 Return to Physiology
Return to Physiology
CARDIOVASCULAR PHYSIOLOGY
Download a copy of this study guide
 Return to top
Return to top
ELECTRICAL CONDUCTION OF THE HEART
MYOCARDIUM DEPOLARIZATION:
- Phase 0: Initial upswing of action potential.
- Na+ Channels open until threshold is reached.
- Phase 1: The potential may repolarize slightly before starting the plateau phase.
- Na+ Channels are inactivated.
- Outward Rectifier K+ Channels open transiently, causing slight
repolarization.
- Membrane potential remains near zero.
- Phase 2: Plateau Phase -- This stage is responsible for prolonging the cardiac
action potential, making it longer than a nerve action potential.
- Ca+2 Channels open, to keep the cells depolarized.
- Phase 3: Repolarization
- Ca+2 Channels close.
- Delayed Rectifier K+ Channels open to effect normal repolarization.
- Phase 4: Diastolic membrane potential.
- Inward Rectifier K+ Channels (different than the ones above) are open, to
maintain resting potential.
- They are open at highly negative membrane potentials (i.e.
hyperpolarization-activated).
SA-NODE DEPOLARIZATION: It is similar to depolarization in the myocardium, except for the following differences:
- Depolarization results from influx of Ca+2 rather than Na+
- There is no plateau phase (no Phase 1 and 2).
- Automaticity: Hyperpolarization-activated cation current is activated at low potentials, resulting in automaticity of the SA-Node.
- Epinephrine increases the rate of rise and acetylcholine decreases the rate of rise of Phase-4 depolarization.
REFRACTORY PERIOD: Cardiac muscle cells have prolonged refractory periods, to
prevent tetany of cardiac muscle.
AUTONOMIC REGULATION of HEARTBEAT:
- Acetylcholine slows heart rate by increasing K+ permeability.
- Norepinephrine speeds heart rate by increasing the rate of rise of the cardiac
action potential during phase 0.
PROPAGATION of ACTION POTENTIAL:
- ATRIAL CONTRACTION: It takes about 70 msec to get from the SA-Node ------>
depolarize the atria ------> to the AV-Node.
- AV-NODAL DELAY: There is a delay in depolarization of about 90msec, once the
impulse reaches the AV-Node.
- The function of this delay is to separate the contraction of the atria (i.e. atrial
systole) from that of the ventricles (ventricular systole), so that more blood
has a chance to fill into the ventricles.
- The AV-Node depends on slow-conducting Ca+2 Channels for depolarization,
which helps to explain its slow rate of depolarization.
- A smaller cell-size also helps to explain the slow rate of conductance.
- BUNDLE OF HIS
- BUNDLE-BRANCHES: Two continuing branches of the Bundle of His.
- Left Bundle Branch: It depolarizes first. Depolarization goes from the left
side of the ventricular septum to the right side, accounting for the Q-Wave.
- Right Bundle Branch: It depolarizes after the left side.
- PURKINJE SYSTEM: Very fast conduction.
- VENTRICULAR MUSCLE
- As depolarization proceeds in the ventricles, it moves from endocardium
------> epicardium.
EKG LIMB LEADS:
 Depolarization occurs toward
the positive side (the positive
sides are labelled to the right,
and the respective negative
sides are unlabeled).
Depolarization occurs toward
the positive side (the positive
sides are labelled to the right,
and the respective negative
sides are unlabeled).- HEXAXIAL SYSTEM: The
positive end of each limb lead is
as follows:
- I: 0
- II: +60
- III: +120: In a normal
ECG, Lead III should
have a net-zero QRS-Complex, as it is
perpendicular to aVR.
- aVR: -150: In a normal
ECG, the aVR lead
should have a completely negative QRS Complex.
- aVL: -30
- aVF: +90
- DIRECTION OF ECG DEFLECTION: A positive deflection on an ECG represents
a depolarization that is traveling toward the positive side of a particular lead.
- Maximal Positive Deflection: Occurs when depolarization vector is in the
exact same direction as the limb lead.
- Zero net deflection: Occurs when depolarization vector is exactly
perpendicular to limb lead.
- Maximal Negative Deflection: Occurs when depolarization vector is in the
exact opposite direction as the limb lead (i.e. in the direction of the negative
end).
ELECTROCARDIOGRAM:
- P-WAVE: Atrial depolarization. P-Wave duration is normally 80 msec.
- PR-INTERVAL: The distance from the beginning of the P-Wave to the
beginning of the Q-Wave.
- PR-Interval is the period from beginning of atrial depolarization to the
beginning of ventricular depolarization.
- PR-Interval is normally 180-220 msec.
- PR-SEGMENT: The distance from the end of the P-Wave and the beginning
of the Q-Wave.
- QRS-COMPLEX: Ventricular Depolarization. QRS Duration is normally 30-100
msec.
- Individual Components:
- Q-WAVE: Depolarization of the septum. On most leads (except III
and aVR) the Q-Wave points downward if it can be seen at all.
Septum depolarization goes from the left side of the septum to the
right side.
- R-WAVE: Depolarization of the ventricles. Sharp upward turn.
- S-WAVE: Return of volt-potential to zero, because all the ventricular
muscle has depolarized and is therefore once again isoelectric.
- Sharp downward turn back to isoelectric point. The S-Wave
may go slightly negative before return back to isoelectric point.
- QT-INTERVAL: From beginning of Q-Wave to end of T-Wave. QT-Interval
is normally 260-490 msec. This is the period from beginning of ventricular
depolarization to the end of repolarization.
- ST-SEGMENT: Short segment from end of S-Wave to beginning of T-Wave.
- ST-INTERVAL: From end of S-Wave to end of T-Wave.
- RR-INTERVAL: Distance between QRS-Complexes, or the distance
between heart beats in a normal sinus rhythm.
- T-WAVE: Repolarization of Ventricles. Atrial repolarization masked by QRS-Complex.
- Repolarization occurs in the opposite direction as depolarization, but the
vector still points in the same direction because the change in voltage also
has an opposite sign.
- In the ventricles, the first tissue to depolarize is the last tissue to repolarize.
READING THE ECG:
- Vertical Direction: 10 mm = 2 big boxes = 1 mV deflection.
- Horizontal Direction:
- 1 mm = 40 msec.
- At standard speed, there are 25 mm, or 5 big boxes, in each second.
- Speeds:
- Standard Speed = 25 mm/sec
- Extra-Sensitivity Speed = 50 msec, at which point all values above must be
doubled.
- Calculating Heart Rate Shortcut:
At standard speed: 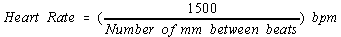
PRECORDIAL LEADS: V1 thru V6 are placed to specific places on the chest, for advanced
ECG diagnostics. V1 is right-most, near the SA-Node, while V6 is leftmost, past the apex
of the heart.
MEAN ELECTRICAL AXIS OF THE HEART:
- Two ways to graphically determine mean electrical axis:
- SHORT WAY: This is only accurate when there is a net QRS-Deflection of
virtually zero (i.e. the R deflection is equal and opposite to the S deflection).
- Determine the lead that has a net zero QRS-Deflection.
- On the hexaxial system, the mean electrical axis points in the
direction that is perpendicular to that lead.
- LONG WAY: This is longer but more accurate.
- Consider any two of the six hexaxial leads. Determine again the Net
QRS-Deflection for each lead.
- Plot that deflection along the appropriate axis on a hexaxial chart.
- Draw a dotted line perpendicular to each of the above plots, and
extend the two lines until the intersect each other.
- The Mean Electrical Axis is the vector that points from the center to
the intersection of those two lines.
- LAB: Different physiological effects on the mean electrical axis:
- INSPIRATION: The diaphragm moves down ------> It pulls the apex of the
heart toward the right (i.e. in a more vertical direction) ------> the mean
electrical axis is more positive (+ more degrees).
- FORCED EXPIRATION: The exact opposite of above. The apex of the heart
gets pushed upward and toward the left horizontal axis ------> the mean
electrical axis is less positive or even negative.
- PREGNANCY: The mean electrical axis would deviate to the left, within
normal limits. The physical presence of the fetus would push up the
diaphragm ------> heart leans toward left.
- LEFT VENTRICULAR HYPERTROPHY: Mean axis deviation toward the left.
- Pulmonary Valve Stenosis: If we assume that it leads to Right Ventricular
Hypertrophy ------> Then we get (potentially severe) right axis deviation.
- INFANCY: Right Axis Deviation, because the infant's right ventricle and left
ventricle musculature are about the same size at birth. Left ventricle
becomes larger within a couple months.
- NORMAL MEAN AXIS: Anywhere between -30 and +110.
- Anything negative of -30 is left axis deviation, as occurs from left
ventricular hypertrophy.
- Anything positive of +110 is right axis deviation, as occurs from right
ventricular hypertrophy.
ECG ABNORMALITIES:
- SINUS BRADYCARDIA: A heart rate slower than 60 SA-Nodal depolarizations per
minute. "Sinus" indicates that the cardiac impulse is originating from the SA-Node
as normal.
- SINUS TACHYCARDIA: Heart rate faster than 100 bpm, originating as normal from
the SA-Node.
- Tachycardia generally means you'll see a shorter RR-Interval (i.e. faster
heart rate).
- SINUS ARREST: No SA-Node depolarization.
- This can be artificially induced by carotid massage, which results in
overstimulation of the Vagus ------> SA-Node hyperpolarized.
- ATRIAL PAROXYSMAL TACHYCARDIA: Faster heart rate resulting from an
ectopic pacemaker in the atrial muscle.
