 Return to Biochemistry
Return to Biochemistry
NITROGEN METABOLISM
Download a copy of this study guide
 Return to top
Return to top
DIABETES
TYPE-I DIABETES -- JUVENILE ONSET: Insulin-Dependent (IDDM) -- The body cannot
make insulin.
- It is caused by an auto-immune response against glutamate decarboxylase, causing
destruction of pancreatic endocrine (beta)-cells as a result.
- TREATMENT: Insulin replacement therapy, with either bovine or porcine insulin, por
humulin.
- KETOACIDOSIS is found especially in this type of Diabetes, due to no insulin at all.
TYPE-II DIABETES: Non-Insulin Dependent (NIDDM) -- There is a problem with insulin-receptors, signal transduction pathways, or pancreatic secretion of insulin, but not production
- TREATMENT: Drugs.
- Sulfonylureas: Close K+ channels on the pancreatic beta-cell surface, allows Ca+
in-flow ------> trigger the release of insulin.
- Metformin: Reduces blood sugar by inhibiting intestinal absorption of glucose
and hepatic gluconeogenesis.
CLINICAL SYMPTOMS: High blood glucose level after having fasted 2 hours. is the differential
diagnosis.
- Ketoacidosis results -- pH of the blood is lower than normal and ketone bodies are
excreted in urine.
- This is due to too much fatty acid mobilization in fat-tissue (high glucagon:insulin
ratio).
- The extra Acyl-CoA's then divert to ketone-body production because the liver,
under the influence of glucagon, is busy with gluconeogenesis.
- Glucose Adducts: High levels of blood-glucose can lead to an irreversible adduct with
the terminal Val of the beta-chain of hemoglobin.
- Glucose can form many other Advanced Glycosylated End-Product proteins. Ruining
of blood proteins in this way can account for many of the secondary symptoms of Diabetes
(i.e. retinopathies etc.)
INSULIN SYNTHESIS: Synthesized by propeptides A, B, and C. C is cleaved away, and A and
B get together to form the final polypeptide.
- Amylin modulates the action of insulin.
Return to top
PROSTAGLANDINS
SYNTHESES OF PROSTAGLANDINS: O2 is required for synthesis.
- Phospholipase-A2 catalyzes Linolenic (Linoleic) Acid ------> Arachidonic Acid.
- This is the rate-limiting step in Prostaglandin Synthesis.
- Prostaglandin H Synthase: Arachidonic Acid ------> Prostaglandin-H2 (PGH2)
- O2 is required for synthesis at this step, too.
- End Products from PGH2:
- Prostacyclin
- Thromboxane
- Other Prostaglandins
SYNTHESIS OF LEUKOTRIENES: Leukotrienes found only in monocytes, neutrophils,
macrophages, and beta-lymphocytes. O2 is required for synthesis.
- 5-Lipoxygenase: Arachidonic Acid ------> Leukotriene-A4 (LTA4)
EICOSANOID STRUCTURE:
- All contain 20 carbon atoms
- All contain additional oxygen atoms.
- Names are based on the type of ring-structure formed.
- Numerical subscripts refer to the parent fatty acid.
PLATELET AGGREGATION:
- Prostacyclin PGI2 inhibits it. It is made in the vascular endothelium.
- Thromboxane TXA2 promotes it. It is made in the platelets. If / when the platelets are
killed, this promotion of clotting is disposed of, at least temporarily.
ASTHMA: Leukotrienes may play a role in asthma, in eliciting an a release of mucous from
neutrophils in the lungs.
THE INFLAMMATORY RESPONSE: Platelets adhere to damages cellular surfaces and eject
their granules into the extracellular space. More platelets flock to the region causing swelling.
Platelet Activating Factor promotes further aggregation.
- Mediators hormonally control and promote the inflammatory response. There are many
mediators (histamines, prostaglandins, leukotrienes) specific to a tissue or circumstance.
- SYMPTOMS of Inflammation:
- Erythema = prolonged period of vasodilation.
- Edema = swelling of extracellular tissue, resulting increased permeability of
vascular tissues.
- Hyperalgesia = pain.
- Fever
- ASTHMA is a chronic inflammatory disease.
- Treated with Steroidal Anti-Inflammatory as an inhaler.
- RHEUMATOID ARTHRITIS is a chronic inflammatory disease.
- Treated with Aspirin as an anti-inflammatory.
STEROIDAL ANTI-INFLAMMATORIES: Cortisol is a very strong anti-inflammatory. It
probably acts by disrupting the action of Phospholipase A2.
NON-STEROIDAL ANTI-INFLAMMATORIES (NSAID's): They inhibit the enzyme
Prostaglandin-H Synthetase in the tissues of the inflamed site. This results in a block of the
conversion of arachidonic acid into prostaglandins, thereby shunting them to leukotrienes.
- Aspirin, Acetylsalicylic Acid: Inhibits the Cyclooxygenase activity of PGH-Synthase
(PGHS) by acetylating a unique Serine residue IRREVERSIBLY.
- The only way to get more PGH-synthesis from these tissues is to make more of the
cyclooxygenase enzyme de novo. Thus aspirin is an effector inhibitor.
- SIDE-EFFECT = aspirin will also block prostaglandin-mediated secretion of
mucous membrane in the intestinal wall, which can result in damage to intestinal
epithelia.
- Other NSAID's are REVERSIBLE.
- ACETAMINOPHEN: A good anti-pyretic and analgesic, but not shown to be a good
anti-inflammatory per se.
Return to top
ALCOHOLISM
METABOLISM OF ETHANOL: The ultimate product of alcohol metabolism is Acetyl-CoA.
- Alcohol Dehydrogenase: Ethanol + NAD+ ------> Acetaldehyde + NADH
- This action requires Zn+2
- OR, at the same time you can do it with Cytochrome P450, utilizing NADPH.
- Acetaldehyde Dehydrogenase: Acetaldehyde + NAD+ ------> Acetate + NADH
- Acetate + ATP + Coenzyme-A ------> Acetyl-CoA
PROLIFERATION OF SMOOTH ER: Increased Cytochrome-P450 in microsomes from chronic
alcohol consumption is associated with a proliferation of smooth endoplasmic reticulum.
ALCOHOL EFFECT ON TISSUES:
- Elevated EtOH can lead to uncontrolled uptake of fatty acids in cells, as the fatty acids can
esterify to the ethanol for uptake.
- Increased NADH from Alcohol Dehydrogenase ------> Increased NADH/NAD+ ratio
------> Increased levels of Lacttate in the blood
- INTERFERENCE WITH LIVER DETOXIFICATION: Lots of alcohol can interfere
with the conjugation of bilirubin in the liver to bilirubin diglucuronide.
- EtOH ------> Lower levels of Direct Bilirubin, which is conjugated water-soluble bilirubin in the blood.
- EtOH ------> Higher levels of Indirect Bilirubin, which is unconjugated
bilirubin stuck to albumin.
- This interference with detoxification leads to fibrotic liver with fatty deposits.
You will also see marked proliferation of smooth-ER prior to this.
ETHANOL AND OTHER DRUGS:
- ACUTE Consumption: Ethanol may increase the effects of other drugs by interfering with
their metabolism in the liver, thereby increasing their effects.
- Barbiturates have a dangerous effect with alcohol because their half-life is
increased in the presence of alcohol.
- The synergistic effect of both EtOH + Barbiturates together is to induce sleep.
- CHRONIC alcohol effect may actually decrease the effects of other drugs.
- Can be seen in acetaminophen toxicity which can lead to hepatic necrosis.
Return to top
PROTEOLYTIC DIGESTION and ABSORPTION OF PROTEINS
PEPSIN: Proteolytic digestive enzyme.
- Pepsinogen is converted to Pepsin by the action of acid (H+).
- The acid is secreted by parietal cells in the intestinal wall. The parietal cells secrete acid
whenever food enters the stomach from the esophagus.
- Auto-Catalytic: Active pepsin acts on pepsinogen to create more of itself.
- CLEAVAGE-SPECIFICITY:
- Pepsin is specific to non-polar amino acids that are adjacent to Phenylalanine,
Tyrosine, or Leucine.
- It cleaves at the carboxyl group adjacent to those residues.
- Pepsin function at an acidic pH -- it uses an interesting catalytic mechanism of interacting
carboxyl groups. The active site is only operational at a pH around 2.
THE ENDOPEPTIDASES: Peptidases that cleave peptides within
- It is a Serine-Protease, i.e. it uses Serine as its active site to cleave proteins.
- Trypsin is activated by ENTEROKINASE, which is secreted in the intestinal
mucosa.
- It converts Trypsinogen ------> Trypsin
- CLEAVAGE-SPECIFICITY:
- Trypsin cuts amino acids that are adjacent to Lysine and Arginine.
- Auto-Catalytic: Activated trypsin acts on trypsinogen to make more of itself.
- Trypsin also acts on Chymotrypsinogen to make Chymotrypsin!
- Chymotrypsinogen is activated by Trypsin.
- CLEAVAGE-SPECIFICITY: It cleaves aromatic and non-polar side-chains. It is
not as specific in its cleavage site as Trypsin. It will cleave any of the following
residues: Trp, Phe, Tyr, Met, Leu
- Elastase is activated by Trypsin, too.
- It converts Proelastase ------> Elastase
- CLEAVAGE-SPECIFICITY: It cleaves residues adjacent to Alanine, Glycine,
and Serine residues.
THE EXOPEPTIDASES: Cut the very last residue off a protein. Generally work on smaller
polypeptides after they have been broken down a bit by the endopeptidases.
- CARBOXYPEPTIDASE: These are exopeptidases that cleave at the carboxy-end.
- They are both activated by Trypsin just like above: Procarboxypeptidase
------> Carboxypeptidase
- They are both metalloproteases which require Zinc for catalysis. This is a
different mechanism than the endopeptidases which are serine proteases.
- These guys are secreted in the pancreas.
- Carboxypeptidase-A: Cleaves neutral and acidic side-chains (on the carboxy
end), such as Alanine, Valine, Isoleucine, Leucine.
- Carboxypeptidase-B: Cleaves basic residues -- Lysine, Arginine
- Every time Trypsin cuts a protein, you are left with a protein piece that has either
Lys or Arg at the carboxy-end! Carboxypeptidase can then take over to remove
that end-piece.
- This in effect gives us a free amino acid of Lysine or Arginine which can then be
absorbed.
- AMINOPEPTIDASE: Exopeptidases that cut at the amino end of a peptide.
- This is secreted by the intestinal mucosa further along the small intestine
(jejunum).
- This is a metalloprotease that requires Zinc and Manganese for catalysis.
- This cuts, usually on smaller peptides, one acid at a time off the amino end.
- DIPEPTIDASE: An Aminopeptidase, similar to above, which cuts two acids at a time
from the amino end of a peptide.
- At this point, the peptides are small enough (two or three residues max) that they can be
absorbed by small intestine and go, through portal circulation, to liver.
SECRETAGOGUES: They stimulate the pancreas to release pancreatic enzymes. They have
specific receptors on the pancreatic acinar (exocrine) cells.
- CHOLECYSTOKININ: Stimulates the alpha-Adrenergic Phosphatidylinositol
intracellular pathway in Pancreatic acinar cells.
- Acetylcholine has the same effect on these cells, through the same pathway, via
neural stimulation.
- Has a sulfonated tyrosine side-chain.
- SECRETIN: Stimulates the beta-Adrenergic Adenylate Cyclase (cAMP) intracellular
pathway
- Between the two above, there is dual stimulation on the pancreatic acinar cells to cause
them to secrete pancreatic enzyme.
- GASTRIN: Has a sulfonated tyrosine side-chain.
OVERALL ACTIVATION-SCHEME OF INTESTINAL PEPTIDASES:
- Food enters stomach, activating parietal cells to secrete HCl
- HCl (i.e. low pH) causes the following:
- Pepsinogen ------> Pepsin, which is also auto-catalytic.
- Gastrin is released
- Histamine is released
- When food reaches small intestine, small intestine releases two secretagogues:
- The Secretagogues cause the following:
- Pancreatic secretion of Trypsinogen, Chymotrypsinogen, and Proelastase.
- Intestinal epithelial secretion of Enteropeptidase
- Enteropeptidase causes Trypsinogen ------> Trypsin, which is also auto-catalytic.
