Chromatographic separation process based on the difference in the surface interactions of the analyte and eluent molecules.
Let us consider a separation of a two component mixture dissolved in the eluent. Assume that component A has the same interaction with the adsorbent surface as an eluent, and component B has strong excessive interaction. Being injected into the column, these components will be forced through by eluent flow. Molecules of the component A will interact with the adsorbent surface and retard on it by the same way as an eluent molecules. Thus, as an average result, component A will move through the column with the same speed as an eluent.
Molecules of the component B being adsorbed on the surface (due to their strong excessive interactions) will sit on it much longer. Thus, it will move through the column slower than the eluent flow.
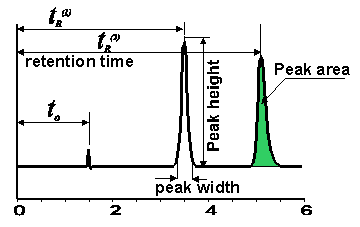
Figure below represents the general shape of the chromatogram for this mixture.
Usually a relatively narrow band is injected (5 - 20 ul injection volume). During the run, the original chromatographic band will be spread due to the noneven flows around and inside the porous particles, slow adssorption kinetics, longitudinal diffusion, and other factors. These processes together produce so called band broadining of the chromatographic zone. In general, the longer the component retained on the column, the more broad its zone (peak on the chromatogram).
Separation performance depend on both component retention and band broadening. Band broadening is, in general, a kinetic parameter, dependent on the adsorbent particle size , porosity, pore size, column size, shape, and packing performance. On the other hand, retention does not depend on the above mentioned parameters, but it reflects molecular surface interactions and depends on the total adsorbent surface.
The easiest way to find the chromatographic retention is to measure the time between the injection point and maximum of the detector response for correspondent compound. This parameter usually called "retention time". Retention time, tR is inversely proportional to the eluent flow rate.
The product of retention time and eluent flow rate, so called "retention volume", is more of a global retention parameter. Retention volume, VR represent the volume of the eluent passed through the column while eluting a particular component.
The second part is equal to the volume of the liquid phase in the column (dead volume, Vo), and it will be the same for any component eluted on this column.
Retention volume is independent of the flow parameters for the particular run, but it depend on the geometrical parameters of the column. VR will be different for the same compound eluted on the different columns packed with the same type of adsorbent.
The more universal and fundamental retention parameter is the ratio of the retention volume and dead volume (k).
k = VR/Vo
Historically, a slightly different retention parameter, called "capacity factor" (k') was introduced by the analogy with the liquid partitioning theory and widely accepted in chromatographic practice.
![]()
Capacity factor is dimentionless and independent on any geometrical parameters of the column or HPLC system. It could be considered to be a thermodynamic characteristic of the adsorbent-compound-eluent system.
After injection, a narrow chromatographic band is broaden during its movement through the column. The higher the column band broadening, the smaller the number of components that can be separated in a given time. In other words, the sharpness of the peak is an indication of how good, or efficient a column is.
The peak width is an indication of peak sharpness and, in general, an indication of the column efficiency . However, the peak width is dependent on a number of parameters (column length, flow rate, particle size). Flow rate is the only parameter which can be changed from run to run on the same column. Thus, it is better to consider a relative value to express column efficiency.
In absence of the specific interactions or sample overloading, the chromatographic peak can be represented by a Gaussian curve with the standard deviation (s). The ratio of standard deviation to the peak retention time (s/tR) is called the relative standard deviation, which is independent on the flow rate.
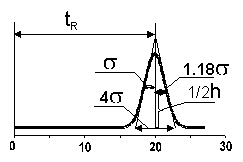
In practice, the square of the reciprocal value is normally used N-(tR/s )2 This has become the accepted expression of column efficiency. The reason for using the second power has come from statistics, and it is related to the fact that not the standard deviation (s) , but its square, the variance (s )2 , is the basic measure of normal distribution. The value N is called the plate number or the number of theoretical plates. Term "theoretical plate" comes form the analogy with the distillation theory.
In practice, it is more convenient to measure peak width either at the base line, or at the half height, and not at 0.609 of the peak height, which actually correspond to (2s).
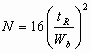
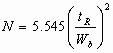
The plate number depends on column length: the longer the column, the larger the plate number. Therefore, the plate height term has been introduced to measure how efficiently column has been packed, h = L/N .