- In the example the P-Wave points downward because the atrial
depolarization starts in the LA, because that is where the tissue is leaky.
- BUNDLE-BRANCH BLOCKS: There is some conduction block in the Bundle of His
(Left or Right Bundle branches), with results as below:
- 1 BLOCK: Partial block. The PR-Interval is longer than normal because
it takes longer to conduct the impulse from SA-Node to AV-Node.
- 2 BLOCK: A QRS-Complex occurs only after every other P-Wave. In other
words, it takes two P-Waves to sufficiently excite the AV-Node to conduct
the impulse to the ventricles.
- 3 BLOCK: There is no temporal relationship between the P-Wave and
QRS-Complex. Atrial and ventricular depolarizations are being controlled
by their own independent pacemakers (the SA-Node and AV-Node
respectively).
- AV-NODAL TACHYCARDIA: Tachycardia, plus the P-Wave is insignificant or
absent.
- This is tachycardia, where the impulse originates from the AV-Node. The
inherent pacemaker of the AV-Node is faster than the SA-Node.
- PREMATURE VENTRICULAR CONTRACTION (PVC): A premature QRS-Complex, or one that occurs without being preceded by a P-Wave.
- That means that the P-Wave didn't start the impulse, but it started
somewhere else.
- Ectopic Pacemaker: With PVC, the impulse originates in the ventricular
muscle itself, due to leaky membranes in the muscle.
- VENTRICULAR FIBRILLATION: Waves of depolarization traveling in multiple
directions all over the ventricular muscle. The pacemaker activity is lost.
- ATRIAL FIBRILLATION: Fibrillation in the atria is not serious in children, but it is
serious in old people.
- That's because in old people, atrial systole contributes a greater relative
blood volume to cardiac output than in children.
CLINICAL LECTURE: WOLF-PARKINSON-WHITE SYNDROME
- Normally, the AV-Node is the only pathway for conduction of the impulse from the
atria to the ventricles.
- Bachman's Bundle: Normally conducts the impulse from Right Atrium to
Left Atrium during atrial systole.
- Moderator Band: Normally conducts the impulse from the right ventricular
septal wall to the right free wall during ventricular systole.
- Lupus Erythematosus: Rare condition associated with pediatric bradycardia.
Usually pediatric heart problems result in Tachycardia -- not bradycardia.
- PEDIATRIC TACHYCARDIAS: They are divided into two types
- Supraventricular Tachycardia (SVT): One where the problem originates
somewhere in the AV-System.
- Ventricular Tachycardia (VT): Problem originates in the ventricular system.
- Wolf-Parkinson-White Syndrome: Extra conductive tissue in the myocardium,
creating an accessory pathway for conduction from atria to ventricles.
- This accessory pathway ultimately results in a Reentry Tachycardia, or a
conduction loop between the normal and accessory pathways.
- The Wolf-Parkinson-White ECG: Shorter PR-Interval due to rapid
conduction of signal to ventricles through accessory pathway.
- This is the ECG when the patient is healthy and no problems are
going on.
- The P-Wave and the QRS-Complex are scrunched together, creating
the appearance of a delta-wave (hump right before QRS), and a
longer overall QRS Complex.
- Reentry Tachycardia: You get it from a unidirectional block in one pathway,
coupled with slowed conduction of an alternative pathway. This results in
continuous impulse conduction, or circus dysrhythmia.
- With WPW, the accessory pathway can get blocked because it hasn't had
the time to repolarize, then the normal pathway provides a mean for
retrograde conduction of depolarization.
- This results in a conduction loop and severe tachycardia.
- TREATMENT: Slow down the conduction through one pathway or the other.
- Use Ca+2-Channel Blockers (such as Verapamil)
- Use Digoxin to increase AV-Nodal sensitivity to ACh.
- Use beta-Blockers to block the normal NorE sympathetic receptors on the
AV-Node and cardiac muscle.
- In severe cases, surgically remove the conductive tissue from the
myocardium.
Return to top
THE CARDIAC CYCLE
VENTRICULAR DIASTOLE:
- ISOVOLUMIC RELAXATION: The very beginning of diastole, right after the aortic
valve closes, during which both valves are closed.
- The ventricular muscle is relaxing as ventricular pressure rapidly decreases.
- Volume remains constant.
- THREE PHASES OF VENTRICULAR FILLING:
- RAPID VENTRICULAR FILLING:
- The Mitral Valve opens, when ventricular pressure falls below atrial
pressure.
- Blood rushes into ventricle, very quickly initially.
- THIRD HEART SOUND (S3): It is turbulent blood flowing past the
ventricular wall during early diastole. It is indicative of pathology.
- SLOW VENTRICULAR FILLING: The later period of diastole. The majority
of blood has already entered the ventricles.
- TOP-OFF PHASE: The blood contributed to ventricles during atrial systole.
- Diastolic Events Associated with the Atria:
- V-Wave: Small increase in atrial pressure associated with the fact that the
mitral valve is closed at the very beginning of diastole.
- Y-Descent: Descent of the V-Wave. Decrease in atrial pressure occurring
when the mitral valve opens, right after ventricular isovolumic relaxation.
- A-Wave: Small rise in atrial pressure, occurring right before systole,
associated with Atrial Systole and cntrxn of atrial muscle.
- FOURTH HEART SOUND (S4): Vibration of mitral valve leaflets during atrial
systole, i.e. during the top-off phase of ventricular filling. This occurs
concurrent with the A-Wave and is indicative of pathology.
- Atrial Fibrillation: There is an age difference in the seriousness of this.
Again, atrial fibrillation isn't a concern with young people but it is with old
people.
- YOUNG: Atrial systole contributes about 20mL to stroke volume
- OLD: Atrial systole contributes about 40mL to stroke volume.
- An Increased heart rate makes the atrial contribution to stroke volume more
significant. Shorter time for ventricular filling ------> The top-off phase
contributes more relative volume to ventricles.
VENTRICULAR SYSTOLE: QRS-Complex occurs and ventricles start contracting.
- FIRST HEART SOUND (S1): The Mitral Valve Closes, as ventricular pressure
exceeds atrial pressure.
- ISOVOLUMIC CONTRACTION: Period of contraction during which both valves are
closed
- Pressure is increasing.
- Volume is constant.
- Systolic Events Associated with the Atria:
- C-WAVE: Small increase in atrial pressure. Occurs during isovolumic
contraction, as the ventricle pushes the mitral valve a little upward toward
the atrium.
- X-DESCENT: The decrease in the C-Wave, due to the change of shape of
the ventricle from prolate spheroid (football-like) to spheroid. This makes the
mitral valve move down and the atrial pressure return to normal.
- Aortic Valve Opens, as ventricular pressure exceeds aortic pressure.
- Ventricle must achieve systolic arterial pressure in order to open the Aortic
valve, so it reaches pressures around 120 mm Hg.
- Ventricular Ejection: 70% of blood is ejected in the first third of systole.
- SECOND HEART SOUND (S2): The aortic valve closes, as ventricular pressure
falls below aortic pressure.
- DICROTIC (AORTIC) NOTCH: When the Aortic Valve closes, there is a temporary retrograde flow of blood against the Aortic valve cusps. This
causes an acute decrease in Aortic pressure at the very beginning of
diastole.
- Two Things act in concert to make the Aortic valve close:
- The left ventricle relaxes so left ventricular pressure decreases.
- The retrograde blood flow against the leaflets actually aids in the
closure of the valve.
HEART SOUNDS: Left Side -vs- Right Side:
- FIRST HEART SOUND (S1):
- The mitral valve (left side) closes before the tricuspid valve (right side),
because the depolarization begins on the left side of the septum.
- On the other hand, the Aortic Valve (left side) opens a little after the
Pulmonary Valve (right side), because there is so much higher volume in the
left side, hence more pressure has to build up before valve will open.
SPLIT SECOND HEART SOUND: During inspiration, You should be able to hear the
pulmonic and aortic valves close separately during the second heart sound (i.e. a "split"
sound).
- Pulmonic Stenosis: In this case the pulmonic valve is not opening well ------>
Wide Splitting during inspiration.
- Aortic Stenosis: Causes paradoxical splitting -- i.e. splitting occurs during
expiration instead of during inspiration.
Return to top
HEMODYNAMICS
|
NORMAL RANGE OF VALUES |
| P-Wave |
~ 80 msec |
| QRS-Wave |
30 - 100 msec |
| P-R Interval |
180 - 220 msec |
| S-T Interval |
230 - 460 msec |
| Q-T Interval |
260 - 490 msec |
| Mean Electrical Axis |
-30 to +110 |
| End Diastolic Volume (LVEDV) |
120 - 140 mL |
| End Systolic Volume (LVESV) |
40 - 60 mL |
| Stroke Volume (SV) |
60 - 100 mL |
| Ejection Fraction |
0.50 - 0.70 |
| Cardiac Output (CO) |
5.0 - 6.0 L / min |
| Cardiac Index |
2.6 - 4.2 L / min / m2 |
| Systolic Pressure |
100 - 140 mm Hg |
| Diastolic Pressure |
60 - 90 mm Hg |
| Systemic Resistance (TPR) |
0.9 PRU, or mm Hg / mL / sec |
| Pulmonary Blood Distribution |
~ 10% total; 500 mL |
| Heart Blood Distribution |
~ 10% total; 500 mL |
| Systemic Arterial Blood Distribution |
~ 10% total; 500 mL |
| Arteriolar Blood Distribution |
~ 5% total; 250 mL |
| Venous Blood Distribution |
~ 65% total; 3250 mL |
| Capillary Hydrostatic Pressure, Pc |
~ 30 mm Hg |
| Capillary Oncotic Pressure, PIp |
~ 25 mm Hg |
| Interstitial Hydrostatic Pressure, Pi |
~ 0 mm Hg |
| Interstitial Oncotic Pressure, PIi |
1 - 10 mm Hg |
| Arterial Compliance |
1 mL / mm Hg |
| Venous Compliance |
20 mL / mm Hg |
STROKE VOLUME = (END DIASTOLIC VOLUME) - (END SYSTOLIC VOLUME)
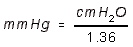
- Cardiac Index is Cardiac Output normalized for body mass.
