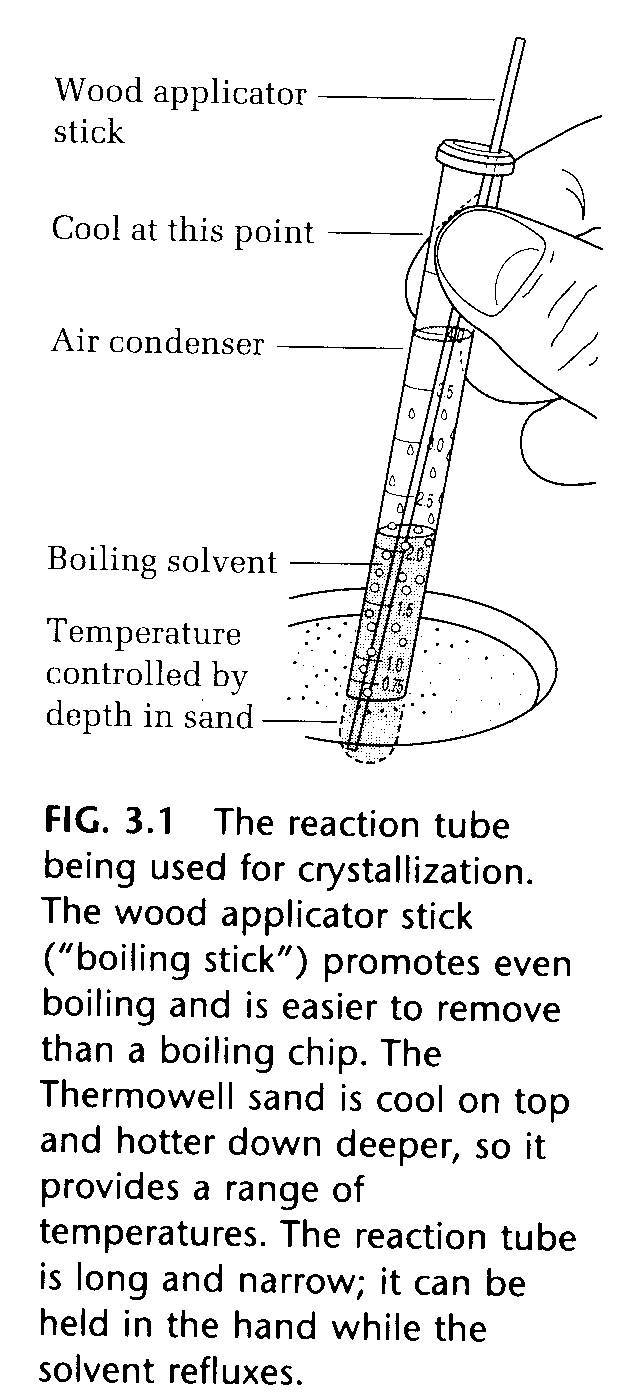
Crystallization: the most important purification method, especially for small-scale experiments.
On both a large and small scale, crystallization is the most important methods for the purification of solid organic compounds. A crystalline organic substance is made up of a three-dimensional array of molecules held together primarily by van der Waals forces. These intramolecular attractions are fairly weak; most organic solids melt in the range of from temperature to 250 degrees C.a
Crystals can be grown from the molten state just as water if frozen into ice, but it is not easy to remove impurities from crystals made in this way. Thus most purifications in the laboratory involve dissolving the material to be purified in the appropriate hot solvent. As the solvent cools, the solution becomes saturated with respect to the substance, which then crystallizes. As the perfectly regular array of a crystal is formed, foreign molecules are excluded, and thus the crystal is one pure substance. Soluble impurities stay in solution because they are not concentrated enough to saturate the solution. The crystals are collected by filtration, the surface of the crystals is washed with cold solvent to remove the adhering impurities, and then the crystals are dried. This process is carried out on an enormous scale in the commercial purification of sugar.
In the organic laboratory, crystallization is usually the most rapid and convenient method for purifying the products of a reaction. Initially, you will be told which solvent to use to crystallize a given substance and how much of it to use; later on, you will judge how much solvent is needed; and finally, the choice of both the solvent and its volume will be left to you. It takes both experience and knowledge to pick the correct solvent for a given purification.
The process of crystallization can be broken into seven discrete steps: choosing the solvent, dissolving the solute, decolorizing and washing the crystals, and drying the product. The process involves dissolving the impure substance in an appropriate hot solvent, removing some impurities by decolorizing and/or filtering the hot solution, allowing the substance to crystallize as the temperature of the solution falls, removing the crystallization solvent, and drying the resulting purified crystals.
Crystallization is initiated at a point of nucleation-a seed crystal, a spick of dust, or a scratch on the wall of the container if the solution is supersaturated with respect to the substance being crystallized (the solute). Super saturation will occur if a hot, saturated solution cools and crystals do not form. Large crystals, which are easy to isolate, are formed by nucleation and then slow cooling of the hot solution.
In choosing the solvent, the chemist is guided by the dictum "like dissolves like." Even the nonchemist knows that oil and water do not mix and that sugar and salt dissolve in water but not in oil. Hydrocarbon solvents such as hexane will dissolve hydrocarbons and other nonpolar compounds, and hydroxylic solvents such as water and ethanol will dissolve polar compounds. Often it is difficult to decide, simply by looking at the structure of a molecule, just how polar or nonpolar it is and therefore which solvent would be best. Therefore, the solvent is often chosen by experimentation.
The best crystallization solvent (and none is ideal) will dissolve the solute when the solution is hot but not when the solution is cold, it will either not dissolve the impurities at all or it will dissolve them very well (so they won't crystallize out along with the solute), it will not react with the solute, and it will be nonflammable, nontoxic, inexpensive, and very volatile (so it can be removed from the crystals).
Some common solvents and their properties are presented in Table 3.1 in order of decreasing polarity of the solvent. Solvents adjacent to each other in the list will dissolve in each other (that is, they are miscible with each other), and each solvent will, in general, dissolve substances that are similar to it in chemical structure. These solvents are used both for crystallization and as solvents in which reactions are carried out.
Water (H20--- Boiling Point (degrees C)---100--- The solvent of choice because it is cheap, nonflammable, nontoxic, and will dissolve a large variety of polar organic molecules. Its high boiling point and high heat of vaporization make it somewhat difficult to remove from the crystals.
Acetic acid (CH3COOH)---Boiling Point (degrees C)---118--- Will react with alcohols and amines. Difficult to remove. Not a common solvent for recrystallization, although used as a solvent when carrying our oxidation reactions.
Dimethyl sulfoxide (DMSO), methyl sulfoxide (CH3SOCH3)---Boling Point (degrees C)---189---Also not a commonly used solvent for crystallization, but used for reactions.
Methanol (CH3OH)---Boling Point (degrees C)---64--- A very good solvent, used often for crystallization. Will dissolve molecules of higher polarity than will the other alcohols
95% Ethanol (CH3CH2OH)---Boling Point (degrees C)---78---One of the most commonly used crystallization solvents. Its high boiling point makes it a better solvent for the less polar molecules than methanol. Evaporates readily from the crystals. Esters may undergo interchange of alcohol groups on recrystallization.
Acetone (CH3COCH3)---Boiling Point (degrees C)---56 An excellent solvent, but its low boiling point means there is not much difference in solubility of a compound at its boiling point and at room temperature.
2-Butanone, methyl ethyl ketone, MEK ( CH3COCH2CH3)---Boling Point (degrees C)---80--- An excellent solvent with many of the most desirable properties of a good crystallization solvent.