The lower the plate height and the higher the plate number, the more efficient the chromatographic column.
Until now, retention and efficiency was discussed separately, but both of these parameters are affecting the separation of the mixture. Retention is developing the separation, and band broadening is destructing it.
Values measured from a chromatogram containing two peaks.
Selectivity is the ratio of the capacity factors of both peaks, or the ratio of its adjusted retention times. Selectivity represents the separation power of particular adsorbent to the mixture of this particular components.
![]()
This parameter is independent of the column efficiency, it only depends on the nature of the components, eluent type, eluent composition, and adsorbent surface chemistry. In general, if the selectivity of two components is equal to 1, then there is no way to separate them by improving the column efficiency.
Resolution is the parameter describing the separation power of the complete chromatographic system relative to the particular components of the mixture.
By convention, resolution (R) is expressed as the ratio of the distance between two peak maxima to the mean value of the peak width at the base line:
![]()
If we approximate peaks by symmetric triangles, then, if R is equal to or more than 1 then components are completely separated. If R is less than 1, then components are overlapped.
By using the expressions for capacity factor and column efficiency the equation for R could be transferred to the form:
![]()
where the dependence of resolution on the column efficiency is represented by the square root of N, which means that increasing the efficiency is not so favourable for resolution improvement.
Resolution can also be expressed in form:
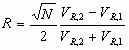
It is well recognized now that column band broadening originates from three main sources:
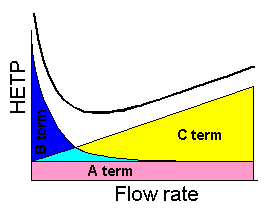
In 1956 J.J. Van Deemter introduced the equation which combined all three sources and represented them as the dependence of the theoretical plate height (HETP) on the mobile phase linear velocity. Originally, it was introduced for gas chromatography, but it happened that the same physical processes occurs in HPLC, and this equation is perfectly fit for liquid chromatography.
The velocity of mobile phase in the column may vary significantly across the column diameter, depending on the particle shape, porosity, and the whole bed structure. This is schematically shown below.
Variation of the zone flows
Band broadening is caused by differing flow velocities through the column, which may be written in form
![]()
where Hp is the HETP arising from the variation in the
zone flow velocity, dp is the particle diameter
(average), and l is the
constant which is almost close to 1.
This shows that Hp may be reduced (efficiency increased) by reducing the particle diameter (which will lead to the increasing of the column back pressure). Coefficient lambda depends on the particle size distribution. The narrower the distribution, the smaller l (which actually lead to decreasing of the column backpessure also).
It is well-known that molecules disperse or mix due to the diffusion. The longitudinal diffusion (along the column long axis) leads to the band broadening of the chromatographic zone. This process may be described by equation:
![]()
It is obvious from the above equation that the higher the eluent velocity, the lower the diffusion effect on the band broadening. Molecular diffusion in the liquid phase is about five orders of magnitude lower than that in the gas phase, thus this effect is almost negligible at the standard HPLC flow rates.
Mass transfer is the most questionable parameter. For the modern types of packing materials it may combine two effects: adsorption kinetics and mass transfer (mainly due to diffusion) inside the particles.
95% of all modern packing materials are the spherical, totally porous rigid particles with average diameter ~5 µm and pore diameter ~100Å. Ratio of the particle to the pore diameter is 500/1. There is no pressure propelled flow inside the particle, and molecules can move there only by diffusion. It can be shown by analogy: if consider a tunnel in the mountain which has diameter of 2 meters and one kilometer length (same ratio 500/1), and if there is a storm outside with 200 km/h wind, there will be almost no wind in 50 meters from tunnel entrance.
Adsorption kinetics is almost negligible compare to the diffusion inside the particles, and band spreading of the peak may be written in form:
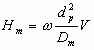
The above equation describes the linear dependence of HETP on the flow rate. The slower the velocity, the more uniformly analyte molecules may penetrate inside the particle, and the less the effect of different penetration on the efficiency. On the other hand, at the faster flow rates the elution distance between molecule with different penetration depth will be high.
Each term discussed above introduces its part in the total band broadening, therefore the sum of all of them will give the total column plate height.
H = Hp + Hd + Hm (11)
or the expanded form will be
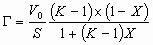
Experimentally measured dependencies of HETP on the eluent flow rate are shown below
HETP dependencies of (a) aniline, (b) benzene, (c) toluene on the eluent flow rate.