- Trypsin causes:
- Chymotrypsinogen ------> Chymotrypsin
- Proelastase ------> Elastase
- Procarboxypeptidases ------> Carboxypeptidases
SYNTHESIS OF ACTIVE SULFATES: Sulfonation is the process of adding a sulfate to a
substance, such as a hormone or drug. In the case of drugs, it makes more soluble so it can be
excreted in the urine. First, the sulfate group must be activated.
- ATP-Sulfurase: SO3-2 + ATP ------> Adenosine-5-Phosphosulfate (AMPS)
- This reaction releases pyrophosphate, and a pyrophosphatase is used to cleave the
high energy and drive the reaction right.
- AMPS-Phosphokinase: Adenosine-5-Phosphosulfate ------> 3-Phosphoadenosine-5-Phosphosulfate (PAPS)
- PAPS is then the active sulfate, which can be transferred to hydroxyl groups to exert
various chemical effects.
- TOTAL ENERGY REQUIRED: 3 ATP
- 2 ATP equivalent from release and cleavage of pyrophosphate in step 1
- 1 ATP required in step two.
PROTEIN TURNOVER: Degrading proteins in the cell and then rebuilding them.
- Cathepsins: A class of acidic LYSOSOMAL degradative enzymes that function
optimally at acidic pH.
- Lysosomes normally act on proteins that are taken up by receptor mediated
endocytosis.
- Calpains: A class of neutral NON-LYSOSOMAL (CYTOSOLIC) degradative enzymes
that function optimally at physiological pH.
- Many of them are metalloproteases, metal-requiring.
- Ubiquitin: This tagging-ligand is often added to the proteins that are destined
(doomed) for degradation by calpains.
- Proteins must be tagged for degradation, so that the Calpains don't attack
just any old protein in the cytosol!
- Attachment of Ubiquitin requires ATP.
- Ubiquitin almost invariably attaches to Lysine residues, but it is not clear
how it knows which proteins to tag on to or what role the Lys residues
might play.
- Proteosome Complex: The ubiquitin-tagged proteins are broken down in very
large proteosome complexes (presumably these are the Calpains).
PROTEIN SYMPORT: Facilitated transport of amino acids is one of two ways that amino acids
can get into cells. The other way is via glutathione.
- Amino Acid comes in as a symport with Na+, with Na+ flowing with its concentration
gradient. So, this transport itself does not require energy.
- Na+ gradient is usually created by a Na+/K+ ATPase pump, which does require energy.
Every ATP yields 3Na+ pumped and 2K+ pumped in.
- The above creates a net negative charge inside the cell, causing Na+ to come back
in by facilitated diffusion.
- There are multiple types of these symport-mechanisms. Different ports were discovered
by protein-saturation experiments and by their deficiency in disease states.
- General Port: Transports neutral amino acids and Citrulline
- Dibasic Port: Transports basic amino acids
- Dicarboxylic Port: Transports acidic amino acids
- Glycine / Imino Port: Transports Gly, Pro, Hydroxyproline
GLUTATHIONE: gamma-Glutamylcysteinylglycine. It is a tripeptide that aids in protein
transport and is essential to RBC-membrane integrity.
- Structure and Prevalence: Glutathione is a tripeptide of Glu-Cys-Gly
- Oxidation States: Glutathione comes in reduced and oxidized states.
- Reduced Form: Abbreviated GSH, to indicate the thiol group on cysteine.
- Oxidized Form: It dimerizes to GS-SG to indicate disulfide bond.
- The reduced form is dominant in the cell, about 100-500x more prevalent than
oxidized form.
- Glutathione Reductase: GS-SG ------> GSH. This reaction requires
NADPH.
- Overall prevalence of Glutathione in cell: 5mM. Very prevalent!
- Cellular Roles of Glutathione:
- Amino Acid Transport (see below)
- Peroxide detoxification
- Methemoglobin Reduction: Glutathione keeps iron in its ferrous (Fe+2) state.
- The ferrous state is necessary for hemoglobin to bind to oxygen.
- Oxidizing agents (as in aging) can cause oxidation of iron to ferric
oxidation state.
- Protein Folding: It aids by providing a means for exchanging disulfide bonds
during protein folding.
- Prostaglandin and Leukotriene Synthesis
- GLUTATHIONE-TRANSPORT OF AMINO ACIDS:
- OUTSIDE CELL: A free amino acid is "glutamylated" from glutathione, so that it
can go through a specific receptor to get through membrane.
- Free Amino Acid + Glutathione ------> gamma-Glutamyl Amino
Acid + Cysteinyl-Glycine
- TRANSPORT:
- The gamma-Glutamyl amino acid goes through a glutamine-specific
transport protein to get through the membrane.
- The Cysteinyl-Glycine gets through the membrane
- INSIDE CELL: The acid is freed, and glutathione is resynthesized.
- gamma-Glutamyl Amino Acid ------> Free Amino Acid + 5-Oxoproline
- 5-Oxoproline ------> Glutamic Acid
- This is the opening of a ring to form free Glutamate
- ATP is required.
- Cysteinyl-Glycine ------> Cysteine + Glycine: Cys and Gly are split
apart so that can be used in the resynthesis of glutathione.
- Cysteine + Glycine + Glutamate ------> Glutathione
- 2 ATP are required, to synthesize the two peptide bonds.
- OVERALL ENERGETICS: 3 ATP are required per amino acid transported via
Glutathione.
- 1 ATP for opening of the 5-Oxoproline ring
- 2 ATP for forming the peptide bonds to resynthesize glutathione.
Return to top
TRANSAMINATION AND GLUTAMATE DEHYDROGENASE
PYRIDOXAL PHOSPHATE: Pyridoxal phosphate acts as the intermediate in a number of
reactions, by forming schiff bases, which involves the condensation (i.e. loss of water) of an
aldehyde function with an amino group, to yield a C=NR structure.
- Transamination
- Decarboxylation of histidine
- Aldol Cleavage of Fructose Biphosphate (aldolase)
- Elimination of beta-carbons in Ser and Thr Dehydratases
OXIDATIVE DEAMINATION via GLUTAMATE DEHYDROGENASE CYCLE: A way to
take the amino group from an amino acid and transfer it to another carbon skeleton, in order to
(1) get rid of the amino group, or (2) create a different amino acid.
- Amino Acid + alpha-Ketoglutarate ------> Keto Acid + Glutamate
- This is catalyzed by Aminotransferase. It is simply the "swapping" of an amino
group for a keto group, between many different amino acids and alpha-Ketoglutarate:
- Glutamate serves as a carrier of nitrogen of here. Hence alpha-Ketoglutarate is
often used for this aminotransferase reaction.
- This reaction is reversible

Pyridoxyl Phosphate is required as an intermediate. Transamination is achieved
via Schiff base.
- Glutamate ------> alpha-Ketoglutarate + NH3
- NAD+ is required. This reaction is an oxidation; NAD+ ------> NADH is
concurrent reduction. The NADH can then be used in electron transport.
- This reaction is catalyzed by Glutamate Dehydrogenase.
- The free ammonia will then must be taken up by some other reaction, such as urea
synthesis.
- alpha-Ketoglutarate is most commonly used in aminotransferase reactions, because
Glutamate Dehydrogenase is available to regenerate the reactant and make it a
cycle. However, other keto acids may also be used.
- OVERALL CYCLE: Two cofactors are required: Pyridoxal Phosphate and NAD+ (but
no ATP!) For every turn of the cycle, we have
- Aminotransferase:
- alpha-amino acid ------> alpha-keto acid
- alpha-Ketoglutarate ------> Glutamate
- Glutamate Dehydrogenase:
- Glutamate ------> alpha-ketoglutarate
- NADH produced
- NH3 produced
- Clinical Significance: Historically, aminotransferase levels were measured in the blood to
diagnose myocardial infarction.
- REVERSIBLE: Glutamate Dehydrogenase is completely reversible.
- Because it is reversible, you can transaminate free ammonia to other carbon
skeletons to make other amino acids.
- Although the reverse reaction uses free ammonia, it is not an important source of
ammonia disposal.
NON-OXIDATIVE DEAMINATION: SERINE AND THREONINE can be deaminated non-oxidatively (i.e. no NAD+ is required).
- Enzyme = Serine-Threonine Dehydratase.
- Serine ------> Pyruvate + NH4+
- Threonine ------> alpha-Ketobutyrate + NH4+
- Cofactor: Pyridoxal Phosphate is required for these reactions, too, and Schiff-Base
formation is involved.
OTHER OXIDATIVE DEAMINATION PATHWAYS:
- L-Amino Acid Oxidase catalyzes Amino Acid ------> Keto Acid
- Concurrent reduction: FMN ------> FMNH2
- FMN is regenerated from FMNH2 using O2, which forms a peroxide.
- The peroxide can then be reduced down to water with catalase (in peroxisomes).
- D-Amino Acid Oxidase only works on D-acids. So in humans it only works achiral
Glycine. It is suspected that these oxidases exist as a defense mechanism against bacteria.

AMMONIA TRANSPORT VIA GLUTAMATES: NH3 can be stored in glutamate or glutamine
for non-toxic storage and transport. Free ammonia is not tolerated in very high concentrations.
- A way of carrying NH3 on a glutamine residue:
- Glutamine Synthetase: Glutamate + NH3 ------> Glutamine
- Glutaminase: Glutamine ------> Glutamate + NH3
- A way of carrying NH3 on a glutamate residue:
- Aminotransferase: alpha-Ketoglutarate + NH3 ------> Glutamate
- Glutamate Dehydrogenase: Glutamate ------> alpha-Ketoglutarate + NH3
- Between the two above, we can carry a total of two NH3's on a glutamine residue.
Return to top
THE UREA CYCLE
SYNTHESIS OF CARBAMOYL PHOSPHATE: This is how we "activate" a free ammonia
before we subsequently make urea.

- Enzyme = Carbamoyl Phosphate Synthetase.
- This reaction occurs in the mitochondrion.
- 2 ATP are required. Basically these are used to "charge" or "activate" ammonia with a
high-energy phosphate bond, before we subsequently start urea synthesis.
- N-Acetylglutamate is absolutely required as a cofactor. This compound also serves a
regulatory role in urea synthesis.
- The rate of carbamoyl phosphate synthesis is dependent on the levels of N-Acetylglutamate in the mitochondria (I'm not sure whether this is a linear
relationship).
- Usually, the free ammonia is derived directly from Glutamate Dehydrogenase, but it could
come from anywhere.
THE UREA CYCLE: 
- REQUIRED STARTING MATERIALS:
- Carbamoyl Phosphate: It donates a free NH3 to urea, per turn of the cycle.
- Ornithine: It is regenerated each turn of the cycle
- Aspartic Acid: It donates an NH3 to urea from aminotransferases, per turn of the
cycle.
- Aspartic Acid has the same carbon skeleton as Oxaloacetate:

- 4 ATP are required per molecule of urea
- 2ATP required to synthesize Carbamoyl Phosphate
- 2ATP required to synthesize Arginosuccinate, via 1 ATP and a
pyrophosphatase
- Carbamoyl Phosphate + Ornithine ------> Citrulline
- This is THE COMMITTED STEP
- One of the two NH3's of urea comes to us from Carbamoyl Phosphate, essentially
the same as starting with a charged free NH3.
- This step occurs in the mitochondria. Citrulline is then transferred from
mitochondria to the cytosol for the rest of the steps.
- Enzyme: Ornithine Transcarbamoylase
- Citrulline + Aspartic Acid ------> Arginosuccinate
- ATP ------> AMP + PPi. Metabolic equivalent of 2ATP required, with
pyrophosphatase driving the reaction forward, making the reaction irreversible.
- Aspartic Acid carries with it the other NH3 of urea. This NH3 has come to us via
Aspartic Acid, courtesy of Aspartate Aminotransferase.
- This step, and all subsequent steps, occurs in the cytosol.
- Arginosuccinate ------> Arginine + Fumarate
- Catalyzed by Arginosuccinase
- Fumarate then goes through the TCA Cycle, in order to regenerate the carbon
skeleton of Aspartic Acid:
- Fumarase: Fumarate ------> L-Malate
- L-Malate Dehydrogenase: L-Malate ------> Oxaloacetate
- Aspartate Aminotransferase: Oxaloacetate + NH3 ------> Aspartic
Acid
- Arginine ------> Urea + Ornithine
- Catalyzed by Arginase. Know this enzyme, because this is the step that
regenerates ornithine.