Ethyl acetate (CH3COOC2H5)---Boiling Point (degrees C)---78--- Another excellent solvent that has about the right combination of moderately high boiling point and yet the volatility needed to remove it from crystals.
Dichloromethane, methylene chloride (CH2CI2)---Boiling Point (degrees C)---40--- Although a common extraction solvent, dichloromethane boils too low to make it a good crystallization solvent. It is useful in a solvent pair with ligroin.
Diethyl ether, ether (CH3CH2OCH2CH3)---Boiling Point (degrees C)---35--- Its boiling point is too low for crystallization, although it is an extremely good solvent and fairly inert. Used in a solvent pair with ligroin.
t-Butyl methyl ether (CH3OC(CH3)3)---52--- A new solvent which is very inexpensive because of its large-scale industrial use as an antiknock agent in gasoline. Does not easily form peroxides. Higher boiling and thus a bit less flammable than diethyl ether. Used extensively for extraction throughout this text. A very good replacement for diethyl ether.
Dioxane (C4H8O2)---Boiling Point (degrees C)---101--- A very good solvent, not too difficult to remove form crystals; a mild carcinogen, forms peroxides.
Toluene (C6H5CH3)---Boling Point (degrees C)---111--- An excellent solvent that has replaced the formerly widely used benzene (a weak carcinogen) for crystallization of aryl compounds. Because of its boiling point it is not easily removed from crystals.
Pentane (C5H12)---Boiling Point (degrees C)---36--- A widely used solvent for nonpolar substances. Not often used alone for crystallization, but good in combination with a number of other solvents as part of a solvent pair.
Hexane (C6H14)---Boiling Point (degrees C)---69--- Similar in all respects to cyclohexane.
Cyclohexane (C6H12)---Boiling Point (degrees C)--- 81--- Frequently used to crystallize nonpolar substances. It is inert and has the correct balance between boiling point and volatility. Often used as part of a solvent pair. Cheaper than hexane.
Petroleum ether---Boiling Point (degrees C)---30-60--- A mixture of saturated hydrocarbons of which pentane is a chief component. Used interchangeably with pentane because it is cheap. Unlike diethyl ether, it is not an ether in the modern chemical sense.
Ligroin---Boiling Point (degrees C)---60-90--- A mixture of saturated hydrocarbons with the properties of hexane and cyclohexane. A very commonly use crystallization solvent. Equivalent to "hexanes."
TABLE 3.2 SOLVENT PAIRS
Acetic acid-water
Ethanol-water
Acetone-water
Dioxane-water
Acetone-ethanol
Ethanol-diethyl ether
Ethyl acetate-cyclohexane
Acetone-ligroin
Ethyl acetate- ligroin
Diethyl ether -ligroin
Dichloromethane-ligroin
Toluene - ligroin
PROCEDURE
Picking a Solvent. To pick a solvent for crystallization, put a few crystals of the impure solute in a reaction tube or centrifuge tube and add a very small drop of the solvent. Allow it to flow down the side of the tube and onto the crystals. If the crystals dissolve instantly at room temperature, that solvent cannot be used for crystallization because too much of the solute will remain in solution at low temperatures. If the crystals do not dissolve at room temperature, warm the tube on the hot sand bath and observe the crystals. If they do not go into solution, add a drop more solvent. If the crystals go into solution at the boiling point of the solvent and then crystallize when the tube is cooled, you have found a good crystallization solvent. If not, remove the solvent by evaporation and try another solvent. In this trial-and -error process it is easiest to try low-boiling solvents first, because they can be removed most easily. Occasionally, no single satisfactory solvent can be found, so mixed solvents, or solvent pairs, are used. Solvent Pairs To use a mixed solvent, dissolve the crystals in the better solvent and add the poorer solvent to the hot solution until it becomes cloudy and the solution is saturated with the solute. The two solvents must, of course, be miscible with each other. Some useful solvent pairs are given in Table 3.2.
2. Dissolving the Solute
Micro scale Procedure
Once a crystallization solvent has been found, the impure crystals are placed in a reaction tube, solvent is added drop wise, the crystals are stirred with a micro spatula or a small glass rod, and the tube is warmed on a steam bath or sand bath with the addition of more solvent until the crystals dissolve. A solution of this type can become superheated, that is, heated above its boiling point without actually boiling. When boiling does suddenly occur, it can happen with almost explosive violence. To prevent this from happening, a wood applicator stick can be added to the solution (Fig. 3.1). Air trapped in the wood comes out of the stick and forms the nuclei on which even boiling can occur. Porous porcelain boiling chips work in the same way. Never add a boiling chips or boiling stick to a hot solution, since it may be superheated and boil over.
To remove impurities (insoluble by-products of reaction, lint, dust, etc.), it is necessary to dilute the solution with excess solvent (the better solvent if a solvent mixture is being used), carry out the filtration neat room temperature, and then evaporate the solvent to a point at which the hot solution is once more saturated with respect to the solute so that crystallization can take place in the usual way. This is described in step 4.
Care must be exercised to use the correct amount of solvent. Observe the mixture carefully as solvent is being added. Allow sufficient time for the boiling solvent to dissolve the solute, and note the rate at which most of the material dissolves. When you believe most of the material has been dissolved, stop adding solvent. There is the possibility that your sample is contaminated with a small quantity of an insoluble impurity which never well dissolve. To hasten the solution process, crush large crystals with a stirring rod, taking care not to break the tube. On a micro scale, there is a tendency to use too much solvent so that on cooling the hot solution little or no material crystallizes. This is not a hopeless situation. The remedy is to evaporate some of the solvent and repeat the cooling process. Inspect the hot solution. If it contains no undissolved impurities and is not colored from impurities, you can simply let it cool, allowing the solute to crystallize (step 5) and then collect the crystals (step 6). On the other hand, if the solution is colored, it must be treated with activated (decolorizing) charcoal and then filtered before crystallization (step 3). If it contains solid impurities, it must be filtered before crystallization takes place (step 4).
