
|
A crystalline face centered cubic solid consisting of point masses
interacting through the Lennard Jones potential and one other model
potential is characterized as an approximation for an isotropic linear
elastic solid, and the linear elastic properties are found. Failure
characteristics are also obtained. This model is then used to study
vibrational modes of objects, as well as the motion of bouncing objects.
|

| B = | 2 ρ ---- 3m |
(dF/dr) (dcc)2 |
| Linear Elastic Properties of Molecular Dynamics Solid: | ||||||
| Property | Symbol | Average value | Unit | Min. value | Max. value | Relation: |
| Mass of one atom | matom | 1.0000000 | kg | (exact) | ||
| Density | ρ | 1.0000000 | kg/m3 | (exact) | ||
| Atomic force constant | dF/dr | 1.0000000 | N/m | (exact) | ||
| Atom center-center distance | dcc | 1.1224621 | m | (exact) | dcc=(sqrt(2)matom/ρ)1/3 | |
| Bulk Modulus | B | 0.8399473 | Pa | (exact) | 2ρ(dcc)2(dF/dr)/(3matom) | |
| Young's Modulus | Y | 1.215 | Pa | 1.172 | 1.261 | (use md4ym.cpp) |
| Poisson's ratio | ν | 0.2589 | - - - | 0.250 | 0.267 | ν=(3B-Y)/(6B) |
| Shear Modulus | G | 0.4826 | Pa | 0.4623 | 0.5045 | G=3BY/(9B-Y) |
| 1st Lamé constant | λ | 0.5182 | Pa | 0.5036 | 0.5317 | λ = B - (2/3)G |
| Shear wave speed | Cs | 0.6947 | m/s | 0.6799 | 0.7103 | Cs=sqrt(G/ρ) |
| Longitudinal wave speed | Cl | 1.2179 | m/s | 1.2068 | 1.2299 | Cl=sqrt((λ+2G)/ρ) |
| Cs/Cl | 0.5704 | - - - | 0.5634 | 0.5775 | ||
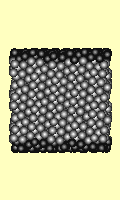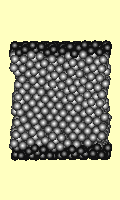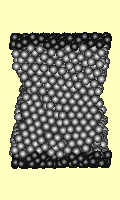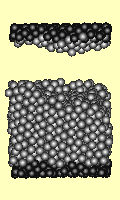
 Failure under shear stress is
depicted (right) for a cube of 1727 atoms. The top and bottom surfaces
(darker atoms) are forced sideways. Strain increases by 0.0001 per iteration,
and the time step per iteration is 0.2 seconds. Every 200th iteration is used
for the animation. The Lennard Jones potential was used.
Failure under shear stress is
depicted (right) for a cube of 1727 atoms. The top and bottom surfaces
(darker atoms) are forced sideways. Strain increases by 0.0001 per iteration,
and the time step per iteration is 0.2 seconds. Every 200th iteration is used
for the animation. The Lennard Jones potential was used.
 The axial stretch mode (shown at left) for a sphere
of radius 6.5 atoms. It has a frequency of 0.041 Hz.
The simulation time step is 0.50 s.
The animation time step is 1.0 s. (fig1m.gif)
The axial stretch mode (shown at left) for a sphere
of radius 6.5 atoms. It has a frequency of 0.041 Hz.
The simulation time step is 0.50 s.
The animation time step is 1.0 s. (fig1m.gif) The wet dog mode (shown at right) for a radius
of 6.5 (1159 atoms). It has a frequency of 0.0364 Hz.
The simulation time step is 0.50 s.
The animation time step is 1.0 s. This is the lowest frequency mode
of vibration. It is reminiscent of the manner in which a dog
shakes its body to dry itself off. (fig1i.gif)
The wet dog mode (shown at right) for a radius
of 6.5 (1159 atoms). It has a frequency of 0.0364 Hz.