CARDIAC OUTPUT = (STROKE VOLUME) x (HEART RATE)
PULSE PRESSURE = (SYSTOLIC PRESSURE) - (DIASTOLIC PRESSURE)

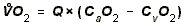
MEAN ARTERIAL PRESSURE = (CO) x (TPR) = (HR) x (SV) x (TPR)
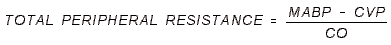
- PERIPHERAL RESISTANCE UNITS (PRU): Units of mm Hg / mL / sec.
- Or, it is TPR as above, where CO is expressed in mL/sec.
RESISTANCE alpha VISCOSITY
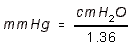
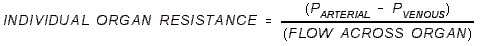
- For the lungs, this resistance is called Pulmonary Vascular Resistance, and the
flow is equal to Cardiac Output.
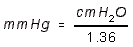
General Trends in Circulation:
- Pressure drop is greatest at the level of the arterioles.
- Velocity of blood is slowest at the capillaries, because they have the largest total
cross-sectional area, given the number of capillaries.
- Turbulence: The higher the velocity of blood flow, the greater the likelihood of
turbulence.
- Turbulence is most likely in large arteries. Never in capillaries and rarely in
venous system.
- Arterial Elasticity (The Windkessel Effect): Arterial Elasticity accounts for a smaller
pulse pressure.
- It relieves a little pressure during systole, since it can give a little.
- It maintains flow during diastole, since it can flex back.
- Thus, atherosclerosis ------> Larger Pulse Pressure.
- THE BASIS OF STEADY BLOOD FLOW: Systole -vs- Diastole
- Systole: More blood is pumped into the arterial tree then flows out of the
arterial tree, so arterial pressure rises.
- Hence volume in arterial tree goes up ------> pressure in arterial
tree goes up to systolic pressure.
- During systole, about half of the blood is stored in the arterial tree,
and the other half is pushed into the capillary beds.
- Diastole: Blood continues to leave the arterial system and no new blood
enters it, so blood pressure goes back down.
- During Diastole, more arterial blood flows into the capillary beds,
providing capillaries with continuous blood flow whether in systole or
diastole.
MEASURING BLOOD PRESSURE / SPHYGMOMANOMETER:
- SYSTOLIC PRESSURE: The first sound you hear -- a rush of blood flowing through
the squeezed artery.
- This happens the instant that the cuff pressure is reduced enough to let
arterial blood squirt through during systole.
- DIASTOLIC PRESSURE: The last sound you hear -- blood is no longer stopped by
the cuff-pressure during diastole.
- Phases:
- Phase I (snapping):
- Phase II (murmur): In hypertensive people, an auscultatory gap can occur
during Phase II.
- Phase III( thumping):
- Phase IV (muffling): The beginning of this muffling is sometimes taken as
the high end of diastole.
- Some people think the muffling sound is a better indicator of diastolic
pressure for children.
- Estimations:
- SYSTOLIC PRESSURE is underestimated by auscultation -- you can't hear
the sound "quick enough" to record the measurement.
- DIASTOLIC PRESSURE is overestimated by auscultation.
- Thus PULSE PRESSURE can be underestimated by auscultation by a
significant amount.
FLOW, VISCOSITY, TURBULENCE, RESISTANCE:
- TURBULENCE: Turbulence is directly related to velocity of fluid. The higher the
velocity, the more likely there is to be turbulence.
- Reynold's Equation tells us the critical velocity at which turbulence will
occur. We can derive three relationships from that equation:
- Turbulence alpha Flow: The higher the flow, the higher the likelihood
of turbulence.
- Turbulence alpha (1 / viscosity): The lower the viscosity, the higher
the likelihood of turbulence.
- Turbulence alpha (1 / diameter): The narrower the radius of the vessel, the higher the likelihood of turbulence.
- Turbulence is indicative of a larger pressure drop (larger DeltaP) across a
region of vessel. Thus turbulence occurs when there is an atherosclerotic
plaque.
- VISCOSITY: Relation between viscosity and turbulence:
- Viscosity of blood is most closely related to hematocrit.
- 20% of blood viscosity if from plasma; 80% is from blood cells.
- ANEMIA: Lower hematocrit ------> Lower viscosity of blood ------> Higher
blood flow ------> Higher likelihood of turbulence.
- FLOW: Relation between flow and radius = flow is inversely proportional to r4.
- RESISTANCE: The resistance to any organ is greater than the sum of all
resistances!
- That's true because the vessels are wired in parallel, and the sum of
resistances in parallel is less than its individual parts.
- Systemic Resistance (TPR) is much greater than Pulmonary Resistance.
- Pulmonary Resistance = Delta Pulmonary Pressures / CO.
BRUIT: Turbulent flow is detected as a bruit which can be heard by the stethoscope.
- Innocent Ejection Murmur: Children can have high velocity of blood flow without
there being any pathology. Bruits are not uncommon.
- Bruits with Anemia: Anemic patients can also have innocent bruits, for two
reasons:
- Lower hematocrit ------> lower blood viscosity ------> higher likelihood of
turbulence.
- Anemics tend to compensate their low hematocrit with a higher cardiac
output.
- Atherosclerotic Plaque: Turbulence can be heard downstream from the plaque.
- Upstream from Plaque: Greater resistance ------> a strong pulse pressure.
- Downstream from Plaque: A bruit can be heard.
STANDING BLOOD PRESSURE: Mean Arterial Pressure goes down when standing,
because of lower venous return.
- Stand up ------> Venous Pressure in feet goes up ------> capillary hydrostatic
pressure goes up ------> fluid flows out of arterial tree and into tissues ------>
venous pooling in the feet ------> venous return decreases ------> CO
decreases ------> lower MABP.
- Venous pressure goes up in feet because of gravity -- DeltaP = gh
- Skeletal Muscle Pump: Tonic contraction of leg muscles while standing aids
venous return, because the veins have valves, so blood is squeezed in only one
direction.
- Thus prolonged standing can lead to incompetent valves in the veins in the
legs.
BLOOD PRESSURE AND THE RESPIRATORY CYCLE:
- INSPIRATION: Systemic blood pressure goes down and pulmonary blood pressure
goes up.
- The Diaphragm moving down has two effects:
- It increases the volume of thoracic airspace and so it decreases
intrathoracic pressure.
- Also the abdominal space becomes smaller, so it increases intra-abdominal pressure.
- The combination of above two effects results in an increased pressure
gradient for venous return from the IVC ------> increased venous return
------> More blood to right atrium and more blood to pulmonary circulation
------> less respective blood in left heart and less CO.
- Thus overall result is the following:
- Lower systemic pressure.
- Higher pulmonary pressure.
- Larger Blood Volume in pulmonary circulation.
- The change in MABP from inspiration normally does not exceed 10 mm Hg.
- EXPIRATION: Has the exact opposite effect.
- Pulmonary pressure decreases.
- Systemic pressure increases.
CENTRAL VENOUS PRESSURE: The pressure going into the right atrium.
- Anything that decreases venous compliance (i.e. sympathetic tone) will increase
venous return ------> Higher CVP.
- ESTIMATING CENTRAL VENOUS PRESSURE: You estimate in cm of water.
- It is approximately equal to the distance from the end of the distended part
(which you can see) to the sternal angle, plus 5, then convert it into mm Hg.

PRESSURES IN PERIPHERY -vs- AORTA:
- Mean Arterial Pressure is slightly higher in the Aorta than in, for example, the radial
artery.
- But, Pulse Pressure is greater in the periphery, i.e. the systolic is higher and the
diastolic is lower.
- This effect in the periphery is due to constructive interference of reflected
waves.
COMPLIANCE: The degree to which a pressure change leads to a corresponding change
in volume. Or, Compliance = DeltaV / DeltaP, or the slope of a pressure-volume curve.
- VENOUS COMPLIANCE is about twenty times more than arterial compliance,
therefore veins can hold a larger volume of fluid at lower pressure.
- Arterial Compliance is about 1 mL / mm Hg
- Venous Compliance is about 20 mL / mm Hg
- EFFECTS OF COMPLIANCE on Blood Pressure:
- Higher Venous Compliance ------> higher capacitance in veins ------> less
venous return ------> lower CVP.
- Lower Venous Compliance (sympathetic influence) ------> lower
capacitance veins ------> more venous return via the one-way valves
------> higher CVP.
- Lower Arterial Compliance results in a higher pulse pressure.
- AGE: Arteries in old people have lower compliance. Thus old
people have higher pulse pressures.
- Pressure-Volume Curve: The analysis of old -vs- young
can be done on the P/V curve.
- The slope of the curve is compliance.
- Pressure is on the X-Axis. Volume is on the Y-Axis.
- Is you plot systolic and diastolic pressure, and look at the corresponding Y-Values, you can calculate the following:
- The difference on the Y-axis (i.e. the volumes corresponding to
systolic and diastolic pressures) is stroke volume.
