What is
Molecular Modeling?
Molecular modeling, also
known as molecular mechanics, is a method to calculate the
structure and energy of molecules based on nuclear motions.
Electrons are not considered explicitly, but rather it is
assumed that they will find their optimum distribution once
the positions of the nuclei are known. This assumption is
based on the Born-Oppenheimer approximation of the Schrödinger
equation. The Born-Oppenheimer approximation states that
nuclei are much heavier and move much more slowly than
electrons. Thus, nuclear motions, vibrations and rotations
can be studied separately from electrons; the electrons are
assumed to move fast enough to adjust to any movement of the
nuclei.

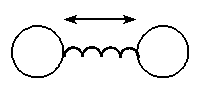
In a very crude sense molecular modeling treats a molecule
as a collection of wieghts connected with springs, where the
weights represent the nuclei and the springs represent the
bonds.
A force field is
used to calculate the energy and geometry of a molecule. It
is a collection of atom types (to define the atoms in a
molecule), parameters (for bond lengths, bond angles, etc.)
and equations (to calculate the energy of a molecule). In a
force field a given element may have several atom types. For
example, ethylbenzene contains both sp3-hybridized
carbons and aromatic carbons. sp3-Hybridized
carbons have a tetrahedral bonding geomtery, while aromatic
carbons have a trigonal bonding geometry. The C-C bond in
the ethyl group differs from a C-C bond in the phenyl ring,
and the C-C bond between the phenyl ring and the ethyl group
differs from all other C-C bonds in ethylbenzene. The force
field contains parameters for these different types of
bonds. Some of these parameters are given below. The total
energy of a molecule is divided into several parts called
force potentials, or potential energy equations. Force
potentials are calculated independently, and summed to give
the total energy of the molecule. Examples of force
potentials are the equations for the energies associated
with bond stretching, bond bending, torsional strain and van
der Waals interactions. These equations define the potential
energy surface of a molecule.
ETOTAL = ESTRETCH
+ EBEND + ES-B + ETORSION +
EvdW + EDP-DP
Below are examples of some
of the force potentials, and parameters one may find in a
force field. These examples are from Allinger's MM2 force
field [(a)"Conformational Analysis. 130. MM2. A
Hydrocarbon Force Field Utilizing V1 and V2
Torsional Terms", Allinger, N. L., J. Am. Chem. Soc.
1977, 99, 8127. (b) Burket, U.; Allinger, N.
L. Molecular Mechanics; American Chemical Society:
Washington, DC, 1982.] and the MMX force field of PCMODEL
["PCMODEL", Gilbert, K., Serena Software:
Bloomington, IN, 1993]
Energy due to Bond
Stretching
 Whenever
a bond is compressed or stretched the the energy goes up.
The energy potential for bond stretching and compressing is
described by an equation similar to Hooke's law for a
spring, except a cubic term is added. This cubic term helps
to keep the energy from rising too sharply as the bond is
stretched. Whenever
a bond is compressed or stretched the the energy goes up.
The energy potential for bond stretching and compressing is
described by an equation similar to Hooke's law for a
spring, except a cubic term is added. This cubic term helps
to keep the energy from rising too sharply as the bond is
stretched.
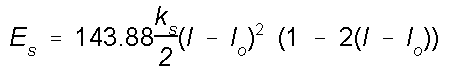
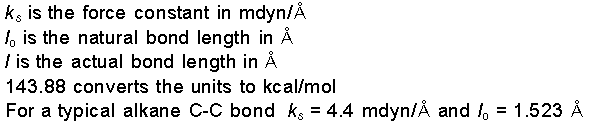
Energy due to Bond Angle
Bending
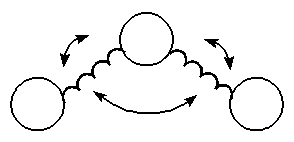
As angles are bent from their norm the energy increases. The
potential function below works very well for bends of up to
about 10 degrees. To handle special cases, such as
cyclobutane, special atom types and parameters are used in
the force field.
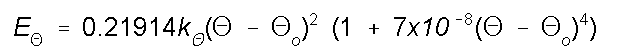
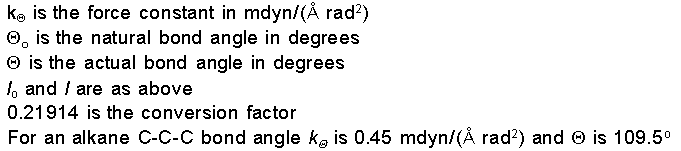
Energy due to Stretch-Bend
Interactions
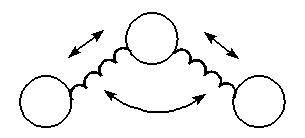
When a bond angle is
reduced the two bonds forming the angle will stretch to
alleviate the strain. To handle phenomena such as this,
cross term potential functions are introduced. Cross term
potential functions take into account at least two terms
such as bond stretching and bond bending.
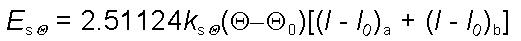
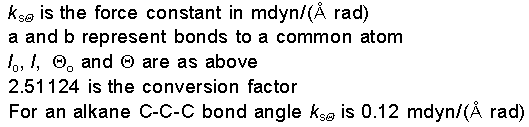
Energy due to Torsional
Strain

