HIV MOLECULAR BIOLOGY
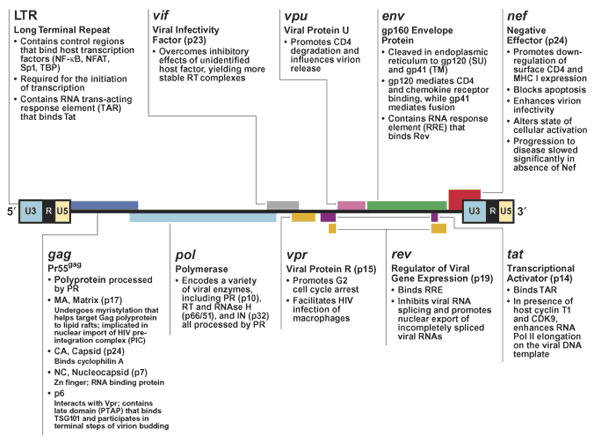
Figure 1 : An overview of the organization of the (approximately) 9-kilobase genome of the HIV provirus and a summary of the functions of its nine genes encoding 15 proteins
Figure 2: Schematic description of early events occurring after HIV infection of a susceptible target cell, including interactions between gp120, CD4, and chemokine receptors (CCR5 or CXCR4) leading to gp41-mediated fusion followed by virion uncoating, reverse transcription of the RNA genome, nuclear import of the viral preintegration complex, and integration of the double-stranded viral cDNA into the host chromosome, thus establishing the HIV provirus
Figure 3: A summary of two different mechanisms potentially underlying post-integration HIV latency contrasted with the central role of Tat in promoting productive infection of target cells.
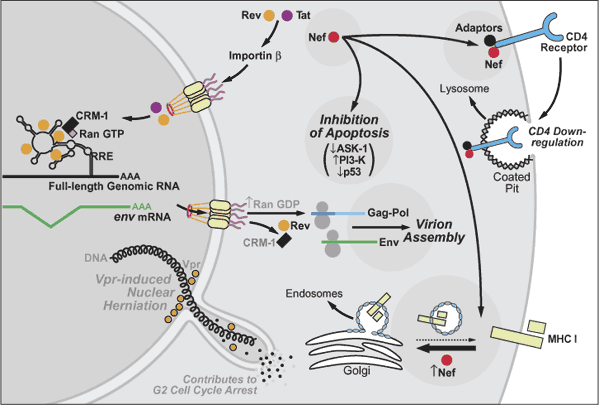
Figure 4: A summary of late events in the HIV-infected cell culminating in the assembly of new infectious virions. Highlighted are the roles of various viral proteins in optimizing the intracellular environment for viral replication including downregulation of CD4 and MHC I and inhibition of apoptosis by Nef, and the induction of G2 cell-cycle arrest by Vpr. A key action of the HIV Rev protein in promoting nuclear export of incompletely spliced viral transcripts that encode the structural and enzymatic proteins as well as the viral genome of new virions is also illustrated.
Figure 5: A summary of late events in the HIV-infected cell culminating in the assembly of new infectious virions. Highlighted are the roles of various viral proteins in optimizing the intracellular environment for viral replication including downregulation of CD4 and MHC I and inhibition of apoptosis by Nef, and the induction of G2 cell-cycle arrest by Vpr. A key action of the HIV Rev protein in promoting nuclear export of incompletely spliced viral transcripts that encode the structural and enzymatic proteins as well as the viral genome of new virions is also illustrated.
Figure 1: An overview of the organization of the (approximately) 9-kilobase genome of the HIV provirus and a summary of the functions of its nine genes encoding 15 proteins
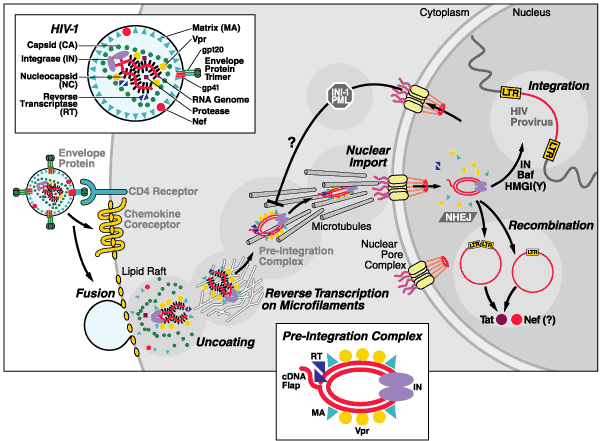
Figure 2 : Schematic description of early events occurring after HIV infection of a susceptible target cell, including interactions between gp120, CD4, and chemokine receptors (CCR5 or CXCR4) leading to gp41-mediated fusion followed by virion uncoating, reverse transcription of the RNA genome, nuclear import of the viral preintegration complex, and integration of the double-stranded viral cDNA into the host chromosome, thus establishing the HIV provirus
BINDING AND ENTRY (chemokine receptors, lipid raft, conformational change, endocytosis of dendritic cell) [Summary]
12 chemokine receptors can function as HIV coreceptors (Fig 2) in cultured cells, but only two are known to play a role in vivo.