- END PRODUCTS:
- Fumarate, which is recycled, via the TCA cycle and transamination, to Aspartate
- Urea
- Ornithine, which is recycled to the first step.
REGULATION OF THE UREA CYCLE:
- Short-Term Regulation (metabolic regulation):
- The primary regulated step is Carbamoyl Phosphate Synthetase.
- Increased levels of Arginine promotes the formation of N-Acetyl Glutamate,
which is required for carbamoyl phosphate synthesis.
- Glutamate + Acetyl-CoA ------> N-Acetylglutamate
- So when Arginine levels goes up, carbamoyl phosphate is made and urea synthesis
occurs.
- Long-Term Regulation (diet)
- In the long-term, a high-protein diet will increase overall levels of urea synthesis.
- People who are protein derived would be ill-equipped to handle a sudden massive
increase in the amount of protein in their diet, because enzyme levels would not be
high enough. That's why you change a diet gradually as a rule of thumb, especially
for the malnourished.
AMMONIA DETOXIFICATION: Ways of getting rid of excess ammonia in the bloodstream.
- From Benzoic Acid: HIPPURATE
- Benzoic Acid + Coenzyme-A ------> Benzoyl-CoA
- 2 ATP are required (coupled with pyrophosphatase), as usual, to make a
CoA derivative.
- Benzoyl-CoA + Glycine ------> Hippurate
- Amino groups are fed in at this step, in the form of Glycine.
- Hippurate is innocuous and is readily excreted in the urine.
- This form of detoxification will only work if we can transaminate an NH3 group
onto glyoxalase (the keto-acid form of glycine). If we can't get the NH3 into
glycine, then we can't get rid of it by this means!
- From Phenylacetate: PHENYLACETYLGLUTAMINE
- Phenylacetate + Glutamine ------> Phenylacetylglutamine
- Coenzyme-A is required as a cofactor, but there is no free Coenzyme-A
intermediate. In other words, it is a concerted reaction.
- 2 ATP are required (coupled with pyrophosphatase), as usual.
- Phenylacetylglutamine is readily excreted in the urine.
CLINICAL CASE-STUDY: HYPERAMMONEMIA
- The patient had a deficiency in Ornithine Transcarbamoylase.
- Blood-levels of NH3 were almost twice normal.
- The enzyme was not completely absent, however. If you gave the patient protein,
it would take him 120 hours to completely metabolize the protein to urea, which is
far longer than normal.
- CLINICAL PRESENTATION: Bizarre behavior: crying, agitation, babbling, loss of
sense of reality.
- Lab results suggest a deficiency in ornithine transcarbamoylase:
- High levels of Alanine, Glutamine, and Orotic Acid (a metabolite of purine
synthesis)
- NH3 of course was way high
- The Carbamoyl Phosphate builds up and must be shunted off to another pathway.
- TREATMENT:
- A high carbohydrate, low protein diet (but not no protein)
- Treat with benzoic acid or phenylacetate for ammonia detoxification.
Return to top
SYNTHESIS AND DEGRADATION OF AMINO ACIDS
GLUCOGENIC: Amino acids whose degradation results in pyruvate or one of the intermediates
of the TCA cycle. That means that those carbons can be used to make glucose via
gluconeogenesis, under the right metabolic circumstances.
KETOGENIC: Amino acids whose degradation results only in Acetyl-CoA or Acetoacetyl-CoA.
These carbon-skeletons can only be funneled into fat-metabolism: aerobic respiration of acetyl-CoA, or fat-synthesis.
ESSENTIAL: The amino acid is required in the diet. We can't synthesize it de novo.
NON-ESSENTIAL: The amino acid can be synthesized de novo from other amino acids or from
metabolic precursors.
SAME CARBON SKELETONS: Each of the below have the same carbon skeletons:
- Glutamate, Glutamine, alpha-Ketoglutarate
- Aspartate, Asparagine, Oxaloacetate
- Alanine, Pyruvate
PVT TIM HALL: A mnemonic for the essential amino acids. Phe, Val, Thr // Trp, Ile, Met //
His*; Arg*; Lue; Lys
| Amino Acid |
Synthesis |
Degradation |
| Leucine |
ESSENTIAL |
KETOGENIC: Leucine ------>
Acetyl-CoA + Acetoacetate
(branched) |
| Lysine |
ESSENTIAL |
KETOGENIC |
| Phenylalanine |
ESSENTIAL -- and it's an essential
precursor to Tyrosine |
GLUCOGENIC + KETOGENIC |
| Tyrosine |
NON-ESSENTIAL: Phenylalanine
------> Tyrosine (Phenylalanine
Hydroxylase) |
GLUCOGENIC + KETOGENIC |
| Tryptophan |
ESSENTIAL |
GLUCOGENIC + KETOGENIC |
| Isoleucine |
ESSENTIAL |
GLUCOGENIC + KETOGENIC:
Isoleucine ------> Acetyl-CoA +
Propionyl CoA ------> Succinyl-CoA (branched) |
| Threonine |
ESSENTIAL |
GLUCOGENIC + KETOGENIC:
Threonine ------> Propionyl-CoA
------> Succinyl-CoA (although it
isn't branched) (Propionyl-CoA
Carboxylase) |
| Aspartate |
NON-ESSENTIAL: Oxaloacetate
------> Aspartate
(aminotransferase) |
GLUCOGENIC: Aspartate ------>
Oxaloacetate (aminotransferase) |
| Glutamate |
NON-ESSENTIAL: alpha-Ketoglutarate ------> Glutamate
(aminotransferase) |
NON-ESSENTIAL: Glutamate
------> alpha-Ketoglutarate
(aminotransferase) |
| Asparagine |
NON-ESSENTIAL: Aspartate
------> Asparagine
(Aminotransferase) |
GLUCOGENIC: Asparagine
------> Aspartate ------>
Oxaloacetate (Asparaginase,
Aminotransferase) |
| Glutamine |
NON-ESSENTIAL: Glutamate
------> Glutamine (Glutamine
Synthetase) |
GLUCOGENIC: Glutamine
------> Glutamate ------> alpha-Ketogglutarate (Glutaminase,
Glutamate Dehydrogenase) |
| Histidine |
NON-ESSENTIAL -- but it is
required in infancy and during
recovery from disease; Glutamate
------> 5-aminoimidazole-4-carboxamide -------> Histidine
(bacterial) |
GLUCOGENIC: Histidine ------>
Glutamate ------> alpha-Ketoglutarate (bacterial, Glutamate
Dehydrogenase) |
| Proline |
NON-ESSENTIAL: Glutamate
------> Proline (reduce the gamma-carboxxyl group of Glutamate) |
GLUCOGENIC: Proline ------>
Glutamate ------> alpha-Ketoglutarate (open ring, oxidize,
Glutamate Dehydrogenase) |
| Arginine |
NON-ESSENTIAL -- but it is
required in infancy and during
recovery from disease; Glutamate
------> Ornithine ------> Arginine
(second step via Arginase) |
GLUCOGENIC: Arginine ------>
Ornithine ------> Glutamate
------> alpha-Ketoglutarate
(Arginase, Glutamate
Dehydrogenase) |
| Glycine |
NON-ESSENTIAL: Serine ------>
Glycine (demethylation with
tetrahydrofolate as cofactor) |
GLUCOGENIC: Glycine ------>
Pyruvate |
| Alanine |
NON-ESSENTIAL: Pyruvate
------> Alanine (aminotransferase) |
GLUCOGENIC: Alanine ------>
Pyruvate (aminotransferase) |
| Serine |
NON-ESSENTIAL: 3-Phosphoglycerate ------> Serine |
GLUCOGENIC: Serine ------>
Pyruvate |
| Cysteine |
NON-ESSENTIAL: Methionine
------> Homocysteine + Serine
------> Cysteine |
GLUCOGENIC |
| Methionine |
ESSENTIAL -- and, it's an essential
precursor to Cysteine |
GLUCOGENIC: Methionine
------> Propionyl-CoA ------>
Succinyl-CoA (although it isn't
branched) (Propionyl-CoA
Carboxylase) |
| Valine |
ESSENTIAL |
GLUCOGENIC: Valine ------>
Propionyl-CoA ------> Succinyl-CoA (branched) (Propionyl-CoA
Carboxylase) |
ASPARAGINASE: The breakdown of asparagine to aspartate. It can be used as a treatment for
Leukemia, by giving Asparaginase and thereby starving metastatic cells of their need for
Asparagine.
PRODUCTS OF AMINO ACID DEGRADATION: All amino acids are ultimately broken down
to one of the following, as described above. Know this list:
- PYRUVATE
- ACETOACETYL-CoA
- ACETYL-CoA
- alpha-KETOGLUTARATE
- SUCCINYL-CoA
- FUMARATE
- OXALOACETATE
DEGRADATION OF BRANCHED AMINO ACIDS: Valine, Isoleucine, Leucine. These three
acids are degraded by parallel mechanisms. All of the below applies to all three.
- Branched Amino Acid ------> alpha-Keto Acid + NH3
- This is called a transamination and is catalyzed by aminotransferases
- The NH3 will then form Carbamoyl phosphate and go onto urea synthesis or purine
synthesis.
- alpha-Keto Acid ------> Acyl-CoA
- This is an Oxidative Decarboxylation very similar to pyruvate dehydrogenase
reaction.
- It uses the same cofactors as PDH-Dehydrogenase and Succinate
Dehydrogenase: Thiamine Pyrophosphate (TPP), Lipoic Acid, NAD+, FAD,
Coenzyme-A are all required.
- CO2 is given off; Coenzyme-A is put on; NAD+ ------> NADH is ultimately the
concurrent reduction.
- Acyl-CoA ------> ------> End Products (Propionyl-CoA, Acetyl-CoA, Acetoacetyl-CoA). These are done by dehydrogenation reactions, similar to beta-oxidation. Specific
products are as follows:
- Valine ------> ------> Propionyl-CoA
- Isoleucine ------> ------> Acetyl-CoA + Propionyl-CoA
- Leucine ------> ------> Acetyl-CoA + Acetoacetyl-CoA
METABOLISM OF PROPIONATE: In the case of Valine and Isoleucine, we need to further
convert Propionyl-CoA to Succinyl-CoA by adding a carbon.
- Propionyl-CoA + HCO3- ------> D-Methylmalonyl-CoA + L-Methylmalonyl-CoA
- Catalyzed by Propionyl-CoA Carboxylase
- It requires biotin as a cofactor.
- 2ATP (coupled with pyrophosphatase) is required, too.
- The reaction produces the racemic mixture of a chiral compound. The products
are driven toward D-product by the action of the next step.
- D-Methylmalonyl-CoA ------> Succinyl-CoA
- Catalyzed by Methylmalonyl-CoA Mutase
- It requires Cobalamin (Vit B12) as a cofactor.
- This is an isomerization reaction: the carboxyl group is rearranged and put on the
end of the molecule.
- Because of this product, Propionyl-CoA can lead to glucose via gluconeogenesis. So, not
all fatty acids are exclusive of glucose. Propionyl-CoA is the exception.
INBORN ERRORS OF AMINO ACID METABOLISM: Problems of nitrogen metabolism can
be caused by a lot of things. Generally nitrogen-metabolism diseases involve mental retardation
and other neurological problems. Tyrosine deficiency (a consequence of PKU) can lead to
albinism.
THE METHYLATION CYCLE: Cysteine is synthesized from Methionine and Serine, via S-Adenosyl Methionine. The methylation cycle consists of methylating and demethylating
methionine repeatedly.
- REQUIRED STARTING REACTANTS:
- Methionine, which is recycled, each turn of the cycle
- 1 ATP, to generate S-Adenosylmethionine, each turn of the cycle.
- Tetrahydrofolate, which donates a methyl group to homocysteine, each turn of the
cycle.
- Vitamin-B12, which is required to resynthesize methionine.
- Methionine ------> S-Adenosyl Methionine
- This is an adenylation reaction. ATP is required -- it is cleaved all the way down
to the Adenosine base (ribose + adenine), releasing both Pi and PPi. Thus the
reaction is very driven.