- The difference on the X-axis is pulse pressure.
MODULATION OF MEAN ARTERIAL PRESSURE: Under a lot of circumstances, it doesn't
change, even when stroke volume and/or pulse pressures do change.
- EFFECT OF STROKE VOLUME: All other factors held constant, a high stroke
volume results in a higher pulse pressure, i.e. higher systolic and lower diastolic,
but MABP remains constant.
- PULSE PRESSURE IS USUALLY DIRECTLY RELATED TO STROKE
VOLUME
- EFFECT OF EXERCISE:
- Increased CO and Stroke Volume
- Compensatory lower vascular resistance (TPR)
- Once again MABP doesn't change (within limits).
- HIGH SYSTOLIC PRESSURE: Tends to occur with higher stroke volume. The
more fluid you pump in one beat, the higher the systolic pressure.
- HIGHER DIASTOLIC PRESSURE: CORRELATES WITH HIGH TPR.
Return to top
MYOCARDIAL PERFORMANCE
General Effects of Autonomic Control on Heart:
- SYMPATHETICS:
- Positive chronotropic effect -- faster heart rate.
- Positive inotropic effect -- greater contractility for the same fiber length.
- PARASYMPATHETICS: Negative chronotropic effect, but no inotropic effect.
PRELOAD: The diastolic filling pressure, or end-diastolic volume.
AFTERLOAD: Ventricular systolic pressure, which is equal to arterial systolic pressure
under normal circumstances.
LAPLACE'S LAW: The stress on the ventricular wall is proportional to the Ventricular
Pressure x Ventricular Radius, where the size of the ventricle is determined by
stretching, i.e. by ventricular volume.
STARLING'S LAW OF THE HEART: Within limits, increases in end-diastolic volume
result in a corresponding increase in stroke volume. Most simplified, within limits, the
volume that comes into the heart goes back out.
- MECHANISM: Increased Filling Volume ------> Stretch Ventricular Muscle
------> Augmented ventricular fiber length ------> greater inotropic state ------>
faster velocity of ejection ------> Greater Cardiac Output.
- Increased fiber length results in more forceful contraction, within limits.
- Optimal muscle fiber length = 2.2 micron. Heart normally works slightly
below this level to give room for optimal filling.
PRESSURE-VOLUME LOOP: P/V graph, with both diastolic and systolic lines plotted on
it. You use this graph to plot the pressure and volume at all points in the cardiac cycle.
- END-SYSTOLIC CURVE: The upper limit to the loop.
- END-DIASTOLIC CURVE: The lower limit to the loop.
- CARDIAC CYCLE in LOOP:
- DIASTOLE:
- ISOVOLUMIC RELAXATION: Volume is constant while pressure goes
straight down.
- VENTRICULAR FILLING: Pressure remains constant while volume
increases.
- SYSTOLE:
- ISOVOLUMIC CONTRACTION: Volume constant while pressure goes
straight up.
- EJECTION: Pressure continues to increase as blood is ejected
from the ventricle. The end-pressure at this point is systolic arterial
pressure.
- The pressure continues to rise during systole because pressure is
rising in the arterial network. You are putting more blood into the
arterial tree then is being put out on the other side. Ventricle must
match that rise in pressure to force blood out.
- AORTIC VALVE CLOSES: At the end of systole, the ventricular
pressure (i.e. fiber length) decreases to the point that the aortic valve
can't stay open, so it closes.
- STROKE WORK: The area of the Pressure-Volume Loop. Mathematically, that
means: Stroke Work = (Stroke Volume) x (Mean Arterial Pressure)
- STROKE WORK is equivalent to stroke volume, but it is normalized for
differences in blood pressure. Thus it is a good indicator of heart
performance.
- Because we have normalized for blood pressure, a shift in the curve for
stroke work means that there must be an increase in the inotropic state.
VENTRICULAR FUNCTION CURVE: A comparison of End-Diastolic Volume (or Pressure
or Fiber Length) and Stroke Volume (or Stroke Work). The curve is essentially a line that
levels off at high values. It is a way of expressing Starling's Law.
- If you plot Stroke Work -vs- LVEDV, you will get the same curve for the same
inotropic state, regardless of blood pressure. So using Stroke Work normalizes
for blood pressure, and it makes the curve represent the inotropic state.
EFFECT OF PRELOAD ON STROKE-WORK:
- Standing at Rest: The least stroke work is performed.
- SUPINE ------> Preload (venous return) increases ------> Fiber-length increases
------>------> Higher Stroke Work.
- PRONE, with LEGS RISEN: Even more pronounced effect as above ------> higher
stroke work.
EFFECT OF AFTERLOAD ON STROKE VOLUME: A higher afterload ------> Higher
systolic pressure must be developed ------> Higher end-systolic volume to achieve that
pressure, but the end-diastolic volume remains the same ------> lower stroke volume.
AUTOREGULATION OF AFTERLOAD: Due to heterometric autoregulation, within
limits, stroke volume will be maintained even in face of a higher blood pressure, but it
takes a few beats for the mechanism to kick in.
- High afterload ------> Lower stroke volume ------> Since pulmonary arterial
pressure hasn't changed, the right heart continues to pump the same stroke volume
as before ------> Pulmonary blood volume increases ------> Higher venous
return back to left atrium ------> Higher preload ------> Higher fiber length +
velocity of ejection ------> ------> Stroke volume returns to normal
- But a new pressure-volume curve is carved out on the P/V-Loop. Stroke-work
overall has increased.
- In compensating for the higher blood pressure, we must use some of our Starling
Reserve -- the extra capacity in the heart to do stroke work, strictly because of the
Starling mechanism.
TOTAL RESERVE: The total stored capacity the heart has to do extra stroke work. It is
equal to Starling Reserve + Inotropic Reserve + Heart-Rate Reserve.
- STARLING RESERVE: The extent to which we can increase Cardiac Output simple
by increasing filling, at the same inotropic state.
- INOTROPIC RESERVE
- HEART-RATE RESERVE
INOTROPIC STATE: It's the contractile force in the muscle, at any particular fiber-length.
That is the same as the Ca+2 concentration in the sarcomeres.
- It increases stroke volume, DUH??
HEART-RATE AND STROKE VOLUME: Heart rate extremes lead to lower stroke volume.
- Bradycardia: Heart-rate slower than 40. Cardiac Output goes way down because
the stroke volume can't increase enough to compensate for the lower heart-rate.
You've reached the maximum of the heart's inotropic state.
- Tachycardia: Heart-rate faster than 180. Cardiac Output goes way down because
there is no longer enough time between beats for sufficient ventricular filling, i.e. the
short diastolic time cuts into the "Fast-Filling Phase" of diastole.
Return to top
VALVULAR DYSFUNCTION
MITRAL INSUFFICIENCY: Insufficiency means the valve can't stay completely closed, so
it is leaky. Mitral Insufficiency causes fluid to reflux into the Left Atrium with each systole,
leading to a chronically high end-diastolic volume ------> left-ventricular hypertrophy.
- Holosystolic Murmur can be heard throughout systole, as turbulent blood flows
through mitral valve.
- Third Heart Sound can be heard during diastole, as there is a large excess of atrial
blood ------> turbulent flow during ventricular filling.
- Large V-Wave is seen: Higher atrial pressure produced during diastole, because
there is higher atrial volume.
MITRAL STENOSIS: Leads to lower filling of the left atrium, as the system backs up. This
leads to overload of blood in the pulmonary system.
- High Pulmonary Arterial Pressure from backup of blood.
- Pulmonary Edema is a likely complication that can result from the
pulmonary hypertension.
- Right Ventricular Hypertrophy also commonly comes from high Pulmonary
hypertension.
- Heart Sounds:
- Pre-Systolic Crescendo Murmur is diagnostic of mitral stenosis. The
murmur results from large increases of pressure during atrial systole,
because of the mitral stenosis.
- Diastolic (S3) Decrescendo Murmur is also heard, as there is a large
pressure difference between atrium and ventricle during diastole. That
pressure difference then becomes smaller (i.e. quieter) as the ventricle fills
and the atrium empties.
AORTIC INSUFFICIENCY: Regurgitation back into left-ventricle, on each systole, leads
to severe left-ventricular hypertrophy (when the insufficiency is severe).
- Dangerously Large Pulse Pressure results from high systolic pressure (due to
compensatory mechanism / inotropic state), and markedly decreased diastolic
pressure (due to low stroke volume).
- High LVEDV ------> Left-Ventricular Hypertrophy which can be severe.
- Heart Sounds:
- Loud Holo-Diastolic Decrescendo Murmur.
AORTIC STENOSIS: Very common in old people.
- Severe Left Ventricular Hypertrophy. The stenosis results in left ventricular
pressure being a lot higher then aortic pressure.
- HEART SOUND: Diamond-Shaped Pansystolic Murmur -- i.e. diamond-shape =
crescendo then decrescendo.
THE RIGHT HEART: Tricuspid and Pulmonic Valve problems are similar to those found
in the left heart.
MEASUREMENT OF CARDIAC OUTPUT (Last few pages of handout):
- Direct Fick Method: You calculate blood flow through the lungs (rate of O2 uptake)
to determine the pulmonary flow. Then you assume that pulmonary blood flow is
equal to systemic blood flow (i.e. CO).
- This assumption is true as long as there are no intracardiac shunts.
- Indirect Fick (Thermal Dilution) Method: Calculation blood flow essentially by
measuring the time that it takes for the flow of blood to neutralize a temperature
difference between injected saline and body temp.