Intramolecular rotations (rotations about torsion or
dihedral angles) require energy. For example, it takes
energy for cyclohexane to go from the chair conformation to
the boat conformation. The torsion potential is a Fourier
series that accounts for all 1-4 through-bond relationships.
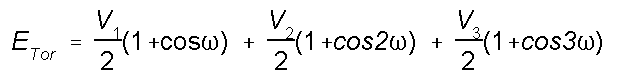
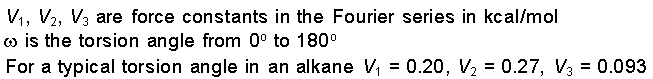
Energy due to van der
Waals Interactions
 The
van der Waals radius of an atom is its effective size. As
two non-bonded atoms are brought together the van der Waals
attraction between them increases (a decrease in energy).
When the distance between them equals the sum of the van der
Waals radii the attraction is at a maximum. If the atoms are
brought still closer together there is strong van der Waals
replusion (a sharp increase in energy). The
van der Waals radius of an atom is its effective size. As
two non-bonded atoms are brought together the van der Waals
attraction between them increases (a decrease in energy).
When the distance between them equals the sum of the van der
Waals radii the attraction is at a maximum. If the atoms are
brought still closer together there is strong van der Waals
replusion (a sharp increase in energy).
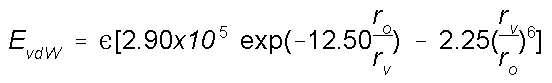

Energy due to
Dipole-Dipole Interactions
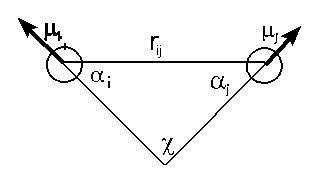
In some force fields electrostatic interactions are
accounted for by atomic point charges. In other force
fields, such as MM2 and MMX, bond dipole moments are used to
represent electrostatic contributions. One can readily see
that the equation below stems from Coulomb's law. The energy
is calculated by considering all dipole-dipole interactions
in a molecule. If the molecule has a net charge (e.g., NH4+),
charge-charge and charge-dipole calculations must also be
carried out.
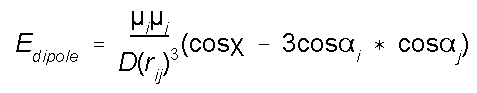
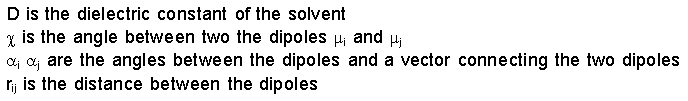
|