MACROSCALE PROCEDURE
Place the substance to be crystallized in an Erlenmeyer flask (never use a beaker), add enough solvent to cover the crystals, and then heat the flask on a steam bath (if solvent boils below 90 degrees C) or a got plate until the solvent boils. Stir the mixture or, better, swirl it (Fig.3.2)
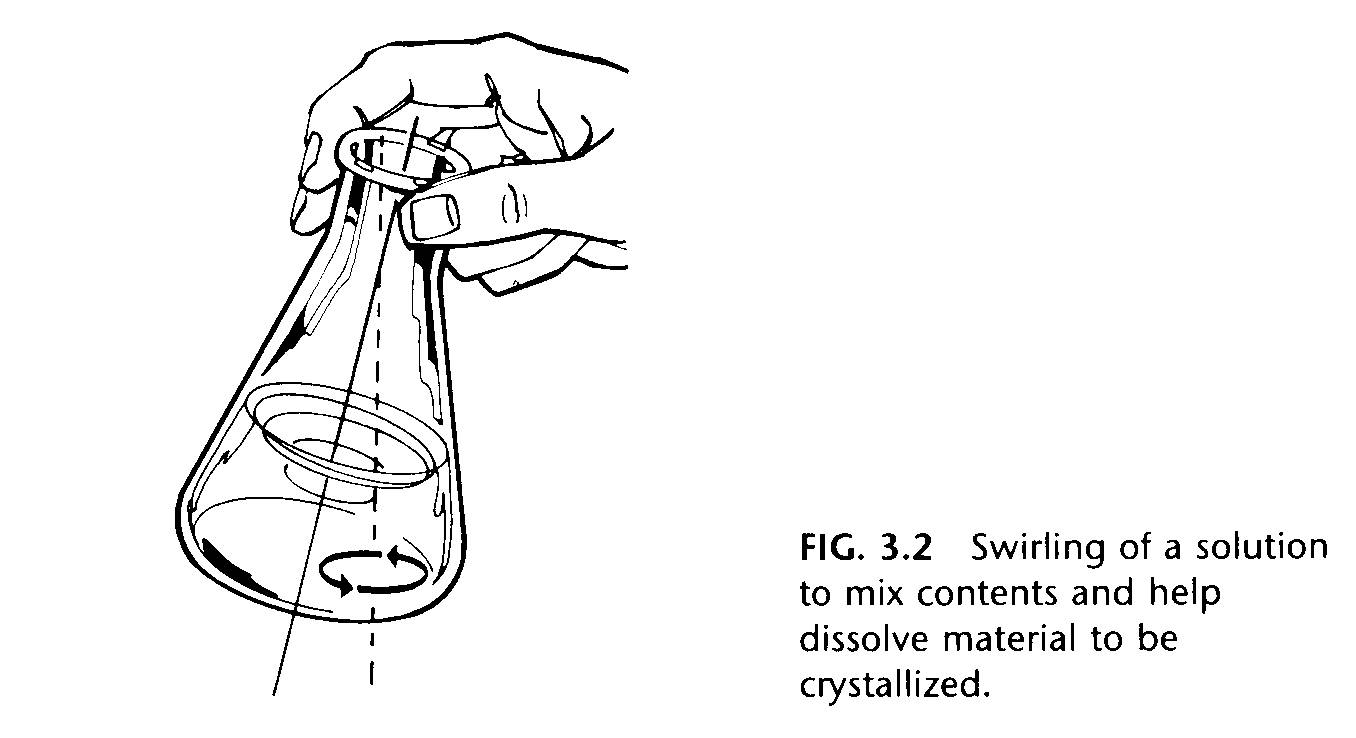
to promote dissolution. Add solvent gradually, keeping it at the boil, until all the solute dissolves. Addition of a boiling stick or a boiling chip to the solution once most of the solid is gone will promote even boiling. It is not difficult to superheat the solution, that is, heat it above the boiling point with no boiling taking place. Once the solution does boil, it does so with explosive violence. Never add a boiling chip or boiling stick to a hot solution. A glass rod with a flattened end can sometimes be of use in crushing large particles of solute to speed up the dissolving process. Be sure no flames are nearby when working with flammable solvents.
Be careful not to add too much solvent. Note how rapidly most of the material dissolves and then stop adding solvent when you suspect that almost all the desired material has dissolve. It is best to err on the side of too little solvent rather than too much. Undissolved material noted at this point could be an insoluble impurity, which never will dissolve. Allow the solvent to boil, and if no further material dissolves, proceed to step 4 to remove suspended solids from the solution by filtration, or if the solution is colored, go to step3 to carry out the decolorization process. If the solution is clear, proceed to step 5, crystallizing the solute.
3. DECOLORIZING THE SOLUTION; USE OF PELLETIZED NORIT
The vast majority of pure organic chemicals are colorless or a light shade of yellow. Occasionally, a chemical reaction will produce high-molecular-weight-by-products that are highly colored. The impurities can be adsorbed onto the surface of activated charcoal simply by boiling the solution with charcoal. Activated charcoal has an extremely large surface area per gram (several hundred square meters) and can bind a large number of molecules to this surface. On a commercial scale, the impurities in brown sugar are adsorbed onto charcoal in the process of refining sugar.
In the past, laboratory manuals have advocated the use of finely powdered activated charcoal for removal of colored impurities. This has two drawbacks. Because the charcoal is so finely divided, it can only be separated from the solution by filtration through paper, and even then some of the finer particles pass through the filter paper. And the presence of the charcoal completely obscures the color of the solution so that adding the correct amount of charcoal is mostly a matter of luck. If too little charcoal is added, the solution will still be colored after filtration, making repetition necessary; if too much is added, it will absorb some of the product in addition to the impurities. We have found that charcoal extruded as short cylindrical pieces measuring about 0.8 X 3 mm made by the Norit Company solves both these problems. It works just as well as the finely divided powder, and it does not obscure the color of the solution. It can be added in small portions until the solution is decolorized, and the size of the pieces makes it easy to remove from the solution.
On both a micro scale and a macroscale, simply add a small amount (0.1% of the solute weight is sufficient) of pellitized Norit to the colored solution and then boil the solution for a few minutes. Be careful not to add the charcoal pieces to a superheated solution; the charcoal functions like hundreds of boiling chips and will cause the solution to boil over. Remove the Norit by filtration as described in step
4. FILTERING SUSPENDED SOLIDS
The filtration of a solution to remove solid impurities or charcoal can be done in a number of ways. Process includes gravity filtration, pressure filtration, decantation, or removal of the solvent using a Pasteur pipette. Vacuum filtration is not used because the hot solvent will cool during the process and the product will crystallize in the filter.
MICROSCALE PROCEDURE
(A) Removal of Solution with a Pasteur Pipette. If the solid impurities are large in size, they can be removed by filtration of the liquid through the small space between the square end of a Pasteur pipette and the bottom of a reaction tube (Fig. 3.3).
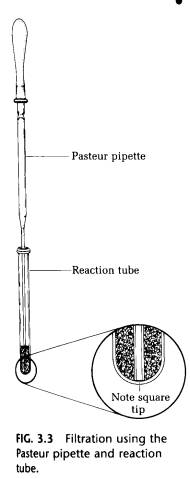
Expel air from the pipette as it is being pushed to the bottom of the tube. Use a small additional quantity of solvent to rinse the tube and pipette. Anhydrous calcium chloride, a drying agent, is removed easily in this way. Removal of very fine material, such as traces of charcoal, is facilitated by filtration of the solution through a small piece of filter paper (3 mm2) placed in the reaction tube. This process is even easier if the filter paper is the thick variety, such as that from which Soxhlet extraction thimbles are made.
(B) Filtration in a Pasteur Pipette. To filter 0.1 to 2 mL of a solution, dilute the solution with enough solvent that the solute will not crystallize out a room temperature. Prepare a filter pipette by pushing a tiny bit of cotton into a Pasteur pipette, put the solution to be filtered into this filter pipette using another Pasteur pipette, and then forces the liquid through the filter using air pressure from a pipette bulb (Fig. 3.4).
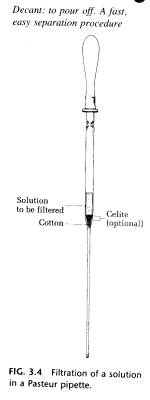
Fresh solvent should be added to rinse the pipette and cotton. The filtered solution is then concentrated by evaporation. One problem encountered with this method is using too much cotton packed too tightly in the pipette so that the solution cannot be forced through it. To remove very fine impurities, such as traces of decolorizing charcoal, a 3- to 4-mm layer of Celite filter aid can be added to the top of the cotton.
(C) Pressure Filtration with Micro Buchner Funnel. The technique applicable to volumes from 0.1 to 5 mL is the use of the micro Buchner funnel. It is made of polyethylene and is fitted with a porous polyethylene frit 6 mm in diameter. This funnel fits in the bottom of an inexpensive disposable polyethylene pipette in which a hole is cut (Fig. 3.5.).