Different components have different dependencies of HETP on the flow rate on the same column. This shows that the components nature, types of the surface interactions and perhaps other parameters have an influence on the column efficiency related to the particular component.
Despite that, we also can notice, that the general dependence of the experimentally measured curves fit well to that described by the above equation. The theoretical graph below highlight the contribution of each terms of the main equation.
The most significant result is that we can find an optimum eluent flow rate
where the column efficiency will be the best.
There are two basic approaches for thermodynamic description of the HPLC retention phenomena, one is based on the partitioning theory and another is based on adsorption.
Partition is a concentrational changes in the system due to the distribution of the components between two (or more) phases.
Adsorption is the concentrational changes in the system in presence of interface with another phase and due to the surface forces.
Phase is a form of matter that is uniform throughout in chemical composition and physical state.
Adsorbent particles are considered to be nonpermeable and nonsoluble for the eluent and analyte molecules. It only introduces surface forces in the system.
Consideration of the HPLC process based on the partitioning theory was transferred from GC (gas chromatography) theory, where we usually have a mobile gas phase and stationary liquid phase, and where a true partitioning occurs.
Usual description of liquid chromatography on the partitioning basis consider the assumption of the existence of the separate liquid phase which is close to the adsorbent surface. Chemically bonded phases are usually considered by this manner. The most popular bonded phase is octadecylsilica, where relatively long (21 Å) alkyl chains are chemically bonded to the silica surface. The main partitioning concept is that analyte molecules can penetrate between these alkyl chains. This process thermodynamically considered as dissolving of the analyte molecules in the surface alkyl phase.
monomolecular layer would not be considered a phase in classical thermodynamics.
Actually an application of the partitioning theory leads to the certain controversy in the description of some HPLC data.
Here, an adsorption thermodynamic theory of HPLC retention will be considered , and we start with the brief description of adsorption from solutions.
A classic description of the adsorption process is based on the Gibbs excess adsorption theory, which basically considers two similar hypothetical adsorption systems with the same volume, temperature, pressure, and adsorbent surface area. The only difference is that the first system does not show any adsorption on the surface (no surface forces), and the second does.
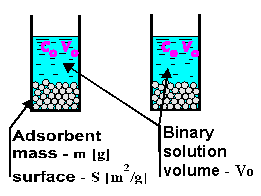
Simple adsorption experiment
After achieving an equilibrium in both systems a concentration of the components in the bulk solution over the adsorbent are measured. The first system obviously will have an original concentration (co) of component A (considering a binary solution), and in the second system different concentration, ce of the same component will be observed.
Excess adsorption is defined as an excess amount of the component concentrated on the adsorbent per unit of the syrface area:
![]()
The dependence of the Gibbs excess adsorption on the equilibrium analyte concentration is usually called adsorption isotherm.
The slope of the adsorption isotherm of the analyte at low concentrations represents a power of the surface molecular interactions. For the simple binary mixture an adsorption isotherm may be described by the equation:
![]()
![]()
where x is an analyte equilibrium concentration in mole fractions, K is the thermodynamic equilibrium constant. K is a measure of interaction energy difference of eluent and analyte molecules with the adsorbent surface, and it may be expressed in the form:
![]()
where DG is the difference of the free Gibbs energy of the analyte and eluent, R is the gas constant, and T is an absolute temperature.
It is assumed that the column is in equilibrium. This means that at any moment and in any part of the column, the conditions are infinitely close to thermodynamic equilibrium.
By considering the analyte mass dynamics in the small cross-sectional part of the column we can write the mass balance equation for the dx part of the column on x distance from the inlet. This mass balance equation has a differential form, and has an exact solution only for binary system. This solution establishes a connection between the analyte retention volume and its excess adsorption isotherm.
In the mass balance equation section the logic of the derivation of the mass balance equation is shown. This information is optional, but as a result we will get:
For the binary system the exact solution of the mass balance differential equation may be written in form:
![]()
This equation is the basic retention equation in adsorption chromatography. It could be used for the thermodynamically based explanation of most of the chromatographic effects.
In general it says, that component retention volume is a sum of the dead volume and product of the adsorbent surface area and the derivative of its excess adsorption isotherm
In the previous sections the thermodynamic theory of chromatographic retention was discussed . Here we will discuss how it is actually working in the real systems, and how the equations are related to the measurable parameters.