- The adenosine base is put on Methionine, and the phosphates are not. The
pyrophosphate is cleaved by a pyrophosphatase to drive the reaction.
- S-Adenosyl Methionine ------> S-Adenosyl Homocysteine + -CH3
- S-Adenosyl Methionine has a formal positive charge.
- It is a strong methylating agent. Methylation restores the neutral charge on the
molecule.
- The methyl group may be donated to any of a number of acceptor-molecules in
this step.
- S-Adenosyl Homocysteine ------> Homocysteine + Adenosine
- This is a hydrolysis reaction. The adenosine is recycled through purine
metabolism.
- Homocysteine + -CH3 ------> Methionine
- Homocysteine here is used to regenerate Methionine, making the methylation
routine a cycle.
- This reaction requires Cobalamin (Vit B12)
- The single carbon fragment is coming from N5-Methyl Tetrahydrofolate. Thus
another essential cofactor is folic acid.
- SYNTHESIS OF CYSTEINE: Instead of regenerating methionine, homocysteine can
also do the following:
- Homocysteine + Serine ------> Cystathionine
- Cystathionine is a thioether (C-S-C linkage)
- Cystathionine ------> Cysteine + alpha-Ketobutyrate
- Cysteine is formed simply by transferring the sulfur to the carbon skeleton
of Serine.
- So, cysteine is formed from both Serine and Methionine.
SYNTHESIS OF TYROSINE: Phenylalanine ------> Tyrosine
- Enzyme = Phenylalanine Hydroxylase.
- O2 is required. This enzyme is a Mixed-function Oxidase, utilizing a mini-electron
transport chain and redox cofactors.
- Tetrahydrobiopterin is a required cofactor.
PHENYLKETONURIA (PKU): Deficiency of Phenylalanine Hydroxylase
- Phenylalanine Hydroxylase is required for the normal breakdown of Phenylalanine, as
well as for Tyrosine synthesis.
- The following build up in the absence of functional Phenylalanine Hydroxylase:
- Phenylalanine
- Phenylpyruvate, aminotransferase product of Phenylalanine, which isn't normally
made.
- Phenyl lactate, reduced phenylpyruvate, which isn't normally made.
- Phenylacetate, oxidized phenylpyruvate, which isn't normally made.
- Clinical Symptoms: Mental Retardation, possible albinism (from tyrosine deficiency).
- Differential Diagnoses:
- Excretion of phenyl ketones in the urine
- High levels of phenylalanine in the blood
- Treatment: PKU screening is done early in life, because the disorder is quite treatable
with diet.
- Diet very low (but not absent) in phenylalanine. Critical period to prevent
retardation is infancy.
- Tetrahydrobiopterin: Up to 3% may have a defect in this cofactor instead of
Phenylalanine Hydroxylase.
- This leads to a more serious condition, because tetrahydrobiopterin is also an
essential cofactor for production of DOPA and the Catecholamines, which are
both also phenylalanine derivatives.
- Supplemental Treatment for Tetrahydrobiopterin-based PKU includes therapeutic
doses of DOPA and hydroxytryptophan.
- L-DOPA is also used to treat people with Parkinson's Disease.
- GARROD'S HYPOTHESIS: It states that "all inherited diseases result from defects
found in a single gene," as opposed to multifactorial etiology of diseases.
- This somewhat holds true for phenylalanine, in that most cases are due to a single
deficiency. Now we know it is not completely accurate for PKU.
- PKU is a recessive inherited disorder.
FOLIC ACID and TETRAHYDROFOLATE:
- Folic Acid is an essential vitamin in the diet.
- Structure: para-aminobenzoic acid base, with multiple glutamates connected to
the carboxyl via peptide bonds.
- Thus Folic Acid is a polyglutamic acid conjugate
- Conjugase in our intestine converts Folic Acid Polyglutamate ------> Folic
Acid Monoglutamate, so that we can absorb it.
- SULFA DRUGS: The old class of antibiotics. They mimic and block para-aminobenzoic acid (PABA), which is required for folic acid synthesis in bacteria,
thus killing the bacteria.
- SYNTHESIS OF TETRAHYDROFOLATE:
- Folic Acid Monoglutamate ------> Dihydrofolic Acid ------>
Tetrahydrofolic Acid
- Two NADPH ------> NADP+ are required. Hence this is a standard
reductive biosynthetic reaction.
- Enzyme = Dihydrofolate Reductase.
- CHEMOTHERAPY: Dihydrofolate Reductase is essential to maintain levels of
Tetrahydrofolate (THF)
- Methotrexate is an analog of dihydrofolate and a great inhibitor of
dihydrofolate reductase. Because THF is required for purine synthesis,
methotrexate stops proliferation of metastatic cells.
- Sulfanilamide (Sulfa drugs) is another drug that acts in the same
way.
- Obvious side effects are that it will stop THF synthesis in other cells, too,
preventing division in intestinal epithelia, skin, and hair, e.g.
- In high doses, methotrexate kills bone marrow.
- UTILIZATION OF TETRAHYDROFOLATE: Once in the fully reduced form, THF is
utilized in various forms as a methylating agent:
- Tetrahydrofolate + -CH3 ------> N5-N10-methylene tetrahydrofolate
- The CH3 in this reaction comes from Serine. Concurrent demethylation
therefore is Serine ------> Glycine
- This (Serine) is the most common source of methyl groups for the
tetrahydrofolate cofactor.
- Methylene-Tetrahydrofolates exist in several different forms having different
oxidation states. Thus THF can donate a methyl group, as it does in purine
synthesis, in several different oxidation states.
- N5-Methyl Tetrahydrofolate: This is the form that donates a methyl group to
the reaction: Homocysteine + CH3 ------> Methionine
- Again, Vit-B12 is required for this reaction.
- METHYL-TRAP HYPOTHESIS explains a Vitamin-B12 deficiency,
saying that methyl groups get "trapped" in the form of N5-Methyl
Tetrahydrofolate because VitB12 is deficient.
- Result of this = THF can't get to other essential forms for lack of
methyl groups, and purine synthesis is impeded as a result.
Return to top
PURINE (ADENINE, GUANINE) METABOLISM
SYNTHESIS OF PURINES: The basic purine is Inosinate (IMP), from which AMP and GMP
are made.
- SYNTHESIS OF THE RIBOSE SUGAR and N-LINKAGE IN PURINES
- Ribose-5-Phosphate + Glutamine ------> 5-Phosphoribosyl-1-Pyrophosphate (PPRP) + Glutamate
- Catalyzed by PPRP-Synthetase, or Ribose-5-Phosphate
Pyrophosphokinase
- Ribose-5-Phosphate came from the Pentose Phosphate Pathway, of course.
- ATP is required, yielding pyrophosphate which is cleaved by
pyrophosphatase. Thus the reaction is irreversible.
- PPRP is also one of the starting points of pyrimidine biosynthesis.
- PPRP ------> 5-Phosphoribosyl-1-Amine
- Enzyme = Amidophosphoribosyl Transferase
- This product has a nitrogen attached to the C-1 Carbon of the ribose. Thus
this product already has the ribose nitrogen-linkage intact. This is the
same linkage that will remain in the final purine structure.
- This is the COMMITTED STEP in the Purine Biosynthesis.
- SYNTHESIS OF THE SMALLER OF THE TWO PURINE RINGS
- 5-Phosphoribosyl-1-Amine ------> Glycinamide Ribonucleotide
- Addition of Glycine, which donates 2 carbons and 1 nitrogen to the
ring
- Glycinamide Ribonucleotide ------> Formylglycinamide Ribonucleotide
- Addition of a single carbon from 10-formyl-tetrahydrofolate.
- So, this carbon comes on as a highly oxidized formate group (single
carboxylic acid), from one of the oxidation states of THF.
- Formylglycinamide Ribonucleotide ------> Formylglycinimadine Ribonucleotide
- Add a lone nitrogen (I presume it comes from free ammonia)
- Formylglycinimadine Ribonucleotide ------> 5-Aminoimidazole Ribonucleotide
- Cyclization to form the imidazole ring (the smaller of the two purine
rings)
- Condensation reaction occurs between the Formate of Step-b and the lone
nitrogen of step-c above.
- SYNTHESIS OF THE LARGER OF THE TWO PURINE RINGS
- 5-Aminoimidazole Ribonucleotide ------> 5-Aminoimidazole-4-Carboxylate
Ribonucleotide
- Addition of a carbon from HCO3-
- 5-Aminoimidazole-4-Carboxylate Ribonucleotide ------> 5-Aminoimidazole-4-N-Succinocarboxamide Ribonucleotide
- Addition of Aspartic Acid
- 5-Aminoimidazole-4-N-Succinocarboxamide Ribonucleotide ------> 5-Aminoimidazole-4-Carboxylate Ribonucleotide
- Removal of the carbon skeleton of Aspartate (i.e. fumarate), leaving
only the alpha-amino Nitrogen
- Aspartate only donates 1 nitrogen to the purine ring -- not its entire
structure.
- 5-Aminoimidazole-4-Carboxylate Ribonucleotide ------> 5-Formamidoimidazole-4-Carboxamide Ribonucleotide
- Addition of a single carbon, from 10-formyl-tetrahydrofolate. Again
the carbon is added in the form of a formyl group.
- 5-Formamidoimidazole-4-Carboxamide Ribonucleotide ------> Inosinate
Monophosphate (IMP)
- Closure of the second (larger) purine ring
- Condensation reaction occurs between the Nitrogen from the Aspartate,
and the single carbon that just came from THF.
SYNTHESIS OF AMP: Replace the carbonyl on the larger purine ring with an amine group.
- Inosinate (IMP) + Aspartic Acid ------> Adenylosuccinate
- The entire Asp residue is put onto the carbonyl of the larger of the two purine
rings.
- Adenylosuccinate ------> Adenylate (AMP) + Fumarate
- Aspartic acid leaves again as fumarate.
- So, again aspartate only donates a nitrogen to change IMP to AMP.
SYNTHESIS OF GMP: Add an amine to the other (non-oxidated) carbon of the larger purine
ring.
- Inosinate (IMP) ------> Xanthylate
- This is an oxidation of the non-carbonyl carbon on the larger purine ring.
- Concurrent reduction: NAD+ ------> NADH
- Xanthylate + Glutamine ------> Guanylate (GMP) + Glutamate
- This is just addition of an NH3 from glutamine. It gets put on the newly oxidized
carbon (keto group) of the purine ring, forming an imine linkage.
- So, in this case the extra NH3 comes from the side chain of glutamine, and
glutamine does not stay on the ring as an intermediate.
- ATP is required.
SUMMARY: WHAT DONATES CARBONS TO THE PURINES?
- RIBOSE and N-LINKAGE:
- Carbons from Ribose-5-Phosphate, from Pentose Phosphate Shunt
- Nitrogen Linkage from Glutamine side-chain nitrogen
- SMALLER PURINE RING (3 carbons, 2 nitrogens)
- 2 carbons and 1 nitrogen from Glycine
- 1 nitrogen from Free NH3
- 1 carbon from 10-formyl-tetrahydrofolate
- LARGER PURINE RING (4 carbons, 2 nitrogens)
- 1 nitrogen from the alpha-amino group of Aspartic Acid
- 1 carbon from HCO3-
- 1 carbon from 10-formyl-tetrahydrofolate
- (the 2 carbons that fuse the two rings together both come from Glycine)
- AMP EXTRA MODIFICATION:
- An extra nitrogen from the side chain of Aspartate
- GMP EXTRA MODIFICATION:
- An extra nitrogen from the side chain of Glutamine
REGULATION OF PURINE BIOSYNTHESIS:
- IMP, AMP, and GMP: They show negative feedback on their own synthesis. They
inhibit steps 1 and 2 of purine synthesis.
- Inhibit formation of PPRP (phosphoribosyl pyrophosphate), which is common to
both purine and pyrimidine synthesis.
- Inhibit formation of Phosphoribosylamine from PPRP -- the committed step.
- SYNERGISTIC: The inhibitory effects of AMP and GMP on these steps are
independent and additive. They bind to separate sites.
- Remember that these reactions are never turned completely off. They are only
slowed down.
- GMP specifically inhibits the conversion of IMP to Xanthylate. Simple negative
feedback.
- AMP specifically inhibits the conversion of IMP to Adenylosuccinate. Simple negative
feedback.