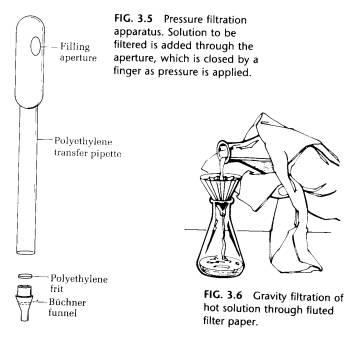
The solution to be filtered is placed in the pipette using a Pasteur pipette. The thumb covers the hole in the plastic pipette, and pressure is applied to filter the solution. It is good practice to place a 6-mm- diameter piece of filter paper over the frit, which can otherwise become clogged with insoluble material.
The glass chromatography column can be used in the same way. A piece of filter paper is placed over the frit. The solution to be filtered is placed in the chromatography column, and pressure is applied to the solution using a pipette bulb. In both procedures, dilute the solution to be filtered so that it does not crystallize out in the apparatus, and use a small amount of clean solvent to rinse the apparatus. The filtered solution is then concentrated by evaporation.
MACROSCALE PROCEDURE
(A) Decantation. On a large scale, it is often possible to pour off (decant) the hot solution leaving the insoluble material behind. This is especially easy if the solid is granular, like sodium sulfate. The solid remaining in the flask and the inside of the flask should be rinsed with a few milliliters of the solution in order to recover as much of the product as possible.
(B) Gravity Filtration. The most common method for the removal of insoluble solid material is gravity filtration through a fluted filter paper (Fig. 3.6.). This is the method of choice for the removal of finely divided charcoal, dust, lint, etc. The following equipment will be needed for this process: three Erlenmeyer flasks on a steam bath or hot plate, on to contain the solution to be filtered, one to contain a few milliliters of solvent and a stem less funnel, and the third to contain several milliliters of the crystallizing solvent to be used for rinsing purposes; a fluted piece of filter paper; a towel for holding the hot flask and drying out the stem less funnel; and boiling chips for all solutions.
A piece of filter paper is fluted as shown in Fig. 3.7.
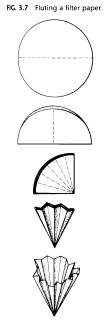
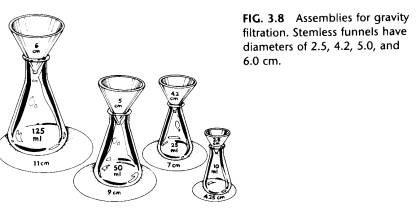
and is then placed in a stem less funnel. Appropriate sizes of Erlenmeyer flasks, stem less funnels, and filter paper are shown in Fig. 3.8.
The funnel is stem less so that the saturated solution being filtered will not have a chance to cool and clog the stem with crystals. The filter paper should fit entirely inside the rim of the funnel; it is fluted to allow rapid filtration. Test to see that the funnel is stable in the neck of the Erlenmeyer flask. If it is not, support it with a ring attached to a ring stand. A few milliliters of solvent and a boiling chip should be placed in the flask into which the solution is to be filtered. This solvent is brought to a boil on the steam bath or hot plate along with the solution to be filtered.
The funnel should be warm in order to prevent crystallization from occurring in the funnel. This can be accomplished in two ways: Invert the funnel over a steam bath for a few seconds. Pick up the funnel with a towel, wipe it perfectly dry, place it on top of the Erlenmeyer flask, and add the fluted filter paper. Alternatively, the stem less funnel is placed in the neck of the Erlenmeyer flask, and solvent is allowed to reflux into the funnel, thereby warming it.
The solution to be filtered should be saturated with the solute at the boiling point. Note the volume and then add 10% more solvent. The resulting slightly dilute solution is not so likely to crystallize out in the funnel in the process of filtration. Bring the solution to be filtered to a boil, grasp the flask in a towel, and pour the solution into the filter paper in the stem less funnel (see Fig. 3.6).
Pour the solution to be filtered at a steady rate into the fluted filter paper. Check to see whether crystallization is occurring in the filter. If the solute does crystallize, add boiling solvent (from the third Erlenmeyer flask heated on the steam bath or hot plate) until the crystals dissolve, dilute the solution being filtered, and carry on. Rinse the flask that contained the solution to be filtered with a few milliliters of boiling solvent, and rinse the fluted filter paper with this same solvent.
Since the filtrate has been diluted in order to prevent it from crystallizing during the filtration process, the excess solvent must now be removed by boiling the solution. The process can be speeded up somewhat by blowing a slow current of air into the flask in the hood or using an aspirator tube to pull vapors into the aspirator (Figs. 3.9 and 3.10). However, the fastest method is to heat the solvent in the filter flask on the sand bath while the flask is connected to the water aspirator. The vacuum is controlled with the thumb (Fig. 3.11).
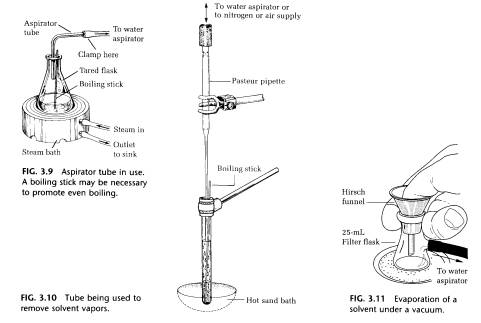
5. CRYSTALLIZING THE SOLUTE
On both a macroscale and a micro scale, the crystallization process should normally start from a solution that is saturated with the solute at the boiling point. If it has been necessary to remove impurities or charcoal by filtration, the solution has been diluted. To concentrate the solution, simply boil off the solvent under an aspirator tube as shown in Fig. 3.9 (macroscale) or blow off solvent using a gentle stream of air or, better, nitrogen in the hood as shown in Fig. 3.10 (micro scale). Be sure to have a boiling chip (macroscale) or a boiling stick (micro scale) in the solution during this process, and then do not forget to remove it before initiating crystallization.
Once it has been ascertained that the hot solution is saturated with the compound just below the boiling point of the solvent, it is allowed to cool slowly to room temperature. Crystallization should begin immediately. If is does not add a seed crystal or scratch the inside of the tube with a glass rod at the liquid-air interface. Crystallization must start on some nucleation center. A minute crystal of the desired compound saved from the crude material will suffice. If a seed crystal is not available, crystallization can be started on the rough surface of a fresh scratch on the inside of the container.
Once it is ascertained that crystallization has started, the solution must be cooled slowly without disturbing the container in order that large crystals can form. On a micro scale, it is best to allow the reaction tube to cool in a beaker filled with cotton or paper towels, which act as insulation so cooling takes place slowly. Even insulated in this manner, the small reaction tube will cool to room temperature within a few minutes. Slow cooling will guarantee the formation of large crystals, which are easily separated by filtration and easily washed free of adhering impure solvent. On a small scale, it is difficult to obtain crystals that are too large and occlude impurities. Once the tube has cooled to room temperature without disturbance, it can be cooled in ice to maximize the amount of product that comes out of solution. The crystals are then separated from the mother liquor (the filtrate) by filtration On a macroscale, the Erlenmeyer flask is set atop a cork ring or other insulator and allowed to cool spontaneously to room temperature. If the flask is moved during crystallization, many nuclei will form and the crystals will be small and will have a large surface area. They will not be so easy to filter and wash clean of mother liquor. Once crystallization ceases at room temperature, the flask should be placed in ice to cool further. Take care to clamp the flask in the ice bath so that id does not tip over.