Return to top
THE MICROCIRCULATION
CAPILLARY FILTRATION AND RESORPTION: STARLING PRINCIPLE FOR CAPILLARY
EXCHANGE: 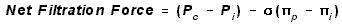
- Filtration: Blood leaving capillary and entering organ. Net flow outward.
- Pc, capillary hydrostatic pressure contributes to this outflow.
- PIi, interstitial oncotic pressure contributes to this outflow. It is the oncotic
(osmotic) pressure created by insoluble proteins in the interstitial space.
- Absorption: Blood leaving organ and entering capillary. Net flow into capillary.
- Pi, interstitial hydrostatic pressure, does not contribute to absorption
under normal circumstances. It is ~ 0.
- PIp, capillary oncotic pressure, is the primary contributor to resorption.
This is the osmotic pressure created by insoluble proteins in the blood.
- sigma, REFLECTION COEFFICIENT: It is equal to the percentage of proteins that
are impermeable to the capillary membrane, i.e. a value between 0 and 1.
- sigma = 1: Proteins are totally impermeable; all of them are "reflected" off the
membrane, thus oncotic pressure has the greatest influence possible on net
filtration.
- sigma = 0: Proteins are completely permeable, hence no proteins are
impermeable and oncotic pressures become zero.
- Low reflection coefficient affects resorption but not so much
filtration, since the capillary oncotic pressure is the only significant
force for resorption.
- RESULT: Edema.
- Lymphatics: Under normal circumstances, filtration is greater than absorption.
Thus more blood is being deposited in organ systems than is being taken up. The
difference is put back into the blood through the lymphatic system.
- The entire blood circulation is turned around through the lymphatic system
every 24 hrs.
- Capillary Hydrostatic Pressure:
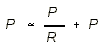
- Ra = arterial resistance is normally much larger than venous resistance.
We can usually safely ignore venous resistance in the calculation.
- Pv = venous pressure
- Pa = mean arterial pressure
- INFLUENCES ON FILTRATION:
- ARTERIOLAR RESISTANCE: Note that local(arteriolar) changes to vascular
tone have an exact opposite effect as systemic (large artery) changes.
- All things constant, increased arterial resistance ------> lower capillary
hydrostatic pressure + lower filtration
- Vasoconstriction or dilation at the level of the arterioles does not affect
MABP.
- Arteriolar Vasoconstriction ------> ------> lower filtration rate.
- Arteriolar Vasodilation ------>------> higher filtration rate.
- VENOUS PRESSURE: Increased Venous Pressure ------> Higher Capillary
Hydrostatic Pressure ------> Increased Net Filtration, because of the
hydrostatic pressure equation:
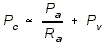
- The capillary pressure must increase in order to achieve filtration in
face of the increased venous pressure.
- ONCOTIC PRESSURE: Negative nitrogen balance or protein malnutrition
(kwashiorkor) will lead to low plasma albumin ------> low plasma oncotic
pressure ------> low or no resorption, which means high net rate of filtration
------> edema, ascites
CAPILLARY PERMEABILITY:
- Three types of capillaries, each having different levels of permeability:
- Continuous: Tight Junctions, as in brain, thymus, retina.
- Fenestrated: Little diaphragms where diffusion can take place, having
somewhat higher permeability. GI-Tract.
- Discontinuous: Liver sinusoids, complete discontinuities in the system.
- Endothelial Cells: When endothelial cells contract, the spaces between them
increase ------> higher capillary permeability.
- Mast Cell Degranulation leads to release of Histamine and Platelet-Activating Factor (PAF).
- This makes the endothelial cell release Calcium from the SR ------> actin-myosin contraction of endothelial cell makes the cell change shape
------> more spaces between the cells.
- Anaphylactic Shock: High capillary permeability leads to low blood pressure. We
can't just give them fluids to increase blood volume, because the fluids leak right
out again.
EDEMA: It can occur from a lot of sources, such as no resorption. Consequences of
edema:
- Impair Exchange of Metabolites: It leads to bigger spaces (longer distance)
between capillaries and the tissues ------> diffusion becomes impossible.
- THE EDEMA POSITIVE-FEEDBACK CYCLE: Edema can compress venules
------> higher venous return ------> higher CVP ------> higher hydrostatic
pressure and filtration rate ------> even more edema.
LYMPHATIC BLOCKAGE: If you block lymphatics, then interstitial fluid along with
interstitial proteins will rise ------> increase interstitial oncotic pressure ------> more
filtration ------> massive edema.
VASCULAR SMOOTH MUSCLE: Anything that increases intracellular Ca+2 concentration
will increase contractility of vascular muscle.
- VASCULAR CONTRACTION:
- Mechanism of Contraction, briefly:
- Calcium binds to Calmodulin
- The Ca+2-Calmodulin Complex then binds to Myosin Light-Chain
Kinase
- This results in myosin being free to interact with actin.
- Vascular Tone: The overall rate of cross bridging is much slower than in
vascular smooth muscle. There is always a baseline level of activity =
vascular tone.
- Norepinephrine will cause vascular contraction by increasing Ca+2 in
smooth muscle, via three pathways:
- NorE can directly open Ca+2-Channels
- NorE can bind to alpha1-Receptors to active the alpha-Adrenergic
Pathway (DAG/IP3) ------> higher intracellular Ca+2
- Voltage-Gated Ca+2 Channels can further open, in response to the
above two.
- VASCULAR RELAXATION: Anything that decreases Ca+2 concentration will cause
relaxation.
- Epinephrine in the blood causes vascular relaxation.
- Epi binds beta2-Receptors to activate beta-Adrenergic Pathway
------> higher levels of cAMP which results in decreased Ca+2 in
cytosol.
- cAMP will facilitate pumping of Ca+2 back into SR.
- ATP: Low levels of ATP will cause vascular relaxation locally, which should
allow greater blood flow, greater perfusion, and hence more ATP to deprived
tissue.
- ATP-Dependent K+-Channels open in response to LOW ATP.
- This leads to Hyperpolarization ------> Vascular Relaxation ------>
greater blood flow to area.
- NO causes relaxation, covered later.
- VASOMOTION: Spontaneous action potentials can cause a cyclic change in
vascular tone.
- Addition of NorEpi increases the rate of firing of those action potentials
------> more vascular tone.
- However, action potentials are not always required to cause sustained
contraction.
ENDOTHELIAL-DERIVED FACTORS: Nitric Oxide
- EXPT: Acetylcholine's (i.e. parasympathetic) effect on vessels depends on the
presence of the endothelial cells.
- Add Ach to vessel with endothelial cells intact ------> relaxation.
- Add Ach to vessel with endothelial cells removed ------> actually leads to
contraction!
- Process of NO-Mediated Vascular Relaxation:
- Ach binds to endothelial cell.
- Ca+2 channels open and Ca+2 pours into endothelium.
- This makes the endothelial cell produce NO from Arginine, by up-regulating
synthesis of the enzyme Constitutive NO-Synthase.
- Endothelium makes NO which diffuses to the underlying vascular smooth
muscle.
- NO then activates Guanylyl Cyclase, which produces cGMP ------> leads
to Ca+2 sequestration and vascular relaxation.
- L-Nitroarginine Methyl Ester (L-NAME): Inhibits NO-Synthase, blocking production
of NO ------> arteriolar constriction ------> lower blood flow to region.
- ISCHEMIA-REPERFUSION: The danger in reperfusing ischemic tissue is that
massive influx of O2 can lead to oxidative free radicals which damage endothelial
cells. The free radicals have two bad effects:
- They react with NO, leading to vasoconstriction and reduced perfusion of the
area.
- They directly damage the endothelial membrane leading to increased
vascular permeability which isn't good (it can lower blood pressure, etc.)
- SEPSIS: Causes vesicle to become less sensitive to vasoconstriction.
Phenylephrine has a lesser effect on septic vessels.
- It leads to higher NO via Inducible NO-Synthase. This is not the same
enzyme as constitutive NO-Synthase.
- The number of vasoconstrictive alpha-Receptors is decreased.
- Basal Ca+2 levels are reduced or Ca+2-channels don't open properly.
- ADHESION MOLECULES: NO protectively prevents expression of adhesion
molecules, so that leucocytes don't stick to vessel wall, which can lead to
microvascular injury.
- Hence we can't use L-NAME as a treatment for Sepsis -- we need the NO to
prevent sticking of blood cells, even if vasodilation is an undesired effect.
- What we need is a drug that blocks only Inducible NO-Synthase (made
during sepsis) and not constitutive NO-Synthase. We don't have that (yet).
ENDOTHELIN-1: Vasoconstrictive agent produced by endothelial cells.
- SLOW-RESPONSE: Endothelin is not stored in vesicles. It is synthesized de novo.
Thus it is a slow (long-term) response.
- SYNTHESES: Preproendothelin ------> Big Endothelin ------> Endothelin.
Multi step synthesis adds to slow response.
- EFFECT: Endothelin causes sustained vasoconstriction. The effect lasts long! It
causes increased levels of Ca+2 and thus increased vascular tone.
- It acts via alpha-adrenergic pathway (PIP/DAG ------> Ca+2)
- It also acts directly on Ca+2-Channels.
- ISCHEMIA REPERFUSION: Endothelin is bad! It can be released along with
inflammatory mediators to cause further vasoconstriction when we want
vasodilation.
LOCAL REGULATION OF BLOOD FLOW:
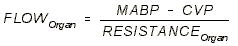
- We control local blood flow by changing local resistance.
- Three factors can change local resistance:
- Endothelial-Derived Factors
- Mechanical Stretch of the vessel itself
- Intrinsic Factors = locally derived metabolites
- Organ-Distribution of Blood Flow: Highest perfusion rates are in liver, kidney, and
skeletal muscle.