CCR5, binds macrophage-tropic, non-syncytium-inducing (R5) viruses, associated with mucosal and intravenous transmission of HIV infection. In up to 13% of individuals of northern European descent, a naturally occurring deletion of 32 base pairs in the CCR5 gene results in a mutant CCR5 receptor that never reaches the cell surface. Individuals homozygous for this mutation (1-2% of the Caucasian population) are completely resistant to HIV infection. Interrupting HIV interaction with CCR5 might form a promising new class of antiretroviral drugs.
CXCR4, binds T-cell-tropic, syncytium-inducing (X4) viruses, during the later stages of disease.
Both CD4 and chemokine coreceptors for HIV are found disproportionately in lipid rafts (Fig 2) in the cell membrane. These cholesterol- and sphingolipid-enriched microdomains likely provide a better environment for membrane fusion, perhaps by mirroring the optimal lipid bilayer of the virus. Removing cholesterol from virions, producer cells, or target cells greatly decreases the infectivity of HIV. Studies currently under way are exploring whether cholesterol-depleting compounds might be efficacious as topically applied microbicides to inhibit HIV transmission at mucosal surfaces.
The binding of surface gp120, CD4, and the chemokine coreceptors produces radical conformational change in gp41. Assembled as a on the virion membrane, this coiled-coil protein springs open, projecting three peptide fusion domains that "harpoon" the lipid bilayer of the target cell. The fusion leads to the release of the viral core into the cell interior. The fusion inhibitors T-20 and T-1249 act to prevent fusion by blocking the formation of these hairpin structures.
HIV virions can also enter cells by endocytosis. Usually, productive infection does not result, presumably reflecting inactivation of these virions within endosomes. However, a special form of endocytosis has been demonstrated in submucosal dendritic cells. These cells, which normally process and present antigens to immune cells, express a specialized attachment structure termed DC-SIGN. This C-type lectin binds HIV gp120 with high affinity but does not trigger the conformational changes required for fusion. Instead, virions bound to DC-SIGN are internalized into an acidic compartment and subsequently displayed on the cell surface after the dendritic cell has matured and migrated to regional lymph nodes, where it engages T cells. Thus, dendritic cells expressing DC-SIGN appear to act as "Trojan horses" facilitating the spread of HIV from mucosal surfaces to T cells in lymphatic organs
CYTOPLASMIC EVENTS (phosphorylation, viral protein Nef + Vif, HIV preintegration complex(PIC), microtubule transport, promyleotic oncogene (PML)) [Summary]
Once inside the cell, the virion undergoes uncoating, likely while still associated with the plasma membrane. This poorly understood process may involve phosphorylation of viral matrix proteins by a mitogen-activated protein (MAP) kinase and additional actions of cyclophilin A and the viral proteins Nef and Vif (Fig 1).
Nef associates with a universal proton pump, V-ATPase, which could promote uncoating by inducing local changes in pH in a manner similar to that of the M2 protein of influenza.
By overcoming destabilizing effects of a recently identified protein termed CEM15/APOBEC3G, Vif stabilizes the reverse transcription complex in most human cells.
After the virion is uncoated, the viral reverse transcription complex is released from the plasma membrane.
Reverse transcription yields the HIV preintegration complex (PIC) (Fig 2), composed of
double-stranded viral cDNA,
integrase,
matrix,
Vpr,
reverse transcriptase,
high mobility group DNA-binding cellular protein HMGI(Y).
The PIC may move toward the nucleus by using microtubules as a conduit. Adenovirus and herpes simplex virus 1 also dock with microtubules and use the microtubule-associated dynein molecular motor for cytoplasmic transport. This finding suggests that many viruses use these cytoskeletal structures for directional movement.