6. COLLECTING AND WASHING THE CRYSTALS
Once crystallization is complete, the crystals must be separated from the ice-cold mother liquor, washed with ice-cold solvent, and dried. Micro scale Procedure
(A) Filtration Using The Hirsch Funnel. When the volume of material to be filtered is larger than about 1.5 mL, then the material is collected on the Hirsch funnel.
The Hirsch funnel in the Williamson/Kontes kit is unique. It is made of polypropylene and has an integral molded stopper, which fits the 25-mL filter flask. It comes fitted with a 20-um polyethylene fritted disk, which is not meant to be disposable, although it costs only about twice as much as an 11-cm piece of filter paper (Fig. 3.12).
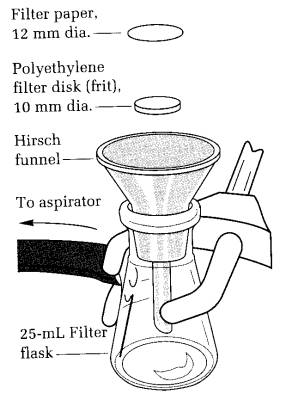
While products can be collected directly on this disk, it is good practice to place an 11- or 12-mm-diameter piece of no. 1 filter paper on the disk. In this way the frit will not become clogged with insoluble impurities. The disk of filter paper can be cut with a cork borer or leather punch. A piece of filter paper must be used on the old-style porcelain Hirsch funnels.
Clamp the clean, dry 25-mL filter flask in an ice bath to prevent it from falling over, and place the Hirsch funnel with filter paper in the flask. Wet the filter paper with the solvent used in the crystallization, turn on the water aspirator (see above), and ascertain that the filter paper is pulled down onto the frit. Pour and scrape the crystals and mother liquor onto Hirsch funnel, and as soon as the liquid is gone from the crystals, break the vacuum at the filter flask by removing the rubber hose. The filtrate can be used to rinse out the container, which contained the crystals. Again, break the vacuum as soon as all the liquid has disappeared from the crystals; this prevents impurities from drying on the crystals. The reason for cooling the filter flask is to keep the mother liquor cold so that it will not dissolve the crystals on the Hirsch funnel when the filtrate used is used to wash crystals from the container onto the funnel. With a very few drops of ice-cold solvent, rinse the crystallization flask. This container should still be ice cold. Place the ice-cold solvent on the crystals and then reapply the vacuum. As soon as the liquid is pulled from the crystals, break the vacuum. Repeat this washing process as many times as necessary to remove colored material or other impurities from the crystals. In some cases, only one very small wash will be needed. After the crystals have been washed with ice-cold solvent, the vacuum can be left on to dry the crystals. Sometimes it is useful to press solvent from the crystals using a cork.
(B) FILTRATION INTO A REACTION TUBE ON THE HIRCH FUNNEL. If it is desired to have the filtrate in a reaction tube instead of spread all over the bottom of the 25-mL filter flask, then the process described above can be carried out in the apparatus shown in Fig. 3,13.
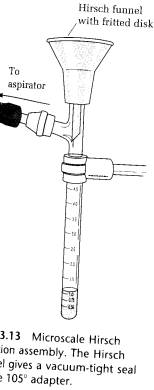
The vacuum hose is connected to the side arm using the thermometer adapter and a short length of glass tubing. Evaporate the filtrate in the reaction tube to collect a second crop of crystals.
Fig. 3.12 Hirsch funnel used for vacuum filtration. The unique design has a removable and replaceable 20-um polyethylene frit. No adapter is needed because there is a vacuum-tight fit to the filter flask. Always clamp the flask.
(C) FILTRATION INTO A REACTION TUBE ON THE MICRO BUCHNER FUNNEL. If the quantity of material being collected is very small, the bottom of the chromatography column is a micro Buchner funnel, which can be fitted into the top of the thermometer adapter as, shown in Fig. 3.14. Again, it is good practice to cover the frit with a piece of 6-mm filter paper (cut with a cork borer).
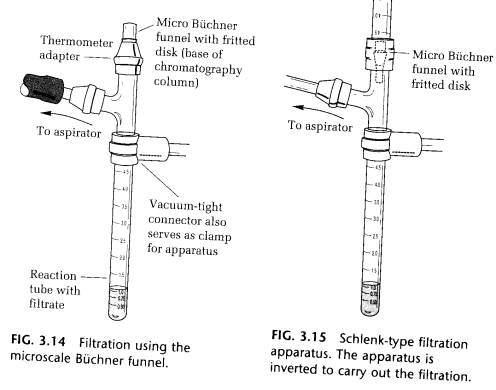
(D) THE MICRO BUCHNER FUNNEL IN AN ENCLOSED FILTRATION APPARATUS. In the apparatus shown in Fig. 3.15, crystallization is carried out in the upper reaction tube in the normal way. The apparatus is then turned upside down, the crystals shaken down onto the micro Buchner funnel, and a vacuum applied through the side arm. In this apparatus, crystals can be collected in an oxygen-free atmosphere. This resembles an Schlenk tube.
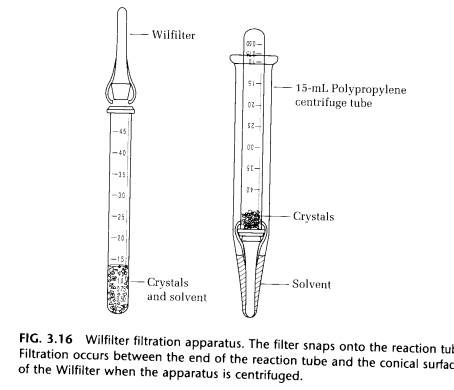
(E) FILTRATION WITH THE WILFILTER. Another method of isolating small quantities of crystals is to centrifuge them. This works particularly well when the crystals are very small and difficult to scrape from the reaction tube. The Wilfilter is snapped over the end of the reaction tube, and the assembly is inverted in a 15-mL polypropylene centrifuge tube and centrifuge for about 60 s. The solvent escapes between the reaction tube and the conical surface of the Wilfilter, leaving the crystals on the top of the Wilfilter and the filtrate in the centrifuge tube. The crystals in this way can be quite dry, depending on the speed and duration of the centrifugation (Fig. 3.16).
(F) FILTRATION USING THE PASTEUR PIPETTE. The most important filtration technique to be used in micro scale organic experiments employs the Pasteur pipette (see Fig. 3.3. About 70% of the crystalline products from the experiments in this text can be isolated in this way. The others will be isolated by filtration on the Hirsch funnel.
The ice-cold crystalline mixture is stirred with the Pasteur pipette, and while air is being expelled form the pipette, it is forced to the bottom of the reaction tube. The bulb is released, and the solvent is drawn into the pipette through the very small space between the square tip of the pipette and the curved bottom of the reaction tube. When all the solvent has been withdrawn, it is expelled into another reaction tube containing the crystals. It is sometimes useful to rap the tube containing the wet crystals against a hard surface to pack them so that more solvent can be removed. The tube is returned to the ice bath, and a few drops of cold solvent are added to the crystals. The mixture is stirred to wash the crystals, and the solvent is again removed. This process can be repeated as many times as necessary.
Fig. 3.17 Matching filter assemblies. The 6.0-cm polypropylene Buchner funnel (right) resists breakage and can be disassembled for cleaning.

MACROSCALE APPARATUS
Filtration on the Hirsch Funnel and the Buchner Funnel. If the quantity of material is small (less than 2g), the Hirsch funnel can be used in exactly the way described above. For larger quantities, the Buchner funnel is used. Properly matched Buchner funnels, filter paper, and flasks are shown in Fig. 3.17. The Hirsch funnel shown in the figure has a 5-cm bottom plate to accept 3.3-cm paper.
