastrin mediated acid secretion by two thirds in patients with duodenal ulcer. Gut 1993; 34:1060
Moss, SF, Calam, J. Acid secretion and sensitivity to gastrin In patients with duodenal ulcer: Effect of eradication of Helicobacter pylori. Gut 1993; 34:888
Konturek, PC, Ernst, H, Konturek, SJ, et al. Mucosal expression and luminal release of epidermal and transforming growth factors in patients with duodenal ulcer before and after eradication of Helicobacter pylori. Gut 1997; 40:463
Hogan, DL, Rapier, RC, Dreilinger, A, et al. Duodenal
bicarbonate secretion: Eradication of Helicobacter pylori and
duodenal structure and function in humans. Gastroenterology 1996;
110:705
Wyatt, JI, Rathbone, BJ, Dixon, MF, et al. Campylobacter pyloridis and acid induced gastric metaplasia in the pathogenesis of duodenitis. J Clin Pathol 1987; 40:841
Walker MM, Dixon MF. Gastric metaplasia: its role in duodenal ulceration. Alimentary Pharmacology and Therapeutics 1996; 10 (Suppl. 1): 119-28.
Graham DY, Lew GM, Evans DG, et al. Effect of triple therapy (antibiotics plus bismuth) on duodenal ulcer healing: a randomized controlled trial. Ann Intern Med 1991;115:266-9
Forbes GM, Glaser ME, Cullen DJ, et al. Duodenal ulcer treated with Helicobacter pylori eradication: seven-year follow-up. Lancet 1994; 343:258-60
Khulusi S, Mendall MA, Badve S, Patel P, Finlayson C, Northfield TC. Effect of Helicobacter pylori eradication on gastric metaplasia of the duodenum. Gut 1995; 36:193-7
Gormally SM, Kierce BM, Daly LE, Bourke B, Carroll R, Durnin MT et al. Gastric metaplasia and duodenal ulcer disease in children infected by Helicobacter pylori. Gut 1996; 38:513-7
Hamlet A, Thoreson AC, Nilsson O, Svennerholm AM, Olbe L. Duodenal Helicobacter pylori infection differs in cagA genotype between asymptomatic subjects and patients with duodenal ulcers. Gastroenterology 1999; 116:259-68
Heikkinen M, Pikkarainen P, Vornanen M, Hollmen S, Julkunen R. Prevalence of gastric metaplasia in the duodenal bulb is low in Helicobacter pylori positive non-ulcer dyspepsia patients. Dig Liver Dis 2001; 33:459-63
Jonkers D, Houben G, de Bruine A, Arends JW, Stobberingh E, Stockbrugger R. Prevalence of gastric metaplasia in the duodenal bulb and distribution of Helicobacter pylori in the gastric mucosa. A clinical and histopathological study in 96 consecutive patients. Ital J Gastroenterol Hepatol 1998; 30:481-3
Hawkey CJ. Risk of ulcer bleeding in patients infected with Helicobacter pylori taking non-steroidal anti-inflammatory drugs. Gut 2000;46:310-1
Azuma T, Konishi J, Ito Y, Hirai M, Tanaka Y, Ito S, Kato T, Kohli Y. Genetic differences between duodenal ulcer patients who were positive or negative for Helicobacter pylori. J Clin Gastroenterol 1995; 21 Suppl 1:S151-4
Kerrigan DD, Read NW, Taylor ME, Houghton LA, Johnson AG. Duodenal bulb acidity and the natural history of duodenal ulceration. Lancet 1989; 2:61-3
Caselli M, Trevisani L, Aleotti A, Bovolenta MR, Stabellini G. Gastric metaplasia in duodenal bulb and Campylobacter-like organisms in development of duodenal ulcer. Dig Dis Sci 1989; 34:1374-8
Collen MJ, Sheridan MJ. Gastric ulcers differ from duodenal ulcers. Evaluation of basal acid output. Dig Dis Sci 1993; 38:2281-6
Yang H, Dixon MF, Zuo J, Fong F, Zhou D, Corthesy I, Blum A. Helicobacter pylori infection and gastric metaplasia in the duodenum in China. J Clin Gastroenterol 1995; 20:110-2
Khulusi S, Badve S, Patel P, Lloyd R, Marrero JM, Finlayson C, Mendall MA, Northfield TC. Pathogenesis of gastric metaplasia of the human duodenum: role of Helicobacter pylori, gastric acid, and ulceration. Gastroenterology 1996;110:452-8