The simulation time step is 0.50 s.
The animation time step is 1.0 s. This is the lowest frequency mode
of vibration. It is reminiscent of the manner in which a dog
shakes its body to dry itself off. (fig1i.gif) The figure on the right was generated by the C++ program
md2graf3.cpp. It shows a ball made of 727 atoms and radius
5.5 falling with initial velocity 0.05 m/s. The ball rebounds with a velocity
of 0.0476 m/s. Some of the energy can be seen to go into
rotational kinetic energy. The simulation time step is 0.50 s,
but each animation time step is 5 times longer, or 2.50 s.
The quadratic potential was used. The floor is infinitely hard.
The figure on the right was generated by the C++ program
md2graf3.cpp. It shows a ball made of 727 atoms and radius
5.5 falling with initial velocity 0.05 m/s. The ball rebounds with a velocity
of 0.0476 m/s. Some of the energy can be seen to go into
rotational kinetic energy. The simulation time step is 0.50 s,
but each animation time step is 5 times longer, or 2.50 s.
The quadratic potential was used. The floor is infinitely hard. The figure on the left was generated
by the same C++ program, and is also a ball of 727 atoms. This time
it falls at 0.10 m/s. The time step is 0.10 seconds, and the
animation time step is 2.50 s. The ball rebounds upwards with
a speed of 0.0359 m/s. This simulation is 52 frames long.
The Lennard Jones potential was used. (fig1f.gif)
The figure on the left was generated
by the same C++ program, and is also a ball of 727 atoms. This time
it falls at 0.10 m/s. The time step is 0.10 seconds, and the
animation time step is 2.50 s. The ball rebounds upwards with
a speed of 0.0359 m/s. This simulation is 52 frames long.
The Lennard Jones potential was used. (fig1f.gif) The picture on the right shows a ball dropped at 0.20 m/s,
with a simulation time step of 0.05 s and animation time step of 2.50 s.
The animation is 75 frames long.
The Lennard Jones potential is used. (fig1g.gif)
The picture on the right shows a ball dropped at 0.20 m/s,
with a simulation time step of 0.05 s and animation time step of 2.50 s.
The animation is 75 frames long.
The Lennard Jones potential is used. (fig1g.gif)|
| On left is an image of a 727 atom ball dropped at 0.40 m/s, also with an animation time step of 2.50 s. The animation is 51 frames long. The Lennard Jones potential is used. The simulation time step was 0.02 s. (fig1h.gif) |
| Material | Density | Young's Modulus | sqrt(Y/ρ) | ν | structure |
| (g/cm3) | (GPa) | (m/s) | |||
| Aluminum | 2.7 | 73 | 5200 | 0.33 | fcc |
| Acrylic | 1.17-1.20 | 3.2 | 1650 | ||
| Brass | 8.4 | 130 | 3930 | ||
| Celluloid | 1.4 | 1.3-1.5 | |||
| Cellulose acetate | 1.22-1.34 | 1.5-2.2 | -- | ||
| Copper | 8.93 | 0.31 | fcc | ||
| Diamond | 3.516 | 0.1-0.29 | diam. | ||
| Glass | 2.60 | 46.2 | 4220 | ||
| Gold | 19.28 | 0.42 | fcc | ||
| Iron | 7.87 | 0.291 | bcc | ||
| Lead | 11.34 | 0.42 | fcc | ||
| LDPE | 0.38 | ||||
| nylon (polyamide) | 1.1 | 1.8 | -- | ||
| nylon 6 | 1.1 | 3.0 | 1650 | 0.35 | |
| polystyrene | 1.04 -1.09 | 1.5-3.5 | -- | 0.33 | |
| polystyrene, impact | 0.33-0.37 | ||||
| rubber (nat. vulc.) | 0.95 | 0.00276 | 54 | ||
| Silver | 10.50 | 0.37 | fcc | ||
| Steel | 7.8 | 210 | 5190 | 0.29 | |
| Teflon | 0.46 | ||||