Place a piece of filter paper in the bottom of the Buchner funnel. Wet if with solvent, and be sure it lies flat so that crystals cannot escape around the edge and under the filter paper. Then with the vacuum off, pour the cold slurry of crystals into the center of the filter paper. Apply the vacuum as soon as the liquid disappears from the crystals; break the vacuum to the flask by disconnecting the hose. Rinse the Erlenmeyer flask with cold solvent. Add this to the crystals, and reapply the vacuum just until the liquid disappears from the crystals. Repeat this process as many times as necessary, and then leave the vacuum on to dry the crystals. NOTE: Clamp the filter flask. Where water is scarce, a large (50-100 mL) plastic syringe is an excellent source for the vacuum needed for the 25-mL filter flask. Use a trap.
THE WATER ASPIRATOR AND TRAP. The most common way to produce a vacuum in the organic laboratory for filtration purposes is by employing a water aspirator. Air is efficiently entrained in the water rushing through the aspirator, so that it will produce a vacuum roughly equal to the vapor pressure of the water going through it (17 mm Hg at 20 degrees C, 5mm Hg at 4 degrees C). A check valve is built into the aspirator, but even so, when the water is turned off, it may back into the evacuated system. For this reason, a trap is always installed in the line (Fig. 3.18).
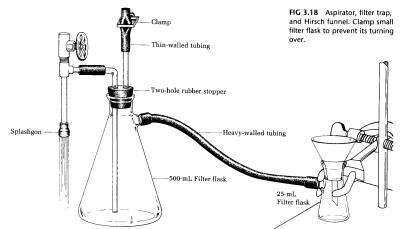
The water passing through the aspirator should always be turned on full force. The system can be opened to the atmosphere by removing the hose from the small filter flask or by opening the screw clamp on the trap. Open the system, then turn off the water to avoid having water suck back into the filter trap. Thin rubber tubing on the top of the trap will collapse and bend over when a good vacuum is established. You will, in time, learn to hear the difference in the sound of an aspirator when it is pulling a vacuum and when it is working on an open system.
Collection a Second Crop of Crystals. Regardless of the method used to collect the crystals on either a macroscale or a micro scale, the filtrate and washings can be combined and evaporated to the point of saturation to obtain a second crop of crystals, hence the necessity for having a clean receptacle for the filtrate. This second crop will increase the overall yield, but the crystals will not usually be as pure as the first crop.
7. DRYING THE PRODUCT
Micro scale Procedure
If possible, dry the product in the reaction tube after removal of the solvent using a Pasteur pipette. This can be done simply by connecting the tube to the water aspirator. If the tube is clamped in a beaker of hot water, the solvent will evaporate more rapidly under vacuum, but take care not to melt the product (Fig. 3.19).
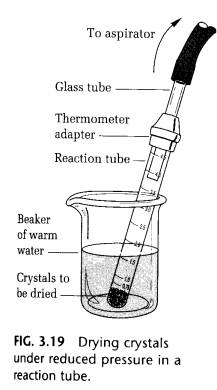
Water, which has a high heat of vaporization, is difficult to remove this way. Scrape the product out onto a watch glass and allow it to dry to constant weight, which will indicate that all the solvent is gone. If the product is collected on the Hirsch funnel, the last bit of solvent can be remove by squeezing the crystals between sheets of filter paper before drying them on the watch glass.
Macroscale Procedure
Once the crystals have been washed on the Hirsch funnel or the Buchner funnel, press them down with a clean cork or other flat object and allow air to pass through them until they are substantially dry. Final drying can be done under reduced pressure (Fig. 3.20).
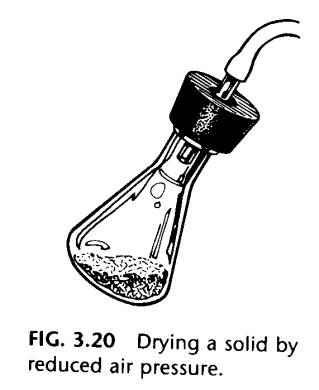
The crystals can then be turned out of the funnel and squeezed between sheets of filter paper to remove the last bit of solvent before final drying on a watch glass.
EXPERIMENTS
1. SOLUBILITY TESTS FOR CRYSTALLIZATION
To test the solubility of a solid, transfer an amount roughly estimated to be about 10 mg (the amount that forms a symmetrical mound on the end of a stainless steel spatula) into a reaction tube or a 10 X 75 mm test tube and add about 0.25 mL of solvent from a calibrated dropper or pipette. Stir with a fire-polished stirring rod (4 mm), break up any lumps, and determine if the solid is readily soluble at room temperature. If the substance is readily soluble in methanol, ethanol, acetone, or acetic acid at room temperature, add a few drops of water from a wash bottle to see if a soled precipitates. If it does, heat the mixture, adjust the composition of the solvent pair to produce a hot solution saturated at the boiling point, let the solution stand undisturbed, and note the character of the crystals that form. If the substance fails to dissolve in a given solvent at room temperature, heat the suspension and see if solution occurs. If the solvent is flammable, heat the test tube on the steam bath or in a small beaker of water kept warm on the steam bath or a hot plate. If the solid completely dissolves, it can be declared readily soluble in the hot solvent; if some but not all dissolves, it is said to be moderately soluble, and further small amounts of solvent should then be added until solution is complete. When a substance has been dissolved in hot solvent, cool the solution by holding the flask under the tap, and if necessary, induce crystallization by rubbing the walls of the tube with a stirring rod to make sure that the concentration permits crystallization. Then reheat to dissolve the solid, let the solution stand undisturbed, and inspect the character of the ultimate crystals.
Make solubility tests on the four test compounds shown on the next page in each of the solvents listed after them. Note the degree of solubility in the solvents, cold and hot, and suggest suitable solvents, solvent pairs, or other expedients for crystallization of each substance. Record the crystal form, at least to the extent of distinguishing between needles (pointed crystals), plates (flat and thin), and prisms. How do your observations conform to the generalization that like dissolves like?
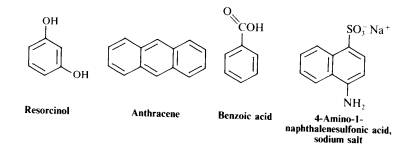
Solvents
Water-hydroxylic, ionic
Toluene-an aromatic hydrocarbon
Ligroin- a mixture of aliphatic hydrocarbons
CLEANING UP
Place organic solvents and solution of the compound in the organic solvents container. Dilute the aqueous solutions with water and flush down the drain.
2. CRYSTALLIZATION OF PURE PHTALIC ACID, NAPHTHALENE, AND ANTHRACENE The process of crystallization can be observed readily using phthalic acid. In the reference book The Handbook of Chemistry and Physics, in the "Physical Constants of Organic Compounds," the entry for phthalic acid gives the following solubility data (in grams of solute per 100 mL of solvent). The superscripts refer to temperature in degrees C. Water 0.54(14) 18(99)
Alcohol 11.71(18)
Ether, etc. 0.69(15) eth., i. chl.
The large difference in solubility in water as a function of temperature suggests this as the solvent of choice. The solubility in alcohol is high at room temperature. Ether is difficult to use because it is so volatile; the compound in insoluble in chloroform (i. chl.).