- Kidneys have the highest Perfusion Index: The ratio of perfusion to organ
size. It measures the relative amount of blood that different organs get per
organ mass.
- OXYGEN UPTAKE:
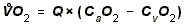
- To increase Oxygen Uptake by tissues, you can therefore increase one of
two things. Most organs increase O2-uptake by a combo of both things.
- Increase O2 extraction. This is how the KIDNEYS primarily get more
oxygen.
- Increase blood flow. This is how the HEART primarily gets more
oxygen. The heart can't increase O2-extraction because it is already
extracting about the maximum amount possible.
- O2-Extraction = Arterial PO2 - Venous PO2
- Oxygen is extracted by simple diffusion.
- To increase oxygen extraction, increase the surface area of
capillaries exposed to tissue.
- Heart-Muscle has a high basal capillary concentration than
skeletal muscle. Thus it has higher oxygen extraction.
- Pre-Capillary Sphincters can be dilated to perfuse more capillaries
in the capillary bed.
- Specific Organs:
- HEART: It has a high oxygen extraction, so the only way to increase
O2 uptake is to increase blood flow.
- KIDNEY: It has a lower oxygen extraction. It can actually increase
O2 extraction to increase O2-Uptake.
AUTOREGULATION:
- Mechanism: Keep constant flow and capillary pressure (i.e. filtration) in the face of
changing systemic pressures.
- Lower local pressure ------> Vasodilate ------> lower resistance ------>
maintain higher flow and higher capillary pressure.
- Higher local pressure ------> Vasoconstrict ------> higher resistance
------> maintain lower flow and lower capillary pressure
- Tissues: Autoregulation works particularly in the kidney, heart, and brain.
- Limits: Autoregulation only works in a limited range of pressures. Vessels won't
change diameter past their minimum and maximum.
MYOGENIC RESPONSE: Sudden stretch of vascular wall can lead to vasoconstriction to
counteract the higher blood-volume. Works in conjunction with the metabolic response to
maintain blood flow.
- There are two types of arterioles:
- One produces Action Potentials to have rhythmic vasoconstriction
(vasomotion)
- The other type does not produce action potentials.
- Both types are still subject to the myogenic response.
- Mechanism:
- AP-Capable Arterioles: Stretch ------> increased frequency of AP-firing
------> higher vascular tone.
- AP-Incapable Arterioles: Stretch ------> depolarization of vascular smooth
muscle ------> Ca+2 influx and higher vascular tone.
METABOLIC RESPONSES: Works in conjunction with the Myogenic Response to
maintain blood flow.
- METABOLIC HYPOTHESIS: Vasodilator Metabolites are made locally in response
to hypoxia and poor blood flow, in order to increase blood flow. The metabolites
are then washed away when blood flow increases again, disposing of their effect.
- HYPOXIA: Hypoxia leads to a decrease in intracellular ATP, which ultimately leads
to vasodilation.
- K+-ATP CHANNELS: They kick K+ out of the cell in exchange for bringing
ATP in. They open in response to low ATP levels.
- Hypoxia ------> low intracellular ATP ------> Open K+-ATP Channels
------> K+ pours out of cell ------> membrane hyperpolarizes ------>
smooth muscle relaxation.
- PROSTACYCLIN (PGI2): Prostacyclin may be released by endothelial cells
in response to hypoxia ------> potent vasodilation in a paracrine manner
no neighboring smooth muscle.
- ACIDOSIS: Acidosis in smooth muscle directly causes hyperpolarization of smooth
muscle membrane ------> vasodilation.
- Acidosis means CO2 levels in tissue are high.
- CO2 <====> H2CO3 <====> H+ + HCO3-
- ADENOSINE: Adenosine is an indicator that the target tissue is out of ATP (as
opposed to the smooth muscle itself).
- Adenosine is membrane-soluble while ATP, ADP, and AMP are not. So
when the compounds gets down to the Adenosine level, it can then leave
the cell to affect the neighboring smooth muscle.
- Adenosine is a potent vasodilator.
- AUTOCOIDS: Histamine, Bradykinin, Serotonin, Prostaglandins, Leukotrienes.
- POTASSIUM: Potassium regulation is especially important in the brain and in
skeletal muscle.
- SMALL AMOUNTS OF K+
- In both tissues, extracellular K+ concentration goes up because of
repeated firing of action potentials.
- This results in release of vasodilator-factors (NO, PGI2) and in
membrane hyperpolarization of vascular smooth muscle.
- HUGE (PHARMACOLOGICAL) INCREASE IN K+ ------> depolarization of
muscle membrane ------> vasoconstriction.
- INTERSTITIAL OSMOLARITY:
ACTIVE HYPEREMIA: Blood flow changes in proportion to changes in metabolic activity
of the organ. Occurs in Skeletal Muscle.
- Lactic Acidosis in skeletal muscle ------> Vasodilation of vasculature.
- In Active Hyperemia, the metabolic activity of the target tissue (i.e. skeletal muscle)
is changing, and that's what causing the vasodilation.
REACTIVE HYPEREMIA: The short-term increase in flow following temporary ischemia
to a region.
- Both myogenic and metabolic effects are playing a role in causing the vasodilation.
- In reactive hyperemia, the metabolic activity of the target tissue does not change,
whereas in active hyperemia, it does.
REGIONAL CIRCULATIONS:
- CEREBRAL CIRCULATION:
- Cerebrospinal Fluid: Normally has a lower protein content than blood.
- Cerebral Vasculatures have very poor sympathetic innervation. Hence in the
Cushing Reflex, massive sympathetics don't cause constriction of vessels
in the cerebrum (which they shouldn't!)
- Regulation of Flow: It is primarily K+-Mediated. We can get higher
extracellular K+ and vascular hyperpolarization by two sources:
- Firing of neurons without repolarization.
- K+-ATPase kicks out K+ in exchange for ATP, at low intracellular ATP
levels.
- The brain is very sensitive to changes in PCO2, but not so much to changes
in PO2.
- CORONARY CIRCULATION: Regulated almost entirely by local factors.
- Increase cardiac work ------> increased coronary blood flow.
- SYSTOLE: Coronary blood flow decreases, as the vessels are squeezed as
the myocardium contracts.
- There may even be some retrograde flow of blood during systole.
- DIASTOLE: Coronary blood flow increases.
Return to top
CARDIOVASCULAR CONTROL MECHANISMS
PARASYMPATHETIC DILATORS: They cause local vascular relaxation.
Parasympathetics do not have an important effect on systemic blood pressure.
- Vasoactive Intestinal Peptide (VIP): This neurotransmitter is released directly
onto the smooth muscle cells to cause relaxation.
- Nitric Oxide (NO):
- The nerve terminals contain Nitric-Oxide Synthase.
- NO, when released by nerve terminals, also acts directly on smooth muscle.
NOCICEPTORS: Sensory receptors to noxious chemicals or toxins. They also cause local
vasodilation.
- Two peptides are released by Nociceptor Nerves:
- Substance P
- Calcitonin Gene-Related Peptide (CGRP)
- TRIPLE RESPONSE OF LEWIS: Wheal and flare response to a local irritant.
- First, a small red area develops.
- This is due to degranulation of mast cells ------> local vasodilation.
- Second, a blanched raised area develops around the small red area.
- Third, a reddened flare (vasodilation) radiates around the irritated region.
- The flare is due to highly branched nociceptor nerves that are
distributed through the skin.
- The Nociceptors release SP and CGRP in the area to cause
vasodilation.
- LOCAL NEURAL RESPONSE: The nociceptor reflex does not go through the CNS!
- If you cut the Dorsal Root (proximal to the cell body), the neural response
still occurs.
- This shows that the reflex signal is independent of the CNS.
- If you cut the peripheral nerve (distal to the Cell Body), then Wallerian
Degeneration occurs and the reflex no longer happens.
- Capsaicin (red-pepper stuff) is an irritant that, if applied to the skin for a period of
time, will overuse and numb the nociceptors. Thus it is a treatment that can prevent
the irritant response.
SYMPATHETIC CONTROL OF VASCULAR MUSCLE: This is the primary short-term
mediator of TPR and hence arterial blood pressure.
- Sympathetics are of course vasoconstrictive, with two possible exceptions:
- beta2-Receptors are vasodilatory. They are most responsive to Epinephrine
(which is not a neurotransmitter), but they are responsive to NorE at huge
doses).
- Dogs and cats have sympathetic cholinergic nerves (like eccrine sweat
glands) that are vasodilatory.
- Norepinephrine: Released from small dense-core vesicles in the sympathetic
varicosity.
- Norepinephrine is released by any depolarization, of any impulse frequency.
- NorE binds to alpha1-Receptors on smooth muscle to increase Ca+2
concentration and effect smooth muscle contraction.
- NorE has a very high affinity for alpha1-Receptors. Epinephrine does
not.
- ATP: Released from small dense core vesicles in the sympathetic varicosity.
- ATP is released by any depolarization. It works at impulse frequencies as
low as 2Hz.
- ATP binds to Purinoreceptors (P-Receptors) to cause depolarization of
the smooth muscle membrane.
- Each ATP dense-core vesicle yields +10mV of depolarization. Two
simultaneous depolarizations (total of +20mV) are required to generate
smooth muscle action potential.
- Neuropeptide-Y (NPY): Released from large dense-core vesicles in the
sympathetic varicosity.
- Neuropeptide-Y is only released by repeated depolarizations (i.e. strong
sympathetic stimulation). It is only released if the impulse frequency is 8Hz
or faster.