As we have seen, the dead volume (Vo) is present in all basic equations and calculated parameters. So, we have to discuss it deeper.
Dead volume is the total volume of the liquid phase in the chromatographic column.
It is obvious from the definition that Vo is the geometrical parameter, and it should not be dependent on the types of analytes and mobile phase. This definition is the most general and it is true if the stationary phase (adsorbent) used is rigid, nonsoluble and nonpermeable. This definition may not be applicable for some ion-exchange stationary phases based on the cross-linked semi-rigid polymers, which may swell or shrink depending on the mobile phase.
In adsorption (normal- or reversed phase) chromatography and in size-exclusion chromatography, dead volume is a property of the column and it has to be measured independently. But, the question - how to measure it - is the most complicated one.
The most obvious way, is to inject a so called "nonretained" component in the column and measure its retention volume. But, what is the "nonretained component", and how is it found?
In the ideal situation, if we could some how mark eluent molecules in such a way that it will not change their properties, and inject them, then their retention will show a true dead volume. It was shown experimentally that even deuturated eluent molecules show some adsorption effect, and could not be used for dead volume measurement.
From the basic retention time equation we can conclude that a compound is not retained if its derivative of the excess adsorption is equal to zero. We have to mention that in general, excess adsorption could be positive (preferential adsorption) and negative.
All these methods may have a deviation from the real dead volume. A 10% deviation is acceptable for analytical purposes.
In physico-chemical applications a true value of Vo should be used.
Two basic methods to measure thermodynamic dead volume have been suggested. All are based on the specific features of the excess adsorption isotherm: that the excess adsorption of the pure component is equal to zero.
If we fill the column with one solvent and displace it with another, than the inflection point of the resulting frontal chromatogram will have retention volume equal to dead volume.
Another method is based on the minor disturbance of the equilibrated chromatographic system. If we equilibrate the system at the certain binary eluent composition and inject a small amount of the same eluent with a small difference in the composition, then the injected "disturbance" will move through the column according to the basic retention equation. By measuring this at different equilibrium concentrations, we will finally have an experimental dependence of the retention volume on the eluent composition for the minor disturbance peaks for the total concentration range.
If we will integrate basic retention equation by concentration we will get:
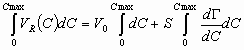
or,
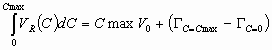
As mentioned before, an excess adsorption of the pure component is equal to zero. Therefore, the rightmost term of the above expression is equal to zero, and the final expression for the dead volume will be:
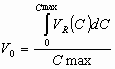
Which is, in fact, an integral average of the dependence of the retention
volume on the concentration.
Temperature effects in HPLC are not as significant as in gas chromatography. First, because we do not have same temperature range. Volatile solvents are not allowed to rise to higher temperatures too much, and the stability of the attached bonded ligands on the adsorbent surface may be influenced by the high temperature. So, the main temperature range is from ambient temperature to 60 or 70 C.
According the equation (23), increasing the temperature will decrease the value of K or k', thus the actual retention time will decrease. For most of the systems these decrease will not exceed 50% of the component reduced retention time at ambient temperature.
Picture below illustrate the influence of the column temperature on the HPLC retention.
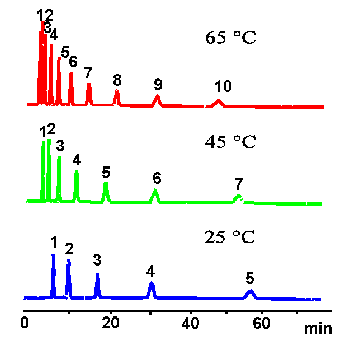
There are two other significant effects of separation under the elevated temperature.
Stabilization of the column under the elevated temperature usually leads to the stabilization of the retention times. Origin of this effect is not well understood yet. Possible explanation is that the solvent viscosity decreased and more uniform stabilized temperature with absence of local temperature fluctuations due to the solvent friction lead to the more uniform adsorption-desorption process.
Another effect is the increase of the column efficiency. At the elevated temperature viscosity of liquids decrease and the diffusion coefficient increase. From the Van Deemter Equation the second term will increase which will lead to the decrease of the efficiency at the very low flow rates (which is not important). The last term will decrease which will lead to the increasing of the efficiency at the common flow rates. It also widens the flow rate range with optimum efficiency.