MICROSCALE PROCEDURE, PHTHALIC ACID
Crystallize 60 mg (0.060 g) of phthalic acid from the minimum volume of water, using the above data to calculate the required volume. First, turn on the electrically heated sand bath. Add the solid to a 10 X 100mm reaction tube, and then, using a Pasteur pipette, add water drop wise. Use the calibration marks found in Fig. 1.18 to measure the volume of water in the pipette and the reaction tube. Add a boiling stick (a wooden applicator stick) to facilitate even boiling and prevent bumping. After a portion of the water has been added, gently heat the solution to boiling on a hot sand bath in the electric heater. The deeper the tube is placed in the sand, the hotter it will be. As soon as boiling begins, continue to add water drop wise until all the solid just dissolves. Cork the tube and clamp it as it cools, and observe the phenomenon of crystallization.
After the tube reaches room temperature, cool it in ice, stir the crystals with a Pasteur pipette, and expel the air from the pipette as the tip is pushed to the bottom of the tube. When the tip is firmly and squarely seated in the bottom of the tube, release the bulb and withdraw the water. Rap the tube sharply on a wood surface to compress the crystals and remove as much of the water as possible with the pipette. Then cool the tube in ice and add a few droops of ice-cold ethanol to the tube in order to remove water from the crystals. Connect the tube to a water aspirator and warm it in a beaker of got water (see Fig. 3.19). Once all the solvent is removed, using the stainless steel spatula, scrape the crystals onto a piece of filter paper, fold the paper over the crystals, and squeeze out excess water before allowing the crystals to dry to constant weight. Weigh the dry crystals, and calculate the percent recovery of product.
MICROSCALE PROCEDURE, NAPHTHALENE AND ANTHRACENE
Following the procedure outlined above, crystallize 40 mg of naphthalene from 80% aqueous methanol or 10mg of anthracene from ethanol. These are more typical of compounds to be crystallized in later experiments in that they are soluble in organic solvents. It will be much easier to remove these solvents from the crystals under vacuum than it is to remove water from phthalic acid. You will seldom have occasion to crystallize less than 30mg of a solid in these experiments.
CLEANING UP Dilute the aqueous filtrate with water, and flush the solution down the drain. Phthalic acid is not considered toxic to the environment. Methanol and ethanol filtrate go in the organic solvents container.
MACROSCALE PROCEDURE
Crystallize 1.0 g of phthalic acid from the minimum volume of water, using the above data to calculate the required volume. Add the solid to the smallest practical Erlenmeyer flask, and then, using a Pasteur pipette, add water drop wise from a full 100-mL graduated cylinder. A boiling stick (a stick of wood) facilitates even boiling and will prevent bumping. After a portion of the water has been added, gently heat the solution to boiling on a got plate. As soon as boiling begins, continue to add water drop wise until all the solid just dissolves. Place the flask on a cork ring or other insulator and allow it to cool undisturbed to room temperature, during which time the crystallization process can be observed. Slow cooling favors large crystals. Then cool the flask in an ice bath, decant (pour off) the mother liquor (the liquid remaining with the crystals), and remove the last traces of liquid with a Pasteur pipette. Scrape the crystals onto a filter paper using a stainless steel spatula, squeeze the crystals between sheets of filter paper to remove traces of moisture, and allow the crystals to dry. Alternatively, the crystals can be collected on a Hirsch funnel. Compare the calculated volume of water with the volume of water actually used to dissolve the acid. Calculate the percent recovery of dry, recrystallized phthalic acid.
CLEANING UP Dilute the filtrate with water, and flush the solution down the drain. Phthalic acid is not considered toxic to the environment.
3. DECOLORIZING A SOLUTION WITH DECOLORIZING CHARCOAL
Into a reaction tube place 1.0mL of a solution of methylene blue dye that has been made up at a concentration of 10mg per 100mL of water. Add to the tube a few pieces (10 or 12) of decolorizing charcoal, shake, and observe the color over a period of a minute or two. Heat the contents of the tube to boiling (reflux), and observe the color by holding the tube in front of a piece of white paper from time to time. How rapidly is the color removed? If the color is not removed in a minute or so, add more charcoal pellets.
CLEANING UP Place the Norit in the no hazardous solid waste container.
4. DECOLORIZATION OF BROWN SUGAR (SUCROSE, C12 H22 011)
Raw sugar is refined commercially with the aid of decolorizing charcoal. The clarified solution is seeded generously with small sugar crystals, and excess water removed under vacuum to facilitate crystallization. The pure white crystalline product is collected by centrifugation. Brown sugar is partially refined sugar and can be decolorized easily using charcoal.
Dissolve 15g of dark brown sugar in 30mL of water in a 50-mL Erlenmeyer flask by heating and stirring. Pour half the solution into another 50-mL flask. Heat one of the solutions nearly to the boiling point, allow it to cool slightly, and add to it 250 mg (0.25 g) of decolorizing charcoal (Norit pellets). Bring the solution back to; near the boiling point for 2 minutes; then filter the hot solution into an Erlenmeyer flask through a fluted filter paper held in a previously heated funnel. Treat the other half of the sugar solution in exactly the same way but use only 50 mg of decolorizing charcoal. In collaboration with a fellow student, try heating the solutions for only 15 s after addition of the charcoal. Compare you results.
CLEANING UP Decant (pour off) the aqueous layer. Place the Norit in the nonhazardous solid waste container. The sugar solution can be flushed down the drain.
5. CRYSTALLIZATION OF BENZOIC ACID FROM WATER AND A SOLVENT PAIR
Crystallize 50mg of benzoic acid from water in the same way phthalic acid was crystallized. Then in a dry reaction tube dissolve another 50-mg sample of benzoic acid in the minimum volume of hot toluene and add cyclohexane to the hot solution drop wise. When the hot solution becomes cloudy and crystallization has started, allow the tube to cool slowly to room temperature; then cool it in ice and collect the crystals. Compare crystallization in water to that in the solvent pair.
CLEANING UP The aqueous solution, after dilution with water, can be flushed down the drain. The toluene and cyclohexane filtrates should be placed in the organic solvents container. 6. RECRYSTALLIZATION OF NAPHTHALENE FROM A MIXED SOLVENT
Add 2.0 g of impure naphthalene to a 50-mL Erlenmeyer flask along with 3 mL of methanol and a boiling stick to promote even boiling. Heat the mixture to boiling over a steam bath or hot plate and then add methanol drop wise until the naphthalene just dissolves when the solvent is boiling. The total volume of methanol should be 4 mL. Remove the flask from the heat and cool it rapidly in an ice bath. Note that the contents of the flask set to a solid mass, which would be impossible to handle. Add enough methanol to bring the total volume to 25 mL, heat the solution to the boiling point, remove the flask from the heat, allow it to cool slightly, and add 30mg of decolorizing charcoal pellets to remove the colored impurity in the solution. Heat the solution to the boiling point for 2 minutes; if the color is not gone, add more Norit and boil again; then filter through a fluted filter paper in a previously warmed stemless funnel into a 50-mL Erlenmeyer flask. Sometimes filtration is slow because the funnel fits so snugly into the mouth of the flask that a backpressure develops. If you note that raising the funnel increased the flow of filtrate, fold a small strip of paper two or three times and insert it between the funnel and flask. Wash the used flask with 2 mL of hot methanol, and use this liquid to wash the filter paper, transferring the solvent with a Pasteur pipette in a succession of drops around the upper rim of the filter paper. When the filtration is complete, the volume of methanol should be 15mL. If it is not, evaporate excess methanol.