THE SALVAGE PATHWAY: Recycling of Hypoxanthine, Guanine, and Adenine, which are
all purines. A salvage pathway exists for pyrimidines too, but it isn't clinically important.
- PPRP + Purine ------> Purine Ribonucleotide
- All we are doing here is putting another ribose-phosphate moiety on a naked
purine base.
- Enzyme for Hypoxanthine and Guanine= Hypoxanthine-Guanine Phosphoribosyl
Transferase (HGPRT)
- Enzyme for Adenine = Adenine Phosphoribosyl Transferase
DEGRADATION OF PURINES: Purines are broken down to URIC ACID for excretion.
- AMP ------> IMP ------> Hypoxanthine
- Hypoxanthine ------> Xanthine
- Xanthine Oxidase catalyzes this step. We need to know this enzyme.
- GMP ------> Xanthine: GMP is degraded directly to Xanthine at this stage.
- Xanthine ------> Uric Acid
- Xanthine Oxidase catalyzes this step, too.
- Uric Acid is then excreted.
GOUT: Buildup and precipitation of uric acid in the joints
- There are multiple causes of Gout:
- Often caused by partial deficiency of HGPRT, the main enzyme of the Salvage
Pathway. No salvage pathway means there is no place for purines to go but to uric
acid ------> buildup of uric acid.
- Loss of feedback inhibition of PRPP-Synthetase, resulting in overproduction of
purines and hence over secretion.
- Kidney damage resulting in reduced secretion uric acid.
- Treatment: Allopurinol: A suicide-inhibitor of Xanthine Oxidase.
- Allopurinol is an analog of Xanthine.
- Xanthine Oxidase reacts with Allopurinol, forming Alloxanthine. Upon reacting,
Xanthine Oxidase renders itself non-functional. Hence allopurinol is a "suicide
inhibitor" of xanthine oxidase.
- Xanthine and Hypoxanthine build up as a side-effect. But they are more soluble
than uric acid.
LESCH-NYHAN SYNDROME: Complete loss of HGPRT and hence the Salvage Pathway
- Partial loss of HGPRT results in Gout. Complete loss is far more serious.
- Symptoms:
- Mental retardation and autistic self-mutilation.
- Gout-like symptoms as well, which can be treated with allopurinol.
- HGPRT is an X-Linked gene. Thus the disease is more common in men.
- De Novo purine synthesis is still available, thus there is no shortage of purines. It is not
clear why the symptoms occur.
- HGPRT is found to be 10-20x more concentrated in the brain. Maybe the brain depends
on that pathway more for some reason.
ANTI-OXIDANTS: Uric Acid, along with Bilirubin, Ascorbic Acid and Glutathione, plays a role
in defense against anti-oxidants.
Return to top
PYRIMIDINE (CYTOSINE, THYMINE, URACIL) BIOSYNTHESIS
CARBAMOYL PHOSPHATE SYNTHETASE II: Forms carbamoyl phosphate to start
pyrimidine synthesis, using a different enzyme and different reaction than is used for urea
synthesis.
- Glutamine + HCO3- ------> Carbamoyl Phosphate + Glutamate
- Glutamine is the nitrogen donor rather than NH3, losing an NH3 to become
glutamate.
- This can occur as a CYCLE -- to make multiple pyrimidines. Just resynthesize
glutamine using more free ammonia.
- SHUNT: If there were a block somewhere in urea synthesis, carbamoyl phosphate could
build up and force this pathway.
- Carbamoyl Phosphate Synthetase II is the primary regulated step of Pyrimidine
Biosynthesis. It is standard negative feedback inhibition: UTP inhibits the enzyme.
|
CARBAMOYL PHOSPHATE
SYNTHETASE I |
CARBAMOYL PHOSPHATE
SYNTHETASE II |
| Synthesis: |
First step in Urea metabolism |
First step in Pyrimidine metabolism |
| Nitrogen Donor: |
Free NH4+, primarily from the
Glutamate Dehydrogenase
reaction. |
Glutamine, which can be
regenerated again from glutamate, to
form a cycle. |
| Tissue
Distribution: |
Liver primarily |
All tissues make pyrimidines |
| Cellular Location: |
Mitochondria |
Cytosol |
PYRIMIDINE SYNTHESIS: SYNTHESIS OF UMP. The first pyrimidine synthesized de novo
is UMP. The other pyrimidines are made off of that. After we have carbamoyl phosphate...
- Carbamoyl Phosphate + Aspartate ------> N-Carbamoylaspartate
- The whole aspartate is added to the carbamoyl phosphate.
- This step occurs in the cytosol.
- This is the COMMITTED STEP of pyrimidine biosynthesis.
- N-Carbamoylaspartate <====> Dihydroorotate
- This is a cyclization reaction, making a ring by forming an amide linkage between
the alpha-amino of aspartate and a carbonyl from carbamoyl phosphate.
- This step also occurs in the cytosol.
- Dihydroorotate <====> Orotate
- This reaction occurs in the mitochondria. Everything from here forward occurs
in the mitochondria.
- It is your basic redox reaction, with dihydroorotate being oxidized to orotate (not
sure what the redox cofactor is).
- Enzyme = Dihydroorotate Dehydrogenase
- Orotate + PPRP ------> Orotidylate + PPi
- This is the step where we add the ribose and form the sugar C1-N linkage. Note
that in pyrimidine synthesis we add it here, whereas in purine synthesis we started
with it in the first place.
- Enzyme = Orotate Phosphoribosyl Transferase. We are adding a ribosyl-phosphate to the orotate structure.
- Orotidylate ------> Uridylate (UMP) + CO2
- Enzyme = Orotidylate Decarboxylase
- Lose the final CO2 off the ring, and all we are left with is the pyrimidine (6-member) ring structure.
ADDING MORE PHOSPHATES: Once the basic nucleotide is formed (such as UMP or IMP),
it can always be converted to the diphosphate and triphosphate level by using phosphates
transferred from ATP, i.e. use a nucleotide kinase.
SYNTHESIS OF CYTIDINE (CTP): Cytidine synthesis occurs at the triphosphate level. It is
the addition of a nitrogen group from the side-chain of glutamine.
- UTP + Glutamine ------> CTP + Glutamate
SYNTHESIS OF DEOXYRIBONUCLEOTIDES: RIBONUCLEOTIDE REDUCTASE. The
reduction involves changing the 2' OH to just an H.
- An ENZYME COMPLEX utilizing a MINI-ELECTRON TRANSPORT CHAIN carries
out the reaction.
- For all nucleotides, this reaction occurs at the DIPHOSPHATE LEVEL -- i.e. CDP, UDP,
ADP, GDP
- Thioredoxin is the key COFACTOR that has oxidized and reduced forms.
- Oxidized form: Disulfide S--S linkage.
- Reduced form: HS-- --SH thiol groups
- Order of Reactions in the chain:
| Reduction |
Oxidation |
| Ribonucleotide ------>
Deoxyribonucleotide |
Thioredoxin (reduced) ------>
Thioredoxin (oxidized) |
| Thioredoxin (oxidized) ------> Thioredoxin
(reduced) |
FADH2 ------> FAD |
| FAD ------> FADH2 |
NADPH ------> NADP+ |
- This reaction is driven by NADPH -- This is a biosynthetic reductive reaction.
SYNTHESIS OF THYMINE: Thymine is made from deoxy-uridine (dUMP). First, some
uridine is converted to deoxyuridine. Then that is used to make thymine.
- dUDP ------> dUMP. First a phosphate must be removed before we make thymine.
- dUMP ------> dTMP
- Enzyme = Thymidylate Synthetase.
- This is methylation reaction. A methyl group is added to uridine to make
thymidine.
- The methylation is done by N5,N10-Methylene Tetrahydrofolate. Dihydrofolate
is left over as a byproduct.
- Thus Dihydrofolate Reductase is required to maintain thymidine synthesis. Every time
we make a thymidine, we use up a THF by oxidizing it to the dihydrofolate form.
Ultimately we must re-reduce it to make more thymidine.
REGULATION OF RIBONUCLEOTIDE REDUCTASE:
- ATP stimulates the production of Deoxy-Cytidine (dCDP) and Uridine (dUDP)
- Extra ATP means have lots of ribonucleotide but not enough deoxyribonucleotide.
Hence it encourages the production of DNA.
- dATP inhibits the production of all the deoxyribonucleotides.
- Three nucleotides inhibit dCDP production: dATP, dTTP, dGTP
- dGTP stimulates dADP synthesis.
- If we have too much dGTP around, relatively, then we need more dATP.
- dTTP stimulates dGDP synthesis. Don't ask me why.
Comparison of Purine and Pyrimidine Biosynthesis:
|
PURINES |
PYRIMIDINES |
| Committed Step |
Formation of PPRP |
Formation of N-Carbamoylaspartate |
| Cellular Location |
Cytosol |
Cytosol and Mitochondria |
| Order of Events |
We start with PPRP -- the
ribose moiety, and build the
purine rings around it |
We start by building the
pyrimidine ring and then add
the ribose to it. |
| Regulation |
IMP, AMP, and GMP inhibit
enzymes at multiple steps |
Carbamoyl Phosphate
Synthetase II is inhibited by
UTP |
| Degradative Product |
Uric Acid. The purine ring-structure is not broken. |
Succinyl-CoA. The
pyrimidine ring-structure is
broken down |
DEGRADATION OF PYRIMIDINES: Pyrimidines are ultimately broken down to Succinyl-CoA
and then recycled.
- Degradation of Thymine:
- Thymidine ------> ------> Methylmalonyl-CoA
- In multiple steps we get Methylmalonyl-CoA
- The nitrogen is lost as NH3 (to go to urea synthesis or transamination)
- Methylmalonyl-CoA ------> Succinyl-CoA.
- I think here a carbon is added, so this is a carboxylation reaction.
- Take Home Lesson: Pyrimidine rings can be broken down to yield succinate.
- Degradation of Uracil: We get beta-Alanine as the product, which ultimately then goes
to Acetyl-CoA.
CHEMOTHERAPY AGENTS: Also see methotrexate above.
- Azaserine: It is a glutamine analog. It would interfere with nucleotide synthesis
wherever glutamine is the nitrogen donor (such as in Carbamoyl Phosphate Synthetase II)
- 5-Fluorocil -- inhibits thymidylate synthetase
- 3-Deazauridine -- Uridine analog with a missing nitrogen. It will stop translation at
uridine residues.
- AZT: An analog of thymidine that is missing TWO hydroxy groups on the ribose sugar
instead of one.
- This prevents it from base-pairing in HIV viral replication and transcription.
- Side-Effects: Other tissues will also use the drug, esp. liver tissue.
- OTHER AIDS DRUGS: Other drugs have recently hit the market: DDC, DDA,
DDI.
Return to top
PORPHYRIN METABOLISM
SYNTHESIS OF HEME: Protoporphyrin IX is the precursor of the heme group found in
hemoglobin and myoglobin.
- Succinyl-CoA + Glycine ------> delta-Aminolevulinate
- Enzyme = delta-Aminolevulinate Synthetase
- This is the COMMITTED STEP and the REGULATED STEP of heme-biosynthesis.
- This is a condensation reaction. Coenzyme-A is given off to drive the reaction,
and a CO2 is given off of glycine.
- delta-Aminolevulinate x 2 ------> Porphobilinogen
- Two of them condense together and cyclize to form porphobilinogen.
- Porphobilinogen x 4 ------> Protoporphyrin IX
- This condensation gives the basic tetrapyrrole structure of heme, with no iron in
the middle yet.
- Protoporphyrin IX, as well as heme, are basically planar.
- Protoporphyrin IX still has side chains hooked to it, of three sorts:
- Propionate group
- Methyl group
- Vinyl group
HEME STRUCTURE AND FUNCTION:
- Coordination Points: The central iron has six coordination points:
- Four of them are to the four nitrogens of the porphyrin ring. This holds the iron in
place.
- One is to a Histidine residue on the globin molecule.
- The final one binds reversibly to O2 or H2O depending on whether oxygen is
currently bound.
- Iron: The central iron of the heme group must remain in the Fe+2 state in order to be
functional.
- It is the job of reducing agents like Glutathione to keep Iron in the ferrous (Fe+2)
state.
- Irons ability to bind oxygen in the heme structure is unique: oxygen can bind
reversibly to it with oxidizing it.