Here we discuss a basic thermodynamic description of the effect of the type and composition of the mobile phase.
In most of the HPLC separations binary eluents are employed. One of the solvents in the eluent is usually inert relative to the surface interactions.
In Reversed-phase HPLC (RP HPLC) one of the eluent components is water, which does not interact with the hydrophobic adsorbent surface. And it does not compete with the analyte for the adsorption sutes.
In normal-phase HPLC (NP HPLC) one of the eluent components is usually hexane, which also does not interact with the very polar silica surface.
Another component of any binary eluent is an active one. It usually called a "modifier" because it can interact with the adsorbent surface and compete with analyte molecules for the adsorption sites. Increasing of the concentration of the "modifier" in the eluent leads to the decreasing of the analytes retention.
As we discussed before, capacity factor is proportional to the thermodynamic equilibrium constant, and the last is an exponent of the free Gibbs energy of the system. In the formula (23) we have a difference of the free energy of analyte and eluent interactions with the surface.
For the binary eluent system we can assume that only the "modifier" can interact with the surface. For RP HPLC it will be an organic component of the eluent, and water is assumed not to interact with the surface.
As a rough approximation we can assume that the total free Gibbs energy of that adsorption system could be considered as a difference of analyte adsorption energy and a product of modifier adsorption energy and its mole fraction. Thus, component retention volume could be roughly estimated by using the equation:
![]()
where Vo is the dead volume of the column, ![]() an.
is the analyte adsorption energy, x - is the mole fraction of the
organic modifier in the eluent, and
an.
is the analyte adsorption energy, x - is the mole fraction of the
organic modifier in the eluent, and ![]() el.
is the adsorption energy of the eluent.
el.
is the adsorption energy of the eluent.
Figure below is shows the experimentally measured dependencies of the retention of alkylbenzenes in reversed phase HPLC mode on C18 column with acetonitrile/water eluent at different compositions. Points are experimental values and a curves was calculated using the above equation. As we can see this simple approximation allows us to describe experimental dependencies pretty well.
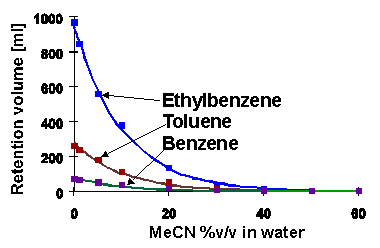
Retention dependencies of alkylbenzenes vs. the eluent composition. Eluent: acetonitrile/water, column: Prodigy-C18 (150x4.6 mm)
This type of the influence of the mobile phase composition and its thermodynamic explanation are true for the chromatographic system with only hydrophobic interactions (dispersive forces). In case of the presence of any specific adsorption sites, the analyte behavior may significantly differ from that described above.
Any specific interactions of the analyte molecules with the eluent molecules also may introduce significant deviations to the analyte retention dependence. Ionizible components usually show a specific behavior.
Organic acids are easily solvated with the water molecules, which block possible interaction of the hydrophobic part of the molecule with the adsorbent surface, and lead to the very early elution of these compounds. For example, benzoic acid is eluted before the dead volume at any composition of acetonitrile/water eluent on the reversed-phase column.
Organic basis usually shows a low retention also due to solvation, but in case of presense of strong acidic accessible adsorption sites on the adsorbent they show a strong retention.
A general approach to the separation of the mixtures containing an ionisible components is to suppress their ionization. Suppression of the ionization decreases a power of the molecular solvation and exposes the hydrophobic (organic) part of the molecule to the surface interaction. Ionization suppression is usually made by the adding a buffer into the solvent, which shift a pH to the certain value.
In the absence of buffer, easy ionizible components are eluted from the column as very broad peaks. According to the Le Chatelier principle, dissolved ionizible component is present in the solution as a mixture of ions and nonionized molecules
[AB] = [A+] + [B-]
According to the above equilibrium, about 50% of all molecules are ionized in the solution . But, the chromatographic behavior of ions and neutral molecules are different. Let us assume that neutral molecules will be retained, so during the run ions will move faster, and at the first moment they will be separated from the neutral molecules. But, according to the above equilibrium, in the absence of the neutral molecules ions will tend to form them, and this new neutral molecule will also be absorbed, and so on. This process will lead to the spreading of the component along the column and causes the appearance of the broad peak.
It does not occurif the equilibrium is shifted due to the presence of
the buffer with the pH at least two units apart of pK of the component.