Recent studies have revealed a mechanism by which the target cell defends against the HIV intruder. Within 30 minutes of infection, select host proteins including the integrase interactor 1 (also known as INI-1, SNF5, or BAF47), a component of the SWI/SNF chromatin remodeling complex, and PML (Fig 2), a protein present in promyelocytic oncogenic domains, translocate from the nucleus into the cytoplasm. Addition of arsenic trioxide sharply blocks PML movement and enhances the susceptibility of cells to HIV infection raising the possibility that the normal function of PML is to oppose viral infection.
NUCLEAR PENETRATION (molecular gymnastics, Matrix vs Vpr shuttle) [Summary]
With a Stokes radius of 28 nm or roughly the size of a ribosome and 3 µm contour length of viral DNA , the PIC is twice as large as the maximal diameter of the central aqueous channel in the nuclear pore and must undergo significant compaction, known as molecular gymnastics.(Fig 2)
The key viral proteins that mediate the nuclear import of the PIC are Integrase, matrix,and Vpr.
Matrix (Fig 2 inset) contains a canonical nuclear localization signal that is recognized by the importins alpha and beta, which are components of the classical nuclear import pathway.
The HIV Vpr (Fig 1) gene product contains at least three noncanonical nuclear targeting signals. Vpr may bypass the importin system altogether, perhaps mediating the direct docking of the PIC with one or more components of the nuclear pore complex.
The multiple nuclear targeting signals within the PIC may function in a cooperative manner or play larger roles individually in different target cells. For example, while Vpr is not needed for infection of nondividing, resting T cells, it enhances viral infection in nondividing macrophages. The finding that both matrix and Vpr shuttle between the nucleus and cytoplasm explains their availability for incorporation into new virions
INTEGRATION (integrase, HMGI(Y), BAF, PIC enter nucleus outcome, NHEJ) [Summary]
Once inside the nucleus, the viral PIC can establish a functional provirus. Integration (Fig 2) of double-stranded viral DNA into the host chromosome is mediated by :
Not all PICs that enter the nucleus result in a functional provirus. Other outcomes are:
In a normal cellular response to DNA fragments, the nonhomologous end-joining (NHEJ) system may form 2-LTR circles to protect the cell.(Fig 2) This system is responsible for rapid repair of double-strand breaks, thereby preventing an apoptotic response.
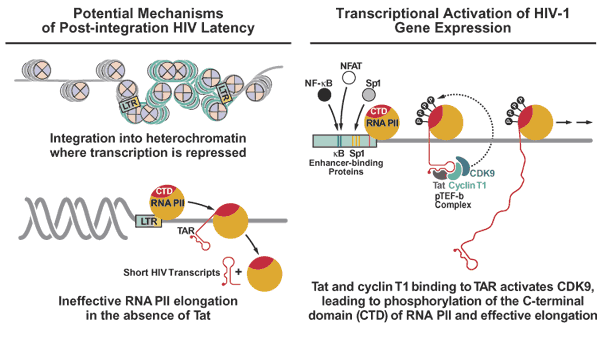
Figure 3 : A summary of two different mechanisms potentially underlying post-integration HIV latency contrasted with the central role of Tat in promoting productive infection of target cells.
TRANSCRIPTION CONTROL (active / latency transcription, chromosomal environment, transcriptional enhancer, TAT function) [Summary]
Integration can lead to
The chromosomal environment likely shapes the transcriptional activity of the provirus. For example, latency might be caused by
However, of the multiple copies of provirus that are usually integrated in a given infected cell, at least one is likely to be transcriptionally active. This fact may explain why the number of latently infected cells (105-106) in infected patients is small.
In the host genome, the 5´ LTR functions like other eukaryotic transcriptional units. It contains downstream and upstream promoter elements, which include the initiator (Inr), TATA-box (T), and three Sp1 sites. These regions help position the RNA polymerase II (RNAPII) at the site of initiation of transcription and to assemble the preinitiation complex. Slightly upstream of the promoter
When these factors engage the LTR, transcription begins, but in the absence of Tat described below the polymerase fails to elongate efficiently along the viral genome.(Fig 3)
Tat significantly increases the rate of viral gene expression. With cyclin T1 (CycT1), Tat binds to the TAR RNA stem-loop structure and recruits the cellular cyclin-dependent kinase 9 (Cdk9) to the HIV LTR. (Fig 3) Within the positive transcription elongation factor b (P-TEFb) complex, Cdk9 phosphorylates the C-terminal domain of RNAPII, marking the transition from initiation to elongation of eukaryotic transcription. Other targets of P-TEFb include negative transcription elongation factors (N-TEF). The high efficiency with which the HIV LTR attracts these negative transcription factors in vivo may explain why the LTR is a poor promoter in the absence of Tat.