BREAKDOWN OF HEME: Heme degradation ultimately leads to bilirubin. Heme breakdown
occurs in the liver.
- Heme ------> Biliverdin
- This is a Mixed-Function Oxidase using the enzyme Heme Oxygenase to cleave
the heme ring-structure.
- Despite the fact that this is an oxygenation, NADPH is required as well!
- Biliverdin ------> Bilirubin
- Enzyme = Biliverdin Reductase
- NADPH is required for this step too: NADPH ------> NADP+ (but this time it
makes more sense because its a reduction reaction).
BILIRUBIN: Derivatives of bilirubin and clinical stuff
- Bilirubin should not build up.
- Buildup of bilirubin in the blood is an indication of liver dysfunction.
- Bilirubin in high levels is non-soluble and toxic. It acts as a neurotoxin.
- JAUNDICE: High levels of bilirubin coupled with hepatitis (liver inflammation)
leads to jaundice.
- Bilirubin is photosensitive. When it builds up in infants, it can be converted to more
soluble, less toxic forms by exposing it to fluorescent light.
- CONJUGATION OF BILIRUBIN: Bilirubin + UDP-Glucuronic Acid ------>
Bilirubin Diglucuronide. This is the soluble, conjugated form of bilirubin.
- BILIRUBIN EXCRETION:
- Glucuronide form is excreted in the feces primarily, and some in the urine as well.
- Urobilinogen is excreted in the urine and is the primary form that gives urine its
color. Urobilinogen is reabsorbed from the GI-Tract, after it has been acted on by
intestinal bacteria.
PORPHYRIAS: Hereditary problems with heme metabolism. Commonly found symptoms =
neurological disturbances and skin photosensitivity
- Acute Intermittent Porphyria: dominant
- Problem with delta-Aminolevulinate Synthetase, the first step in heme-synthesis.
Or it could happen with an error in the second step of heme-synthesis --
production of Porphobilinogen (Uroporphyrinogen I Synthetase)
- Symptoms: Neurological disturbances
- Congenital Erythropoietic Porphyria: recessive
- Skin sensitivity and fragile erythrocytes
- Hereditary Coproporphyria -- dominant, + same screwed up enzymes as above.
- Neurological disturbances and skin photosensitivity.
- Hereditary Protoporphyria -- dominant
- Ferrochelatase is altered enzyme -- the enzyme that puts the iron in heme.
- Lead Poisoning: This disorder is also associated with lead poisoning, which alters
delta-aminolevulinate metabolism.
- TREATMENT: Injection of hematin -- preformed heme with the iron already present.
REGULATION OF HEME SYNTHESIS: Negative feedback. Heme inhibit delta-aminolevulinate synthetase, which is the committed step in the pathway.
ANTI-OXIDANTS: Here are nitrogen metabolic intermediates that serve as anti-oxidants.
- Reduced glutathione (with SH at the end)
- Uric Acid -- One theory of evolution says that we live long because we have high (almost
to the precipitation point) concentrations of uric acid.
- Ascorbic Acid (Vit. C)
- Bilirubin has anti-oxidant properties
Return to top
MISCELLANEOUS STUFF
SYNTHESIS OF S-ADENOSYL METHIONINE (SAM): Review. Once again, S-Adenosyl
Methionine is formed from Methionine + ATP, where the Adenylyl Group of the ATP goes onto
the Methionine, and all three phosphates are given off as Pi + PPi.
METHYL-TRANSFERS: Some of the methylation reactions that S-Adenosyl Methionine
partakes in. Remember that THF is required to recycle Homocysteine back to SAM, for each
time it methylates.
- METHYLATION AT A NITROGEN: The CH3 group is added to a N.
- Norepinephrine ------> Epinephrine
- Guanidoacetate ------> Creatine
- Phosphatidylethanolamine ------> Phosphatidylcholine
- Lysine or Guanine ------> N-Methyl Lysine or N-Methyl Guanine
- METHYLATION AT AN OXYGEN:
- Synthesis of Melatonin
- Norepinephrine ------> Normetanephrine (Ephedrine)
- METHYLATION AT CARBON:
- Cytosine ------> 5-Methyl Cytosine
- OTHER TRANSFERS REACTIONS:
- Aminopropyl group of SAM is transferred in the synthesis of polyamines (see
below)
- Adenosyl group of SAM may be transferred in the conversion of vitamin B12
(Cobalamin) to storage form.
CHOLINE: Phosphatidylcholine is a Quaternary Amine with a positive charge -- R-N+(CH3)4.
So to completely convert ethanolamine to choline, it takes 3 SAM.
- Serine ------> Ethanolamine
- Ethanolamine ------> Choline
POLYAMINES: Polyamines are highly positively-charged species that serve to condense
negatively-charged DNA in sperm. They are made from Ornithine, from the Urea cycle.
- Ornithine ------> Putricine
- Decarboxylation reaction.
- Putricine ------> Spermidine
- This reaction involves an Aminopropyl Group Transfer from a derivative of
SAM.
- Spermidine ------> Spermine
- Same as above -- we use SAM to transfer yet another Aminopropyl group -- to
give us a highly aminated (and therefore very positively charged) compound.
- ROADKILL: Polyamine formation occurs with dead, decaying tissue. It smells bad.
- TISSUE DISTRIBUTION: Prostate, seminal vesicles, and sperm itself have highest
concentration of polyamines. Brain tissue has almost none.
CREATINE: Creatine Phosphate is the form in which high-energy bonds are stored in muscle.
- SYNTHESIS: Creatine is formed by methylating the dipeptide Arg-Gly.
- Arginine-Glycine ------> Creatine via S-Adenosyl Methionine (SAM)
- Creatine Kinase: Creatine + ATP ------> Creatine Phosphate + ADP. This is the
longer-term storage form of high energy bonds in muscle. ATP can only stick around for
so long.
- EXCRETION:
- Muscle has a high concentration of creatine phosphate. The amount of creatine we
find in our system is therefore proportional to muscle mass and to muscle activity.
- Creatine <====> CREATININE. Creatinine is a cyclized form of creatine, and
the form in which we excrete it.
- In a healthy individual, the amount of creatinine excreted in the urine is constant
over a given period, and is proportional to muscle mass.
CARNITINE: Carnitine was the acyl-intermediate in mitochondria, necessary for beta-oxidation.
- Lysine ------> Epsilon Trimethyl-Lysine.
- This again is the transfer of 3 methyl groups using 3 SAM. Like choline, all three
of the CH3's go onto a nitrogen to make a quaternary amine.
- Epsilon Trimethyl-Lysine ------> Carnitine
CERAMIDE: The basic component of sphingomyelin and of the gangliosides.
- Palmitoyl-CoA + Serine ------> Sphingosine
- Sphingosine is the base-structure of ceramide, without any acyl groups added.
- Sphingosine + Acyl-CoA ------> Ceramide + CoA
- Acylate the sphingosine, using a charged acyl-group, to make ceramide.
- Ceramide ------> Sphingomyelin -- Add a choline as the polar head group, to make
sphingomyelin.
- Ceramide ------> ------> Gangliosides -- Add UDP-Galactose (and other sugars) to
make gangliosides
GAMMA-AMINOBUTYRIC ACID (GABA): GABA, a neurotransmitter implicated in
depression, is made from glutamate.
- Glutamate Decarboxylase: Glutamate ------> GABA + CO2. GABA is simply a
decarboxylated form of glutamate.
- Chinese-Restaurant Syndrome: MSG can be hazardous because of its associated to
GABA. People who are sensitive to MSG are though to get too high levels of GABA in
the brain for a short time.
- DEGRADATION of GABA: GABA ------> ------> Succinate. GABA can be
degraded down to succinate.
CATECHOLAMINES: Dopamine, Epinephrine, Norepinephrine.
- Tyrosine ------> DOPA (Dihydrophenylalanine)
- This is a hydroxylation reaction -- simply add another hydroxyl group.
- DOPA ------> Dopamine
- This is a Decarboxylation
- Dopamine ------> Norepinephrine
- Hydroxylation of the benzylic carbon.
- Norepinephrine ------> Epinephrine
- This is a SAM-Methylation. Epinephrine is methyl-norepinephrine.
- PARKINSON'S DISEASE: Deficiency of dopamine neurotransmitter, from cell death i
the substantia nigra.
- Treatment = huge doses of dopamine along with an inhibitor that prevents rapid
breakdown of dopamine, in hopes of getting lots to the brain.
- The treatment only treats the symptoms. It does not cure the disease.
- FORMS OF BREAKING DOWN the Catecholamines: They are rapidly inactivated and
excreted by two enzymes. So, we need a continuous supply of them.
- Norepinephrine ------> 3,4-Dihydroxyphenylglycolaldehyde, via Monoamine
Oxidase
- Norepinephrine ------> 3-O-Methylepinephrine, via Catechol-O-Methyltransferase.
SEROTONIN: Neurotransmitter comes directly from tryptophan, via a two-step process:
decarboxylation and hydroxylation
HISTAMINE: Intercellular mediator, comes directly from histidine, via a single
decarboxylation.
MELANIN: Comes from Tyrosine
COENZYME-A: Comes from pantothenic acid, plus cysteine and ATP.
NICOTINIC ACID: You can make nicotinic acid, and hence NAD, from Tryptophan via its
degradative pathway.
Return to top
INTEGRATED METABOLISM
Generalities:
- ATP is the universal currency of energy.
- ATP is generated by the oxidation of metabolic fuels
- Glycolysis
- Oxidative Phosphorylation
- NADPH is the major electron donor in REDUCTIVE BIOSYNTHETIC pathways.
- NAD+ is the major electron acceptor in OXIDATIVE DEGRADATIVE pathways.
- Biological molecules are constructed from a small set of building blocks.
- Synthetic and Degradative pathways are almost always distinct from each other.
Principles of Metabolic Regulation:
- Substrate Supply: An enzymatic reaction cannot proceed in the absence of sufficient
substrate.
- Allosteric Interactions. They can be either positive or negative. They are not all-or-none
interactions.
- Covalent Modifications = primarily phosphorylation. This is completely on-or-off per
molecule, but still overall you are not affecting every substrate so the change is still at
least somewhat gradual one way or the other.
- Enzyme-Levels
- Compartmentalization
- Organ Specialization
GLUCOSE: Glucose-6-Phosphate has key metabolic branch points.
- Glucose-6-P <====> Glucose-1-P <====> Glycogen: Glycogenesis, Glycogenolysis
- Glucose-6-P ------> 6-Phosphogluconolactone (PPP Shunt)
- Glucose-6-P <====> Fructose-6-P <====> Pyruvate (Glycolysis)
PYRUVATE: Key metabolic branch points
- Pyruvate <====> Oxaloacetate (Anaplerosis) ------> Glucose-6-P
(Gluconeogenesis)
- Pyruvate <====> Lactate (Lactate Dehydrogenase, regeneration of NAD+)
- Pyruvate ------> Acetyl-CoA (Pyruvate Dehydrogenase)
ACETYL-CoA: Primary branch points
- Acetyl-CoA ------> HMG-CoA (Sterol Biosynthesis)
- HMG-CoA ------> Cholesterol ------> Hormones etc.
- HMG-CoA ------> Ketone Bodies
- Acetyl-CoA ------> CO2 (Oxidative Phosphorylation)
- Acetyl-CoA <====> Fatty Acids (Lipogenesis, Lipolysis)
FATTY ACID BIOSYNTHESIS: REGULATION
- Regulated Step = Acetyl-CoA Carboxylase: Acetyl-CoA ------> Malonyl-CoA
- INHIBITED by Palmitoyl-CoA, i.e. by its own product.
- STIMULATED by Citrate
- Citrate generally indicates lots of energy to spare, hence its time to do fat
synthesis.
GLYCOLYSIS: REGULATION
- Regulated Step = Phosphofructokinase (PFK): Fructose-6-Phosphate ------>
Fructose-1,6-Biphosphate
- STIMULATED BY -- these two indicate a deficiency of energy
- Fructose-2,6-Biphosphate
- AMP
- INHIBITED BY -- once again...