Because the filtrate is far from being saturated with naphthalene at this point, it will not yield crystals on cooling; however, the solubility of naphthalene in methanol can be greatly reduced by addition of water. Heat the solution to the boiling point and add water drop wise from a 10-mL graduated cylinder using a Pasteur pipette (or use a precalibrated pipette). After each addition of water, the solution will turn cloudy for an instant. Swirl the contents of the flask and heat to redissolve and precipitated naphthalene. After the addition of 3.5 ml of water, the solution will be almost saturated with naphthalene at the boiling point of the solvent. Remove the flask from the heat and place it on a cork ring or other insulating surface to; cool, without being disturbed, to room temperature.
Immerse the flask in an ice bath along with another flask containing methanol and water in the ratio of 15:3.5. this cold solvent will be used for washing the crystals. The cold crystallization mixture is collected by vacuum filtration on a small Buckner funnel (50 mm) (Fig. 3.21).
The water flowing through the aspirator should always be turned on full force. In collecting the product by suction filtration, use a spatula to dislodge crystals and ease them out of the flask. If crystals still remain in the flask, some filtrate can be poured back into the crystallization flask as a rinse for washing as often as desired, since it is saturated with solute. To free the crystals from contaminating mother liquor, break the suction and pour a few milliliters of the fresh cold solvent mixture into the Buckner funnel and immediately reapply suction. Repeat this process until the crystals and the filtrate are free of color. Press the crystals with a clean cork to; eliminate excess solvent, pull air through the filter cake for a few minutes, and then put the large, flat, plate like crystals out on a filter paper to dry. The yield of pure white crystalline naphthalene should be about 1.6g. The mother liquor contains about 0.25g, and about 0.15 g is retained in the charcoal and on the filter paper.
CLEANING UP Place the Norit in the nonhazardous solid waste container. The methanol filtrate and washing are placed in the organic solvents container.
7. PURIFICATION OF AN UNKNOWN
Bear in mind the seven-steps crystallization procedure:
1. Choose the solvent.
2. Dissolve the solute.
3. Decolorize the solution (if necessary).
4. Filter suspended solids (if necessary).
5. Crystallize the solute.
6. Collect and wash the crystals.
7. Dry the product.
You are to purify 100mg (microscale) or 2.0g (macroscale) of an unknown provided by the instructor. Conduct test for solubility and ability to crystallize in several organic solvents, solvent pairs, and water. Conserve your unknown by using very small quantities for solubility tests. If only a drop or two of solvent is used, the solvent can be evaporated by heating the test tube on the steam bath or sand bath, and the residue can be used for another test. Submit as much pure product a possible with evidence of its purity (i.e., the melting point). From the posted list, identify the unknown.
CLEANING UP Place decolorizing charcoal, if used, and filter paper in the nonhazardous solid waste container. Put organic solvents in the organic solvents container, and flush aqueous solutions down the drain.
NOTE: Small (less than 200mg) samples of solids are most conveniently submitted in 1 � X 1 �-in (4 X 4 cm) polyethylene zip lock bags. The bag can be stapled to a card or to the appropriate page in the notebook.
CRYSTALLIZATION PROBLEMS AND THEIR SOLUTIONS
Induction of Crystallization
Occasionally, a sample will not crystallize from solution on cooling, even though the solution is saturated with the solute at elevated temperature. The easiest method for inducing crystallization is to add to the supersaturated solution a seed crystal that has been saved from the crude material (if it was crystalline before recrystallization was attempted). In a probably apocryphal tale, the great sugar chemist Emil Fischer merely had to wave his beard over a recalcitrant solution and the appropriate seed crystals would drop out, causing crystallization to occur. In the absence of seed crystals, crystallization often can be induced by scratching the inside of the flask with a stirring rod at the air-liquid interface. One theory holds that part of the freshly scratched glass surface has angles and planes corresponding to the crystal structure, and crystals start growing on these spots. Often crystallization is very slow to begin, and placing the sample in a refrigerator overnight will bring success. Other expedients are to change the solvent (usually to a poorer one) and to place the sample in an open container where slow evaporation and dust from the air may help induce crystallization.
OILS AND "OILING OUT"
Some saturate solutions, especially those containing water, when they cool, deposit not crystals but small droplets referred to as oils. Should these droplets subsequently crystallize and be collected, they will be found to be rather impure. Should the temperature of the saturated solution be above the melting point of the solute when it starts to come out of solution, the solute will, of necessity, be deposited as an oil. Similarly, the melting point of the desired compound may be depressed to a point such that a low-melting eutectic mixture of the solute and the solvent comes out of solution. The simplest remedy for this latter problem is to lower the temperature at which the solution becomes saturated with the solute by simply adding more solvent. In extreme cases it may be necessary to lower this temperature well below room temperature by cooling the solution with dry ice.
CRYSTALLIZATION SUMMARY
1. Choosing the solvent. "Like dissolves like." Some common solvents are water, methanol ethanol, ligroin, and toluene. When you use a solvent pair, dissolve the solute in the better solvent and add the poorer solvent to the hot solution until saturation occurs. Some common solvent pairs are ethanol-water, diethyl ether-ligroin, toluene-ligroin, and t-butyl methyl ether-hexane.
2. Dissolving the solute. To the crushed or ground solute in an Erlenmeyer flask or a reaction tube, add solvent and heat the mixture to boiling. Add more solvent as necessary to obtain a got, saturated solution.
3. Decolorizing the solution. If it is necessary to remove colored impurities, cool the solution to near room temperature and add more solvent to prevent crystallization from occurring. Add decolorizing charcoal in the form of pelletized Norit to the cooled solution, and then heat it to boiling for a few minutes, taking care to swirl the solution to prevent bumping. Remove the Norit by filtration, and then concentrate the filtrate.
4. Filtering Suspended Solids. If it is necessary to remove suspended solids, dilute the hot solution slightly to prevent crystallization from occurring during filtration. Filter the hot solution. Add solvent if crystallization begins in the funnel. Concentrate the filtrate to obtain a saturated solution.
5. Crystallizing the Solute. Let the hot saturated solution cool spontaneously to room temperature. Do not disturb the solution. Then cool it in ice. If crystallization does not occur, scratch the inside of the container or add seed crystals.
6. Collecting and Washing the Crystals. Collect the crystals using the Pasteur pipette method or by vacuum filtration on a Hirsch funnel or a Buchner funnel. If the latter technique is employed, wet the filter paper with solvent, apply vacuum, break vacuum, add crystals and liquid, apply vacuum until solvent just disappears, break vacuum, add cold wash solvent, apply vacuum, and repeat until crystals are clean and filtrate comes through clear.
7. Drying the Product. Press the product on the filter to remove solvent. Then remove it from the filter, squeeze it between sheets of filter paper to remove more solvent, and spread it on a watch glass to dry.
1. A sample of naphthalene, which should be pure white, was found to have a grayish color after the usual purification procedure. The melting point was correct and the melting point range small. Explain the gray color.
2. How many milliliters of boiling water are required to dissolve 25g of phthalic acid? If the solution were cooled to 14 degrees C, how many grams of phthalic acid would crystallize out?
3. What is the reason for using activated carbon during a crystallization?
4. If a little activated charcoal does a good job removing impurities in a crystallization, why not use a lot?
5. Under what circumstances is it wise to use a mixture of solvents to carry out a crystallization?
6. Why is gravity filtration and not suction filtration used to remove suspended impurities and charcoal from a hot solution?
7. Why is a fluted filter paper used in gravity filtration?
8. Why are stem less funnels used instead of long-stem funnels to filter hot solutions through fluted filter paper?
9. Why is the final product from the crystallization process on a macroscale isolated by vacuum filtration and not by gravity filtration?