** Because murine CycT1 contains a cysteine at position 261, the complex between Tat and murine P-TEFb binds TAR weakly. Thus, Tat transactivation is severely compromised in murine cells.
Fig 4 : A summary of late events in the HIV-infected cell culminating in the assembly of new infectious virions. Highlighted are the roles of various viral proteins in optimizing the intracellular environment for viral replication including downregulation of CD4 and MHC I and inhibition of apoptosis by Nef, and the induction of G2 cell-cycle arrest by Vpr. A key action of the HIV Rev protein in promoting nuclear export of incompletely spliced viral transcripts that encode the structural and enzymatic proteins as well as the viral genome of new virions is also illustrated.
VIRAL TRANSCRIPT (splicing, Rev, RanGTP,exportin) [Summary]
Transcription of the viral genome results in more than a dozen different HIV-specific transcripts.
These viral transcripts encode the structural, enzymatic, and accessory proteins and represent viral genomic RNAs that are needed for the assembly of fully infectious virions.
Incomplete splicing likely results from suboptimal splice donor and acceptor sites in viral transcripts. Rev, may inhibit splicing by its interaction with alternate splicing factor/splicing factor 2 (ASF/SF2) and its associated p32 protein.
Transport of the incompletely spliced viral transcripts to the cytoplasm depends on an adequate supply of Rev. Rev:
The nuclear export of this assembly (viral RNA transcript, Rev, and CRM1/exportin 1) depends critically on another host factor, RanGTP. Ran
Outbound cargo is only effectively loaded onto CRM1/exportin-1 in the presence of RanGTP. However, when the complex reaches the cytoplasm, GTP is hydrolyzed to GDP, resulting in release of the bound cargo. The opposite relationship regulates the nuclear import by importins alpha and beta, where nuclear RanGTP stimulates cargo release.
For HIV infection to spread, a balance between splicing and transport of viral mRNA species must be achieved. If splicing is too efficient, then only the multiply spliced transcripts appear in the cytoplasm. Although required, the regulatory proteins encoded by multiply spliced transcripts are insufficient to support full viral replication. However, if splicing is impaired, adequate synthesis of Tat, Rev, and Nef will not occur. In many non-primate cells, HIV transcripts may be overly spliced, effectively preventing viral replication in these hosts.
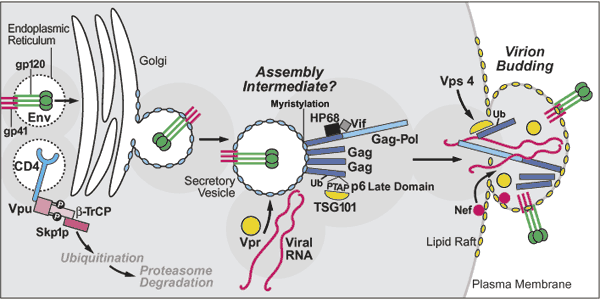
Fig 5 : A summary of late events in the HIV-infected cell culminating in the assembly of new infectious virions. Highlighted are the roles of various viral proteins in optimizing the intracellular environment for viral replication including downregulation of CD4 and MHC I and inhibition of apoptosis by Nef, and the induction of G2 cell-cycle arrest by Vpr. A key action of the HIV Rev protein in promoting nuclear export of incompletely spliced viral transcripts that encode the structural and enzymatic proteins as well as the viral genome of new virions is also illustrated.
HIV REPLICATION ( Nef, low CD4 expression, Vpr ) [Summary]
In contrast to Tat and Rev, which act directly on viral RNA structures, Nef modifies the environment of the infected cell to optimize viral replication. (Fig 4) The absence of Nef in infected monkeys and humans is associated with much slower clinical progression to AIDS. This virulence caused by Nef appears to be
Nef impairs immunological responses to HIV.
** However, Nef does not decrease the expression of HLA-C,which prevents recognition and killing of these infected cells by natural killer cells.
Nef also inhibits cell apoptosis by binding to
Hence, Nef prolongs the life of the infected host cell, thereby optimizing viral replication.
Rev-dependent expression of Vpr (Fig 1) induces the arrest of proliferating infected cells at the G2/M phase of the cell cycle. Since the viral LTR is more active during G2, this arrest likely enhances viral gene expression. These cell-cycle arresting properties involve localized defects in the structure of the nuclear lamina that lead to dynamic, DNA-filled herniations that project from the nuclear envelope into the cytoplasm. (Fig 4) Intermittently, these herniations rupture, causing the mixing of soluble nuclear and cytoplasmic proteins.