GLUCONEOGENESIS: REGULATION
- Regulated Step = Fructose-1,6-Biphosphotase: Fructose-1,6-Biphosphate ------>
Fructose-1-Phosphate
- INHIBITED BY AMP and Fructose-2,6,-Biphosphate (not enough energy is around for
gluconeogenesis)
- STIMULATED BY Citrate and ATP
PENTOSE PHOSPHATE PATHWAY: REGULATION
- Regulated Step: Glucose-6-Phosphate Dehydrogenase: Glucose-6-Phosphate ------>
6-Phosphogluconolactone ------> 6-Phosphogluconate
- INHIBITED by NADPH
- STIMULATED by NADP+, which means we need to make more NADPH.
THE HORMONES:
- GLUCAGON = generally catabolic
- PROMOTE Gluconeogenesis in liver, glycogenolysis, lipolysis
- INHIBIT Glycogenesis, Lipogenesis
- EPINEPHRINE = generally catabolic -- the "hormone of starvation"
- STIMULATES Lipolysis in particular, plus the other catabolic processes
- STIMULATES the release of glucagon from the pancreas
- CORTISOL = both catabolic and anabolic -- the "hormone of the defense state"
- PROMOTE GLycogenesis, Gluconeogenesis, and Proteolysis.
- INSULIN = generally anabolic
- PROMOTE Glucose absorption by cells, amino acid transport into cells,
Glycogenesis, Lipogenesis
- It does NOT promote Glucose-Uptake, per se, in the lens of the eye and brain,
where glucose is essential at all time anyway.
- INHIBITS Gluconeogenesis and Glycogenolysis
FUTILE CYCLES: Irreversible reactions, catalyzed by different enzymes going in each
direction. The purpose to them is that you can go back and forth, accomplishing nothing but
generating heat from lost high-energy bonds (usually ATP).
- PEP <====> Pyruvate
- Fructose-1-Phosphate <====> Fructose-1,6-Biphosphate
- Lots and lots of them
CORI CYCLE: Hard-working muscles release lactate.
- MUSCLE = Glycolysis: Glucose-6-Phosphate ------> Pyruvate ------> Lactate
- LACTATE than travels from skeletal muscle to liver.
- LIVER = Gluconeogenesis: Lactate ------> Pyruvate ------> Oxaloacetate
------> Glucose-6-Phosphate
GLUCOSE-ALANINE CYCLE: Alanine is released by muscles that are not necessarily working
so hard. Object = Take an AMINO GROUP from muscle and send it to liver for urea synthesis.
- MUSCLE: Glucose-6-Phosphate ------> Pyruvate ------> Alanine
- The alanine step is accomplished via transamination of pyruvate. Both of them
have the same carbon-skeleton!
- ALANINE then travels in venous blood to liver.
- LIVER: Alanine ------> Pyruvate ------> ------> Glucose-6-Phosphate
- The nitrogen group is removed from Alanine here to contribute to urea synthesis
AMINO ACID BALANCE: After eating (i.e. in the post absorptive state):
- There is more glutamine and alanine being released from the skeletal muscle into the
blood.
- Glutamine is released significantly, because it is a carrier of amino groups out of
the tissues.
- Alanine is released significantly because of the glucose-alanine cycle.
- That means there is much more of these amino acids in venous blood then in arterial
blood.
Return to top
BIOCHEMICAL NUTRITION
FOOD ENERGY UTILIZATION:
- Nondigestible Energy (Feces) 1-9%
- Digestible Energy 90%
- HEAT (Futile Cycles) 50%
- SPECIAL DYNAMIC EFFECT = energy required to digest food 10%
- UTILIZED ENERGY for storage / metabolism 25-40%
METABOLIZABLE ENERGY: For proteins, not all that is digested is metabolizable, as some is
lost as NH3 groups in urea. Ethanol is pure calories and nothing else.
METABOLIC REQUIREMENTS:
- HYPERMETABOLISM: A higher than basal level of metabolism is required (i.e. more
energy is used) under some circumstances:
- Burns covering >40% of body surface
- Severe infection
- Multiple or long-bone fractures
- Postoperative recovery
- HYPOMETABOLISM: Up to 40% less than basal metabolism of energy can be utilized
to adapt to a state of mild starvation.
PROTEIN TURNOVER: About 250g of protein each day are turned over (i.e. synthesized or
degraded) from one form to another
- 100g Equilibrium with free amino acids
- 70g Protein consumed in dividing cells in GI-Epithelia
- 50g Turnover associated with skeletal muscle. For folks who exercise a lot this will be
more
- 30g Red (8g) and White (20g) blood cells
- 25g LIVER -- It is the key tissue that is synthesizing new plasma proteins. The
principle proteins the liver synthesizes for excretion are as follows:
- Albumin -- by far the most predominant
- Transferrin
- Fibrinogen
NITROGEN BALANCE: Nitrogen Balance = Nintake - Nloss
- Nitrogen Loss: Nloss = urine (90% is urine) + feces + skin + fluids
- NITROGEN BALANCE = 0 ------> healthy adult
- NITROGEN BALANCE > 0 (take in more than excrete)
- Healthy growing infants
- Pregnancy and lactation
- Recovery from illness
- NITROGEN BALANCE < 0 (take in less than excrete)
- Inadequate protein in diet
- Trauma or surgery
- Metabolic stress
|
KWASHIORKOR |
MARASMUS |
| Primary Deficiency: |
PROTEIN deficiency, due to
no protein in diet or
inadequate absorption. |
OVERALL ENERGY
deficiency |
| Description: |
"A response to some kind of
stress" |
"A slow adaptation to
starvation" |
| Edema |
EDEMA is present -- due to
lost oncotic pressure in
blood vessels, in turn due to
hypoalbuminemia |
Absent |
| Hypoalbuminemia |
LOW ALBUMIN is the
cardinal symptom. Liver
shuts down albumin
production to conserve on
protein which is lacking. |
Absent |
| Fatty Liver |
PRESENT -- Low protein in
diet means lots of
carbohydrates. This will yield
a fatty liver (due to no
albumin transport) and maybe
hepatomegaly. |
Absent |
| Insulin Level |
MAINTAINED -- hormonal
levels are responding to stress
but staying within normal
limits |
LOW -- body is in catabolic
state to get all energy out of
stores (whatever is left) |
| Epinephrine Level |
NORMAL -- hormone levels
remaining within normal
limits |
HIGH -- catabolic attempt to
scrape every last bit of energy
out of body stores |
| Muscle Wasting |
Absent or mild |
YES -- may be severe, as
muscle is catabolized for
essential bodily functions |
| Body Fat |
Some loss |
ABSENT |
| RESPONSE-TIME |
ACUTE -- very severe
catabolic response in a short
time |
GRADUAL -- mildly
catabolic response to
starvation can take a long
time |
| Hypopigmentation |
Can be present -- amino acids
(tyrosine) are required to
generate pigmentation.
Hence patient may be pale. |
|
Return to top
WATER-SOLUBLE VITAMINS
THIAMINE: 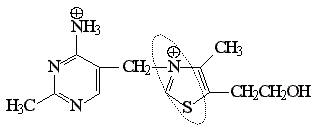
- FUNCTION: Active-Aldehyde
Transfers (as in TPP)
- SPECIFIC REACTIONS:
- Oxidative Decarboxylations
(Pyruvate Dehydrogenase,
alpha-Ketoglutarate
Dehydrogenase)
- Transketolase Reaction in the Pentose Phosphate Pathway
- Functions in Sodium Channels too, in neural activity.
- CHEMISTRY:
- It has a very acidic center that readily ionizes to give a carbanion.
- ACTIVE FORM: THIAMINE PYROPHOSPHATE -- it is phosphorylated twice by
ATP to get to its active form.
- GROSS DEFICIENCY: WERNICKE'S ENCEPHALOPATHY
- Classic Triad of Symptoms:
- Ophthalmoplegia -- paralysis of eye muscles
- Ataxia -- lost muscular coordination
- Mental Confusion
- Biochemical Deficiency: alpha-Ketoglutarate Dehydrogenase can't operate
------> alpha-Ketoglutarate builds up -------> It gets shunted to amino acid
metabolism ------> too much glutamate.
- Transketolase and Pyruvate Dehydrogenase also require TPP but their
levels don't seem so effected by deficiency.
RIBOFLAVIN: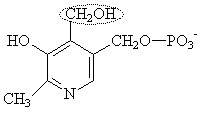
- ACTIVE FORMS
- FMN: Add one phosphate to the terminal
OH-Group.
- FAD: Add one phosphate to terminal OH-group, plus an AMP
- CHEMISTRY: The two nitrogens can be oxidized /
reduced between the double bond form and NH
form.
- FUNCTION: Redox Reactions
- SPECIFIC REACTIONS
- Electron Transport Chain
- Succinate Dehydrogenase
PANTOTHENIC ACID:
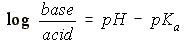 ACTIVE FORM: 4-Phosphopantetheine
ACTIVE FORM: 4-Phosphopantetheine
- Add a cysteine to the terminal
COOH via an amide linkage. This is
required for the high-energy thiol
bond.
- Add a Phosphate to the terminal OH on the other end.
- COENZYME-A: 4-Phosphopantetheine (above) + ADP.
- FUNCTION: Acyl Activation and Transfer. Coenzyme-A activates acyl groups by
giving them a high-energy thioester bond.
- SPECIFIC REACTIONS:
- Acylation during fatty acid synthesis and oxidation
- Lots of other examples dealing with fats especially.
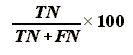 ASCORBIC ACID:
ASCORBIC ACID:
- SPECIFIC REACTIONS:
- Covalent Hydroxylation Reactions -- those that are
not dependent on Cytochrome-P450, i.e. not
cholesterol.
- Formation of hydroxylysine and
hydroxyproline in collagens
- Catecholamines formation
- Tryptophan ------> Serotonin
- Lysine ------> Carnitine
- Anti-Oxidant
- Uptake of iron in intestinal tract.
- CHEMISTRY: The two OH's can be oxidized to two keto groups, making
Dehydroascorbic Acid. Hence Ascorbic Acid is an excellent anti-oxidant, as it gets
oxidized instead of some other vulnerable substance.
BIOTIN:
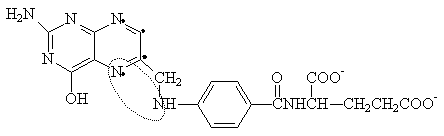 ACTIVE FORM: Biotin is linked to a Lysine
residue on the protein.
ACTIVE FORM: Biotin is linked to a Lysine
residue on the protein.- FUNCTION: ATP-Dependent Carboxylations
- SPECIFIC REACTIONS:
- Pyruvate ------> Oxaloacetate
- Acetyl-CoA ------> Malonyl-CoA
- Propionyl-CoA ------> Methylmalonyl-CoA
- CHEMISTRY: TWO STEP process
- Biotin accepts the CO2 from the donor. This step requires ATP.
- Biotin transfers the CO2 to the recipient. This step doesn't require ATP.
- Avidin from raw eggs prevents absorption of biotin.
 PYRIDOXINE:
PYRIDOXINE:
- COENZYME-FORM: Pyridoxyl Phosphate (pictured)
- FUNCTION: Forms stable, yet transient Schiff-Base
Complexes with amines.
- SPECIFIC REACTIONS: Lots
- Amino Acid Metabolism, virtually all phases of
it.
- Glycogen Phosphorylase -- breakdown of glycogen to glucose.
 NICOTINIC ACID:
NICOTINIC ACID:
- COENZYME FORMS: The carboxyl group is first converted to an
amide, then things are added to the nitrogen.
- NAD: Add Ribose-5-Phosphate + AMP.
- NADP: Add Ribose-5-Phosphate + ADP
- CHEMISTRY: It is an electron carrier. 1 proton and 2 electrons can be added to the
active center, making it non-aromatic.
- FUNCTION: Redox Reactions
- SPECIFIC REACTIONS: LOTS
COBALAMIN: VITAMIN B-12
- CHEMISTRY / STRUCTURE:
- Corrin ring center, similar to porphyrin ring.
- Cobalt chelated to the center, chelated to the following:
- 4 nitrogens around ring
- Benzamide down below
- A VARIABLE ligand, X
- VARIABLE ligand, X = Cyano in dietary form, and Methyl in Coenzyme form
- ACTIVE FORMS:
- X = Methyl, as in THF
- X = 5-Deoxyadenosyl
- SPECIFIC REACTIONS: Exactly two
- Methylmalonyl-CoA ------> Succinyl CoA REARRANGEMENT
- Methyl Transfer of an N-5-methyl-tetrahydrofolate to Homocysteine, to
regenerate Methionine.