Harris AW, Gummett PA, Walker MM, Misiewicz JJ, Baron JH.: Relation between gastric acid output, Helicobacter pylori, and gastric metaplasia in the duodenal bulb. Gut 1996; 39:513-20
Noach LA, Rolf TM, Bosma NB, Schwartz MP, Oosting J, Rauws EA, Tytgat GN. Gastric metaplasia and Helicobacter pylori infection. Gut 1993; 34:1510-4
Savarino V, Mela GS, Zentilin P, Lapertosa G, Ceppa P, Vigneri S et al. 24-hour gastric pH and extent of duodenal gastric metaplasia in Helicobacter pylori-positive patients. Gastroenterology 1997; 113:741-5
Savarino V, Mela GS, Zentilin P, Mele MR, Bisso G, Pivari M et al. Effect of Helicobacter pylori eradication on 24-hour gastric pH and duodenal gastric metaplasia. Dig Dis Sci 2000; 45:1315-21
Amarapurkar DN, Parikh SS, Prabhu SR, Kalro RH, Desai HG. Is gastric metaplasia essential for duodenal ulcer? J Clin Gastroenterol 1993 Oct;17(3):204-6
Kim N, Lim SH, Lee KH, Choi SE:.Long-term effect of Helicobacter pylori eradication on gastric metaplasia in patients with duodenal ulcer. J Clin Gastroenterol 1998; 27:246-52
Shaoul R, Marcon P, Okada Y, Culz E, Forstner G. The pathogenesis of duodenal gastric metaplasia: the role of local goblet cell transformation. Gut 2000; 30:397-403
Rudnicka L, Bobrzynski A, Stachura J. Short-term eradication therapy for Helicobacter pylori does not reduce the incidence of gastric metaplasia in duodenal ulcer patients. Pol J Pathol 1997;48:103-6
Noshiro M, Kusugami K, Sakai T, Imada A, Ando T, Ina K, Nobata K, Morise K, Kaneko H, Ito M, Nishio Y. Gastric metaplasia in the duodenal bulb shows increased mucosal interleukin-8 activity in Helicobacter pylori-positive duodenal ulcer patients. Scand J Gastroenterol 2000; 35:482-9
Pugh S, Jayaraj AP, Bardhan KD. Duodenal mucosal histology and histochemistry in active, treated and healed duodenal ulcer: correlation with duodenal prostaglandin E2 production. J Gastroenterol Hepatol 1996; 11:120-4
Khulusi S, Hanby AM, Marrero JM, Patel P, Mendall MA, Badve S, Poulsom R, Elia G, Wright NA, Northfield TC.Expression of trefoil peptides pS2 and human spasmolytic polypeptide in gastric metaplasia at the margin of duodenal ulcers. Gut 1995; 37:205-9
Kaise M, Echizen H, Umeda N, Ishizaki T. Catecholamine concentrations in biopsied gastroduodenal tissue specimens of patients with duodenal ulcer. Dig Dis Sci 1993; 38:1866-73
Harris AW, Walker MM, Smolka A, Waller JM, Baron JH, Misiewicz JJ. Parietal cells in the duodenal bulb and their relation to Helicobacter pylori infection. J Clin Pathol 1996; 49:309-12
Chang CC, Pan S, Lien GS, Chen SH, Cheng CJ, Liu JD, Cheng YS, Suk FM. Investigation of the extent of gastric metaplasia in the duodenal bulb by using methylene blue staining. J Gastroenterol Hepatol 2001;16:729-33
Mertz H, Kovacs T, Thronson M, Weinstein W. Gastric metaplasia of the duodenum: identification by an endoscopic selective mucosal staining technique. Gastrointest Endosc 1998; 48:32-8
Urakami Y, Sano T. Endoscopic duodenitis, gastric metaplasia and Helicobacter pylori. J Gastroenterol Hepatol 2001; 16:513-8
Futami H, Takashima M, Furuta T, Hanai H, Kaneko E. Relationship between Helicobacter pylori infection and gastric metaplasia in the duodenal bulb in the pathogenesis of duodenal ulcer. J Gastroenterol Hepatol 1999; 14:114-9
Pan S, Lien GS, Liao CH, Chen SH.: Gastric metaplasia of regenerating duodenal mucosa and deformity of duodenal bulb: a correlative study. J Gastroenterol Hepatol 1996; 11:108-12
Chang CC, Pan S, Lien GS, Chen SH, Fang CL, Liu JD, Cheng YS,
Suk FM.: Relationship of duodenal ulcer recurrence to gastric
metaplasia of the duodenal mucosa and duodenal bulb deformity. J
Formos Med Assoc 2001; 100:304-8
Table 1. The possible role of some factors in pathogenesis of duodenal ulcer
|
|||
|
|
|||