- NPY binds to its own Y-Receptor.
- COTRANSMISSION: NorE, ATP, and NPY have additive effects.
- At high impulse frequencies, NPY facilitates the release of additional NorE.
- The summation of signals will lead to stronger contraction of vascular
smooth muscle, up to a point.
MODULATION OF SYMPATHETIC NEUROTRANSMITTERS: Cotransmission
principles are based on increased likelihood that a dense-core vesicle will fuse with the pre-synaptic membrane. The higher the impulse frequency, the more likely that is to occur.
- AUTORECEPTORS: Simple negative feedback. There are receptors for NorE,
ATP, and NPY. When the respective hormones bind, they inhibit further release of
the neurotransmitter (i.e. they decrease the likelihood of vesicle fusion).
- HETERORECEPTORS: These are pre-synaptic receptors that bind to other
substances to inhibit or excite release of neurotransmitters.
- Inhibitory Heteroreceptors:
- Acetylcholine binds to muscarinic receptors on the sympathetic
varicosity to inhibit the release of NorE.
- Prostaglandins, Serotonin, and Histamine can all bind to inhibitory
heteroreceptors as well.
- Excitatory Heteroreceptors: ANGIOTENSIN II will bind to excitatory
receptors to promote further release of NorE ------> vasoconstriction.
AUTONOMIC TONE: Heart rate and vascular tone is determined by the relative amounts
of sympathetic and parasympathetic continual stimulation.
- PARASYMPATHETIC TONE: The Vagus Nerve (CN X).
- Vagal tone for the heart and abdomen originates from:
- Nucleus Ambiguus (NA)
- Dorsal Motor Nerve of CN X (DMV)
- Parasympathetics have the following general effects on CV-System:
- They increase venous compliance ------> lower venous return.
- They indirectly decrease systemic resistance by inhibiting
sympathetics ------> lower blood pressure.
- Vagal Tone on heart slows down the heart-rate at the SA-Node.
- SYMPATHETIC TONE:
- In the brain, sympathetics originate from the C1 AREA, which is the
Reticular Formation of the closed medulla.
- From there, the pathway is Reticular Formation ------>
Intermediolateral Column of Thoracic spinal cord.
- Sympathetics have the following general effects on the CV-System:
- They decrease venous compliance ------> higher venous return
- They directly increase systemic resistance ------> higher blood
pressure
- They indirectly speed heart rate by inhibiting Vagal Tone on the SA-Node.
- Miscellaneous Drugs that Affect Heart Rate:
- Chlorisondamine: Nicotinic blocker -- it blocks pre-ganglionics of
both sympathetics and parasympathetics.
- RESULT = a slight increase in HR.
- Atropine: Blocks muscarinic receptors -- i.e. no parasympathetics.
- RESULT = substantially increase HR.
- Propanolol: It is a beta-Blocker -- it blocks beta1-Sympathetic receptors on
the heart.
- VAGAL TONE ON THE HEART:
- Parasympathetics (CN X) decrease heart rate by slowing the rate of rise of
autodepolarization on the SA-Node. That is, it directly decreases heart rate.
- Sympathetics increase heart rate by inhibiting the release of
parasympathetics, i.e. they increase heart rate indirectly.
VASCULAR BEDS: There are six main vascular beds in the body. Going from supine to
upright lowers blood pressure, so blood is conserved for the organs that really need it.
| VASCULAR
BED |
SNS
DENSITY |
TONE
(SUPINE) |
TONE
(UPRIGHT) |
NOTES |
| Cerebral |
Moderate |
Low |
Low |
High metabolic
requirements; no change |
| Coronary |
Low |
Low |
Low |
No Change |
| Cutaneous |
High |
High |
High |
Skin doesn't get much
blood either way (not much
change) |
| Skeletal
Muscle |
Moderate |
Low |
Moderate |
+ |
| Splanchnic
(Mesenteric) |
High |
Low |
HIGH |
+++ Blood is pulled away
from the GI-System |
| Renal |
High |
Low |
HIGH |
+++ Renal blood flow
(urine prod.) is cut down. |
BARORECEPTOR REFLEX: Short-term modulation of blood-pressure.
- MODE OF ACTION: Baroreceptor firing increases parasympathetic tone and inhibits
sympathetic tone.
- They decrease heart-rate via increase in vagal tone on the heart.
- They decrease blood pressure via inhibition of sympathetic tone on the
vessels.
- MODE OF STIMULATION: Baroreceptors are stretch receptors. They are
stimulated by high volume and or pressure in the region.
- Three Baroreceptors:
- Two Atrial Receptors -- detect "low" (venous) pressures
- Locations:
- At junction of SVC and RA.
- At junction of pulmonary veins and LA.
- It detects high venous return to the RA and goes off as a result
------> increase venous compliance ------> decrease venous
return.
- One Aortic Arch Receptor -- modulates "high" (arterial) pressures.
- Carotid Sinus: Two high-pressure baroreceptors at the bifurcation of the
Common Carotid Artery, bilaterally.
- BARORECEPTOR PATHWAY: The baroreceptor impulse is sent to the Nucleus
of the Tractus Solitarius (NTS). It has two outputs in response to the impulse:
- EXCITATORY IMPULSE is sent to the Vagal Nuclei (Dorsal Motor N and
the N Ambiguus) ------> higher parasympathetic tone
- INHIBITORY IMPULSE is sent to the C1-Area ------> lower sympathetic
tone.
- Short-Term Modulation of Blood-Pressure:
- STANDING UP: Blood pools to feet ------> much lower venous return to
heart.
- The drop in venous return can be as much as 500 mL. That's quite
a bit.
- Baroreceptors stop firing (i.e. are down-regulated) in response to
standing up, so that sympathetics are dis-inhibited (turned on), and
b.p. goes back up.
- IF PRESSURE FALLS: Baroreceptors are turned off and sympathetics
increase ------> faster heart rate and vasoconstriction.
- IF PRESSURE RISES: Baroreceptors are turned on ------> higher activity
on NTS ------> slower heart rate and vasodilation.
- Limitations: The Baroreflex is only short-term.
- Autoregulatory Escape: Certain tissues can override the CNS baroreflex
if they have been vasoconstricted for too long.
- Baroreceptors do not determine blood pressure. They only modulate it.
- They are a buffering system. They operate best between 180 mm Hg and
60 mm Hg.
Bainbridge Reflex: An exception to baroreceptor regulation, where increased stretching
actually increases the inotropic state of the heart, i.e. turns on sympathetics.
- It occurs when the Left Atrium is stretched, indicating high preload on the heart.
CHEMORECEPTORS: They have the exact opposite effect as Baroreceptors.
- Two locations: One in the Aortic Region and one at the bifurcation of the Carotid,
called the Carotid Body.
- Stagnant Hypoxia: Chemoreceptors respond to low O2 levels. The cells have a
higher metabolic rate, and when they run out of O2 they fire.
- CHEMORECEPTOR REFLEX: It is the same pathway, but the exact opposite effect
as the baroreceptors. They turn on sympathetics and turn off parasympathetics.
- Reflex again goes back to the Nucleus of Tractus Solitarius (NTS)
- Afferent signals stimulate the C1 Area (sympathetics) and inhibit the DMV
of the Vagus.
- RESULTS: Typical sympathetic CV effects.
- Arterial vasoconstriction in the splanchnic beds (alpha1) to divert blood to
the brain and heart.
- Venous vasoconstriction to increase venous return.
- Faster heart-rate from inhibition of Vagus.
- CUSHING REACTION: Happens from high CSF pressure to the point that it
occludes cerebral vessels.
- This rather quickly causes massive sympathetic outflow and a huge increase
in MABP.
- Note that this can occur even when systemic b.p. was normal. All that is
required is occlusion of cerebral blood flow due to CSF pressure.
THE SYMPATHO-ADRENAL SYSTEM: Intermediate and long-term modulation of blood
pressure.
- Sympathetic Receptors:
- alpha1-Receptor: Primary vasoconstrictor found in VASCULAR SMOOTH
MUSCLE
- alpha2-Receptor: Also found in vascular smooth muscle.
- beta1-Receptor: Found in HEART AND KIDNEYS.
- Increases heart rate via innervation of SA-Node.
- Increases inotropic state via innervation of myocardial muscle.
- Stimulates release of Renin from the kidneys.
- beta2-Receptor: VASODILATOR found in VASCULAR SMOOTH MUSCLE
- Epinephrine is the primary ligand to bind to these receptors ------>
vasodilation ------> lower TPR.
- Sympathetic Neurotransmitters / Neurohormones:
- Norepinephrine:
- Binds to alpha1 and alpha2 Receptors (vasoconstriction)
- Binds to beta1-Receptors (Positive inotropy and chronotropy)
- Epinephrine:
- Binds primarily to beta1 and beta2 receptors: positive inotropic /
chronotropic on heart and VASODILATORY
- Only at high doses, it also binds to alpha-receptors, which will tend to
counteract or even override the vasodilatory effect of the beta2-Receptors.
ANTI-DIURETIC HORMONE (ADH): It increases Na+-retention in the kidney ------> more
water retention ------> high blood volume. It is a "long-term," slow-responding effect.
- CAUSE of Release: ADH is stimulated to be released by lower baroreceptor firing.
Not sure of the exact pathway -- but somehow that leads to posterior pituitary being
stimulated to release ADH.
- EFFECTS:
- Intermediate Effect: There are ADH receptors on arteries and veins. ADH
causes vasoconstriction.