VIRAL ASSEMBLY (plasma membrane, ViF, molecular chaperone, CEM15/APOBEC3G) [Summary]
New viral particles are assembled at the . (Fig 5) Each virion consists of
Several proteins participate in the assembly process, including Gag polyproteins and Gag-Pol, as well as Nef and Env. A human ATP-binding protein, HP68 (previously identified as an RNase L inhibitor), likely acts as a molecular chaperone, facilitating conformational changes in Gag needed for the assembly of viral capsids.
In primary CD4 T lymphocytes, Vif plays a key but poorly understood role in the assembly of infectious virions. In the absence of Vif, normal levels of virus are produced, but these virions are noninfectious, displaying arrest at the level of reverse transcription in the subsequent target cell. Heterokaryon analyses of cells formed by the fusion of nonpermissive (requiring Vif for viral growth) and permissive (supporting growth of Vif-deficient viruses) cells have revealed that Vif overcomes the effects of CEM15/APOBEC3G :
VIRAL BUDDING (Gag protein, TSG 101, ubiquitination, multivesicular body) [Summary]
The Gag polyproteins (Fig 1) are subject to myristylation, and thus associate preferentially with cholesterol- and glycolipid-enriched membrane microdomains. Virion budding occurs through these specialized regions in the lipid bilayer, yielding virions with cholesterol-rich membranes. This lipid composition likely favors release, stability, and fusion of virions with the subsequent target cell.
The budding reaction involves the action of several proteins, including the "late domain" sequence (PTAP) present in the p6 portion of Gag.(Fig 5) The p6 protein also appears to be modified by ubiquitination. The product of the tumor suppressor gene 101 (TSG101) binds the PTAP motif of p6 Gag and also recognizes ubiquitin through its ubiquitin enzyme 2 (UEV) domain.
**The TSG101 protein normally associates with other cellular proteins in the vacuolar protein sorting pathway to form the ESCRT-1 complex that selects cargo for incorporation into the multivesicular body (MVB). The MVB is produced when surface patches on late endosomes bud away from the cytoplasm and fuse with lysosomes, releasing their contents for degradation within this organelle.
In the case of HIV, TSG101 appears to be "hijacked" to participate in the budding of virions into the extracellular space away from the cytoplasm
HIV entry into cell mediated through
After entry, viral uncoating release viral reverse transcription complex which produce HIV preintegration complex (PIC). Two viral protein
PIC ‘docked’ on microtubules (cytoskeletal structure), traveling towards nucleus.
Host promyelolytic oncogen (from nucleus to cytoplasm) might
prevent PIC movement
Molecular gymnastics occur as PIC is 2x larger than nuclear pore. Viral proteins involved
Integration (viral DNA into hos chromosome) to form provirus helped by
Integrase
Not all become functional provirus
NHEJ (non homologous endjoining) system involved in DNA fragment repair
Chromosomal environment induce latency:
heterochromatin
low transcriptional enhancer
nuclear factor [kappa]B (NF-[kappa]B) - rate elongation / initiation replication
nuclear factor of activated T cells (NFAT) – form active complex
Ets family members
lack of viral protein, Tat
binds cyclin T1 (Cyc T1) forming cellular cyclin-dependent kinase 9 (Cdk9) needed for effective elongation
Murine cell impair Tat function
Transcription produces spliced transcripts Nef, Tat, Rev
Transporting these from nucleus o cytoplasm required:
Viral protein Rev
Binds RRE (Rev Response Element, located in Env gene)
Has leucine-riched nuclear export system
Host factor RanGTP
Gradient RanGTP (nucleus) vs RanGDP (cytoplasm) determine
direction movement
Exportin 1 – ‘cargo'
Tat, Rev acts on viral RNA
Nef acts on environment of infected cells
Nef virulence:
CD4 expression on cell surface. Helped by
Gp120 – stop CD4 movement to cell surface by binding to them
Vpu – enhance CD4 proteosomal degradation
inhibit cell apaptosis, prolonging life of infected cell, hence optimizing viral replication
Fortunately, Nef is no expressed in infected human cell which
slow progression to AIDS.
Vpr arrest replication of infected cell by inducing DNA-filled
herniation to rupture, producing mixing nucleus and cytoplasm protein
Assembled in plasma membrane
Vif overcomes host CEM15/APOBEC3G
for effective assembly
Budding needs enriched cholesterol and lipid environment.
Gag proteins involved by ‘hijacking’ TSG101 through ubiquitination to form MVB
ANIMATED MOLECULAR BIOLOGY of HIV CYCLE
![]()