- DEFICIENCY:
- Can lead to ANEMIA
- Can be caused by lack of intrinsic factor in the intestine (that is what absorbs
Vitamin B12 in intestine)
- CNS and peripheral neuropathies can also result.
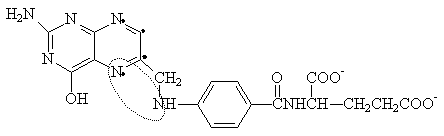 FOLATE:
FOLATE:
- DIETARY
FORM: Has one
or two
glutamate
residues stuck to
it.
- ACTIVE FORM:
- More Glutamates are added
- 4 Hydrogen are put on the spots indicated.
- FUNCTION: One-Carbon transfers, in the form of Tetrahydrofolate.
- SPECIFIC REACTIONS: Purine and pyrimidine metabolism. Amino acid synthesis.
- DEFICIENCY:
- ANEMIA (Megaloblastic or pernicious) -- abnormal DNA structure due to
insufficient nucleotide synthesis leads to chromosomal abnormalities in the RBC's.
- Has been linked to neural tube defects. Adequate amounts must be taken during
pregnancy.
- Homocystinemia is elevated homocysteine, which can result from no folate.
Return to top
FAT-SOLUBLE VITAMINS
 VITAMIN-A: RETINOL
VITAMIN-A: RETINOL
- Pro-Vitamin-A is beta-CAROTENE
which is a polyisoprene and is twice
as long as retinol with a cyclic group
on both sides.
- SYNTHESIS OF ACTIVE FORM:
- 15,15'-Dioxygenase cuts beta-Carotene ------> Two Retinal (aldehyde form)
- Retinal ------> Retinol, which is Vitamin-A
- Retinal ------> Retinoic Acid
- Configurations:
- beta-Carotenes are all in trans-configuration.
- Retinoic Acid can be trans or 9-cis
- Processing of Vitamin-A: Retinol is carried in an esterified form in chylomicrons.
- INTESTINAL ABSORPTION:
- Conversion to Retinol occurs in the intestine.
- Retinyl Esters can be formed by esterifying Retinol to long chain fats.
- Retinyl Esters are then absorbed into Chylomicrons
- TRANSPORT: Chylomicrons take some of the Vit-A to extra-hepatic tissues
and the rest goes to liver.
- STORAGE: Liver stores Vitamin-A in Stellate Cells.
- MOBILIZATION: Liver releases Vitamin-A coupled to a protein called
Prealbumin
- FUNCTIONS:
- Retinal (aldehyde) -- VISUAL CYCLE. Interconversion of trans <====> 11-cis forms is necessary for maintenance of rods and cones.
- Retinoic Acid (all trans and 9-cis) acts as a steroid hormone to affect cellular
differentiation, especially:
- Morphogenesis
- Reproduction
- Immune Response
- RETINOIC ACID RECEPTORS: Steroid-hormone receptors. At least two
classes identified
- RAR Receptor: Binds both all-trans and 9-cis forms.
- RXR Receptor: Binds only 9-cis form.
- VITAMIN-A TOXICITY: An excess of all-trans Vitamin-A in the diet induces the beta-isoform of the RAR-Receptor to high levels ------> teratogenic birth defects.
- beta-CAROTENE is NOT TOXIC!! -- only Vitamin-A is toxic. beta-Carotene is
probably not toxic because the initial Dioxygenase (cutting) step is regulated.
- DEFICIENCY:
- Causes: Insufficiency in diet, or malfunctioning absorption or bad RAR-Receptors.
- Symptoms: Skin lesions, night blindness, corneal ulcerations and conjunctivitis,
poor bone remodeling.
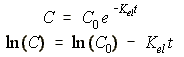 VITAMIN E: alpha-TOCOPHEROL
VITAMIN E: alpha-TOCOPHEROL
- FUNCTION: Only
one function -- as an
anti-oxidant.
- It will protect poly-unsaturated fats,
cholesterol, and rods
and cones, from radical oxidation.
- Importantly, it is a fat-soluble anti-oxidant whereas Ascorbic Acid is water-soluble. So
Vit.E gets into membranes.
VITAMIN K: PHYLLOQUINONE (K1), MENAQUINONE (K2)
- FUNCTION: The Quinone structure of Vitamin-K is reversibly oxidized in forming
gamma-Glutamylcarboxylate. The reaction occurs on a glutamate residue on a protein
- gamma-Glutamylcarboxylase catalyzes Glutamate Residue ------> gamma-Carboxyglutamate
- Vit-K (reduced) ------> Vit-K (oxidized) concurrently.
- The reaction requires Manganese
- The gamma-Glutamylcarboxylate can chelate calcium and that is why it is
important.
- The enzymes which use these calcium-chelating residues are CLOTTING
FACTORS and enzymes involved in bone-mineralization and demineralization.
- ANTI-COAGULANTS: Many of them inhibit the reduction of Vitamin K, from Vit-K
Keto ------> Vit-K Alcohol. Hence Vit-K cannot be recycled, and calcium-dependent
clotting factors cannot function.
- Dicumarol, specifically, inhibits the reduction of Vitamin K, to recycle it.
 VITAMIN D: CHOLECALCIFEROL (D3)
VITAMIN D: CHOLECALCIFEROL (D3)
- SYNTHESIS of 1,25-(OH)2-D3, or 1,25-DIHYDROXYCHOLECALCIFEROL:
Vitamin-D is not strictly a vitamin because
we can synthesize it ourselves.
- SKIN: Cholesterol ------>
Cholecalciferol is a non-enzymatic
cleavage catalyzed by UV-LIGHT.
- LIVER: 25-Hydroxylase puts a
hydroxyl group on the side chain.
- KIDNEY: 1alpha-Hydroxylase puts a hydroxyl at the 1alpha position.
- DIETARY Vitamin-D3: It does not have the hydroxyl groups on it.
- FUNCTION OF VITAMIN-D3: It is stimulated by Parathyroid Hormone, and it
promotes the release of Calcium into the blood.
- 1alpha-Hydroxylase in the Kidney is tightly regulated. Parathyroid hormone will
allow that reaction to occur, so Vitamin-D gets to its final form.
- ACTION on INTESTINE: Vitamin-D promotes uptake of more calcium and
phosphate in the intestine.
- ACTION on LONG BONES: Will stimulate osteoclasts to break down
hydroxyapatite and release calcium into blood.
Return to top
MINERALS
MACROMINERALS:
- CALCIUM: Major mineral cation of bone, hydroxyapatite.
- FUNCTIONS: Blood blotting, muscle contraction, nerve transmission, Calcium-Dependent Calmodulin
- PARATHYROID HORMONE: Releases calcium from blood and stimulates
Secretion of Vitamin-D in kidney to uptake more calcium. It takes calcium from
the bone via osteoclasts.
- CALCITONIN: Takes calcium from blood and deposits calcium and phosphate
onto bone, when blood-calcium levels are high.
- gamma-Carboxyglutamate residues (Vitamin-K dependent) require calcium as
cofactors, often found in clotting.
- PHOSPHOROUS: Major mineral anion of bone, hydroxyapatite.
- Also important in forming a phosphate buffer system in blood.
- SODIUM: Principle cation in extracellular fluids. Essential to nerve transmission.
- POTASSIUM: Principle cation in intracellular fluids. Essential to nerve transmission.
- CHLORIDE: Gastric secretions, membrane potentials.
- MAGNESIUM: Complexed with ATP, found in bone and teeth.
MICROMINERALS:
- ZINC: Cofactor in lots of reactions
- SPECIFIC REACTIONS
- Carbonic Anhydrase: CO2 ------> HCO3 in blood.
- Carboxypeptidases and other Zinc Metalloproteases
- Superoxide Dismutase
- Aldolase in glycolysis.
- FNXN: Wound healing.
- TRANSPORT: It is transported bound to albumin and alpha2-Macroglobulin.
- STORAGE: It is stored complexed to the protein Metallothionein.
Metallothionein has sulfur groups to chelate zinc in place, found in most cells.
- COPPER:
- FNXN: Associated with OXYGENASES. Aids in oxygenation and hydroxylation
of residues.
- SPECIFIC REACTIONS:
- Cytochrome Oxidase in the electron transport chain.
- Dopamine Hydroxylase (catecholamines)
- Tyrosinase (melanin)
- Superoxide Dismutase
- TRANSPORT: CERULOPLASMIN is a dedicated copper-transport protein.
- SELENIUM: Same family as O and Si.
- Glutathione Peroxidase: helps dispose of reactive oxygen species.
- Thyroid Synthesis: Also it is a cofactor for an enzyme involved synthesis of
thyroid hormone.
- Selenocysteine: Selenium takes the place of oxygen on a Serine residue, to form
selenocysteine. It is incorporated during translation, in an ATP-dependent step, in
response to a STOP CODON!
IRON: THE MAJOR MICROMINERAL
- IRON UTILIZATION CYCLE
- Red Blood Cells lyse and get phagocytosed. Hemoglobin is disposed as bilirubin,
while iron is recycled.
- Phagocytes release iron into the blood.
- Transferrin picks the iron up and sends it back to the BONE MARROW
- Bone Marrow then utilizes it to make more red blood cells.
- TRANSPORT: TRANSFERRIN = Major plasma protein synthesized / secreted by the
liver. It has a fairly short half-life and can thus be used to monitor liver function.
- STORAGE:
- FERRITIN: Primarily found in liver and bone-marrow. These are two
important storage tissues for iron.
- In storage, iron is actually converted to Ferric Oxyhydroxide Crystal
(Fe(III)OOH)
- This allows Ferritin to polymerize around iron in a ring-like fashion and
make sure it is not floating around free which we don't want, as it could
promote reactive oxygen species.
- HEMOSIDERIN: Found in many tissues, represents long-term storage of iron.
- IRON UPTAKE by CELLS: Receptor-Mediated Endocytosis.
- Iron-Transferrin Complex is taken up through clathrin coats.
- The Endosome is acidified and only the Fe+3 is released in the ferric state.
- Both the Transferrin and Receptor are recycled back to membrane.
- REGULATION OF IRON-LEVELS: Levels of Ferritin and of the Transferrin-Receptor are regulated.
- THE PLAYERS:
- Iron-Response Element (IRE) is a region of mRNA on both Ferritin and
Transferrin-Receptors.
- It is located upstream (5'-end) in Ferritin, so that binding to it stops
translation.
- It is located downstream (3' end) in the Transferrin-Receptor, so
that binding to it stabilizes the mRNA and allows it to translate
more protein.
- Iron-Response Element Binding-Protein (IRE-BP): ACONITASE.
The protein that binds to IRE to control levels of Ferritin has been shown
to be Aconitase, from the TCA cycle.
- IRON LEVELS TOO LOW:
- Iron-Response Element binds to Ferritin mRNA ------> Stop
translation of Ferritin. This will hopefully mobilize more iron out of
storage.
- Iron-Response Element binds to Transferrin Receptor ------>
Stabilize it ------> Translate more receptor. This will increase the rate
of iron-endocytosis by cells.
- IRON LEVELS TOO HIGH: Don't make any Iron-Response Element. Ferritin
levels go down due to their half-life. Transferrin receptors at normal levels.
- IRON DEFICIENCY: IRON-DEFICIENT ANEMIA
- Commonly seen in premature infants, and commonly caused by chronic disorders
of the GI-Tract (ulcers etc.)
- SYMPTOMS: LOW HEMATOCRIT + ANEMIA. Not much hemoglobin being
made.
- First, less iron storage
- Then, the bone marrow will start to make fewer red blood cells for lack of
iron to put in hemoglobin.
- Iron-Deficient Anemia is defined as hemoglobin concentration more than
two standard deviations below normal.
- HEMOCHROMATOSIS: Too much iron.
- Symptoms:
- High levels of Hemosiderin and Ferritin.
- Oxidative damage to cardiac muscle / heart tissue is biggest concern.
- Causes: May be genetic.
- beta-Thalassemia can cause it. If we don't have much beta-globin, then
less iron will be mobilized, putting a potential overload in the blood stream.
- Treatment: Drugs which chelate excess iron.
Return to top