- Long-Term Effect: ADH increases blood volume via increased Na+-Retention in the kidney.
RENIN-ANGIOTENSIN SYSTEM:
- Renin Release from Kidney:
- The Juxtaglomerular Apparatus detects low renal blood flow. It will
stimulate release of Renin from the kidney.
- Sympathetic innervation of kidney will also stimulate release of Renin.
- Biosynthetic Pathway of Angiotensin II:
- Renin, from the kidney, circulates in the blood stream.
- Angiotensinogen ------> Angiotensin I.
- This conversion occurs in the bloodstream.
- This conversion is catalyzed by Renin from the kidney.
- Angiotensin I ------> Angiotensin II (active form)
- This conversion occurs in the lungs.
- This conversion is catalyzed by Angiotensin Converting Enzyme
(ACE).
- ACE-INHIBITORS are common drugs to battle hypertension by
preventing synthesis of Angiotensin II.
- EFFECTS OF ANGIOTENSIN II:
- It binds heteroreceptors on sympathetic varicosities to cause increased
release of NorE onto the vasculature ------> higher arterial resistance.
- It stimulates the release of Aldosterone from adrenal medulla. Aldosterone
goes to kidney where it causes Na+-retention and thus increased plasma
volume.
- It also directly affects the kidneys to decrease urine production and increase
plasma volume.
ATRIAL NATRIURETIC PEPTIDE (ANP): It causes increased Na+-Excretion (opposite
effect as ADH) in the kidney.
- It is found in granules in atrial muscle.
- RELEASE: Stretch of Atrial Muscle means there is high preload ------>
mechanical release of ANP-granules from Atrium ------> to kidney to increase
urine production and decrease plasma volume.
Carotid Sinus Syndrome: Hypersensitivity of the Carotid Sinus, in some old people.
Turning their head to the right stimulates parasympathetics and makes them pass out.
Return to top
HYPOTENSION AND HYPERTENSION
Classifications of Shock: Shock means low blood volume.
- HYPOVOLEMIC SHOCK: Shock from loss of fluid, either blood or vomit, diarrhea,
etc.
- VITAL SIGNS:
- Low CVP: Veins in neck will be flat.
- High heart rate, breathing rate, and TPR, at least initially.
- Low urine output and low cardiac index.
- SHOCK FROM TISSUE INJURY
- CARDIOGENIC SHOCK: Pump failure, either intrinsic or extrinsic.
- VITAL SIGNS:
- High CVP: Veins in neck will be distended.
- High heart rate, breathing rate, and TPR, at least initially.
- Low urine output and low cardiac index.
- CARDIAC TAMPONADE: Fluid in the pericardial sac. Heart failure can
easily result from tamponade, which leads to cardiogenic shock.
- Cardiac Index increases initially, then decreases.
- Peripheral Resistance is initially low and then increases.
- ETIOLOGY: Systemic blood infection can start from compensatory
vasoconstriction of the GI-Tract (due to low systemic blood pressure)
------> GI-Ischemia ------> High permeability in intestinal wall ------>
bacteria enter blood.
- FOUR STAGES OF SEPTIC SHOCK:
- STAGE 1: Trauma, inflammation, or infection, leading to
hypovolemia and tissue injury.
- STAGE 2:
- CO is markedly increased
- TPR is reduced.
- STAGE 3:
- CO begins returns to near normal.
- TPR is markedly reduced.
- Hypotension
- Lactic Acidosis
- STAGE 4: Irreversible Stage, some say. All of above, except cardiac
output is subnormal.
- VASOGENIC / NEUROGENIC SHOCK: Collapse of nervous system and loss of
sympathetic tone in blood vessels ------> severe hypotension.
- PROGRESSIVE (IRREVERSIBLE) SHOCK can result from any of the above forms
of shock. This is characterized by:
- Ischemia to gut and kidney
- High capillary permeability
- Marked vasodilation, as mediated by local factors such as bradykinins,
serotonin, NO.
- INTERDEPENDENCE: One type of shock begets another. Especially septic shock
can result from any of the other types of shock.
HYPERTENSION: Defined as worse than 140 / 90.
- ETIOLOGY: Hypertension is always explained by a combination of either higher
preload (blood volume) or Higher TPR.
- STRESS ------> Higher sympathetic tone ------> higher TPR.
- GENETIC PREDISPOSITION to high levels of Angiotensin II
- EXCESS SODIUM UPTAKE ------> Excess water-retention in kidney
------> higher blood volume
- SYMPTOMS:
- Atrial Natriuretic Peptide (ANP) may be released as a compensatory
mechanism, from stretch of atrial muscle ------> Higher Na+ excretion and
lower blood volume.
- Increased Inotropic State in early hypertension shifts the Systolic Curve up,
while maintaining the same EDV ------> More stroke work and bigger
stroke volume.
- Continual higher preload will lead to an increased basal level of Ca+2
------> higher basal TPR.
- CONC: Whether the hypertension starts from high blood volume or
high TPR, ultimately it will manifest as high TPR.
- This further perpetuates vasoconstriction.
- Higher basal levels of Endothelin will lead to further vasoconstriction.
- Structural Hypertension: Hypertrophy of vascular muscle, from hypertension.
- PROLONGED HYPERTENSION:
- Left Ventricular Hypertrophy ------> Higher End-Diastolic Pressure.
- Decrease in the Inotropic State in later hypertension: The Ventricular
Function Curve therefore shifts downward -- A greater end-diastolic pressure
is required to achieve the same stroke volume.
- Baroreceptors are down-regulated: With chronic hypertension,
baroreceptor-firing becomes less than normal. They are essentially
desensitized to the hypertensive condition.
- TREATMENT:
- beta-Blockers: Slow heart-rate and decrease contractility ------> Decrease
CO
- ACE-Inhibitors -- decrease basal levels of Angiotensin II ------> Decrease
TPR
- Ca+2-Channel Blockers: Decrease contractility of vascular smooth muscle
------> Decrease TPR
- Diuretics -- decrease blood volume ------> Decrease CO
- alpha-Blockers: Decrease vascular tone (sympathetic influence on alpha1-receptors) ------> Decrease TPR
CONGESTIVE HEART FAILURE: From chronic hypertension.
- Four Progressive Stages of CHF based on activity-tolerance: Class I has no
limitations on activity, and in Class IV, symptoms are present even at rest.
- SYMPTOMS:
- LEFT-SIDE CHF SYMPTOMS: Pulmonary Hypertension leading to
Pulmonary Edema.
- RIGHT-SIDE CHF SYMPTOMS: Central Venous Hypertension leading to
Peripheral Edema.
- NEGATIVE INOTROPY: NOREPINEPHRINE in blood is HIGH, but NorE in
the Heart is low. The Heart is in a low inotropic state with CHF.
- There are fewer actin-myosin cross-bridges being made.
- This leads to a LOWER LEVEL of SYSTOLE on the pressure-volume
curve.
- NEGATIVE LUSITROPY: Incomplete relaxation of myocardial muscle.
- There are high basal levels of Ca+2 in the myocardial cytoplasm.
- There are low levels of Ca+2 being stored in the myocardial SR,
because Ca+2-ATPase Channels are fewer.
- This leads to a HIGHER LEVEL of DIASTOLE
- LOWER SYSTOLE + HIGHER DIASTOLE = HEART-FAILURE. Look at the
area in the curve now, and it is much lower stroke-work.
- ORTHOSTATIC HYPOTENSION: High levels of Epinephrine make TPR go
markedly down, which especially shows up when standing.
- Epinephrine can also get stored in sympathetic nerve-terminals,
because of perpetually high circulating levels of Epi. This leads to
vasodilation when we should have vasoconstriction!
- Baroreceptor malfunction also contributes to orthostatic hypotension.
- COMPENSATORY MECHANISMS: High Levels of ANP are found in late-stage
CHF.
|
DOG BLOOD PRESSURE DEMO |
| PROCEDURE |
MABP |
HEART
RATE |
TPR |
SV |
CO |
| Acetylcholine |
DOWN |
DOWN |
DOWN |
UP
Higher
preload
from longer
diastolic
filling |
UP |
| Phenylephrine (alpha-agonist) |
UP
No effect
on pulse
pressure |
DOWN
Baroreceptors
compensate
for higher
TPR |
UP
This is the
primary
effect |
SAME
Increased
afterload
but also
preload |
DOWN
Because of
lower heart
rate |
| Isoproterenol
(beta-Agonist) |
DOWN
From
lower TPR |
UP
beta1-Receptors |
DOWN
Vasodilation beta2-Receptors |
UP |
UP |
| Epinephrine
(beta+alpha
Agonist) |
UP
Systolic
increases
but not
diastolic: |
UP
beta1-Receptors
on heart |
DOWN
beta2 at
low doses;
maybe
some
alpha1 at
high doses |
UP
|
UP |
| Carotid
Massage |
DOWN
Baroreceptors |
DOWN
Baroreceptors |
DOWN |
DOWN |
DOWN |
| Carotid
Occlusion |
UP
Inhibition
of
Baroreceptors
No change
in pulse
pressure |
UP
Inhibition of
Baroreceptors
|
UP
Because
Diastolic
Pressure
went up |
SAME |
UP |
| Nitroglycerin
(NO) |
DOWN |
SAME
Or slightly
up |
DOWN |
UP
From higher
EDV |
UP |
RIGHT VAGAL STIMULATION: Acts primarily on the SA-Node, hence it will cause a
decrease (or arrest) of heart-beat.
LEFT VAGAL STIMULATION: Acts primarily on the AV-Node, causing an atrioventricular
heart-block when stimulated.
Return to top