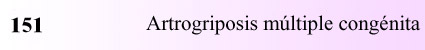|
9
ARTROGRIPOSIS
MÚLTIPLE CONGÉNITA
Artrogriposis significa
flexión permanente de una articulación. Se hace el diagnóstico de
artrogriposis múltiple congénita cuando dos o más articulaciones en más de
una extremidad están permanentemente contraídas al nacer (Figura 151.1).
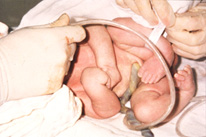
Figura 151.1.—Artrogriposis
múltiple congénita.
 La
articulación asume una determinada posición por fuerzas de motilidad
desiguales. Este fenómeno es el resultado de debilidad muscular o
hipotonía segmentaria, o de una postura asimétrica sostenida. La debilidad
muscular o la hipotonía segmentaria, produce un desequilibrio entre los
músculos agonistas y antagonistas que actúan en una articulación. La
postura asimétrica sostenida se establece por restricción, cuando el
volumen uterino está reducido o existen bandas cutáneas que limitan los
movimientos. La
articulación asume una determinada posición por fuerzas de motilidad
desiguales. Este fenómeno es el resultado de debilidad muscular o
hipotonía segmentaria, o de una postura asimétrica sostenida. La debilidad
muscular o la hipotonía segmentaria, produce un desequilibrio entre los
músculos agonistas y antagonistas que actúan en una articulación. La
postura asimétrica sostenida se establece por restricción, cuando el
volumen uterino está reducido o existen bandas cutáneas que limitan los
movimientos.
Constituye un hecho fundamental determinar la causa de la artrogriposis
múltiple congénita, pues ella establece el pronóstico y la frecuencia de
recidivas de la enfermedad. Son muchas las causas de esta afección en los
neonatos. El texto de Smith sobre características reconocibles de
malformación humana cataloga la artrogriposis congénita múltiple como un
rasgo presente en más de 100 síndromes.
El diagnóstico diferencial de la artrogriposis múltiple congénita se lleva
a cabo analizando la distribución, y la presencia de dismorfismo facial y
otros hallazgos concomitantes.
La
artrogriposis múltiple congénita abarca las articulaciones distales o
proximales. La artrogriposis distal es más frecuente que la artrogriposis
proximal. A menudo en neonatos con artrogriposis distal, la mano asume una
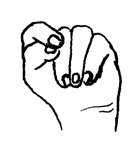
posición muy típica (Figura152.1).
Esta posición de la mano es frecuente en los síndromes de las trisomías 13
y 18, en los síndromes I y II de Pena-Shokeir, y en el síndrome de Smith-Lemli-Opitz.
La artrogriposis proximal suele afectar los hombros, codos, caderas y
rodillas. La artrogriposis proximal se observa en neonatos con amioplasia
congénita.
|
 |
 |
Figura 152.1.— Mano de la trisomía
18. El dedo medio está debajo del índice y parcialmente debajo del cuarto
dedo. El dedo pulgar está total o parcialmente debajo del índice.
La combinación de artrogriposis múltiple congénita
con dismorfismo facial ocurre en: trisomías 13 y 18; y en los síndromes de
Pena-Shokeir I y II, Antley-Bixer, Sheldon-Fremann, Beals, Smith-Lemli-Opitz
y Zellweger.
 Las
causas de la artrogriposis pueden tener un origen neurológico o no
neurológico. Las artrogriposis de origen neurológico se producen por
anormalidades del sistema nervioso central o periférico. Las artrogriposis
de origen no neurológico se producen por anormalidades cartilaginosas o
por restricción física de los movimientos. Por lo tanto las artogriposis
pueden estudiarse clasificándolas como artrogriposis debidas a
anormalidades cartilaginosas, restricción física de movimientos y
disfunciones neurológicas. Las
causas de la artrogriposis pueden tener un origen neurológico o no
neurológico. Las artrogriposis de origen neurológico se producen por
anormalidades del sistema nervioso central o periférico. Las artrogriposis
de origen no neurológico se producen por anormalidades cartilaginosas o
por restricción física de los movimientos. Por lo tanto las artogriposis
pueden estudiarse clasificándolas como artrogriposis debidas a
anormalidades cartilaginosas, restricción física de movimientos y
disfunciones neurológicas.
ANORMALIDADES
CARTILAGINOSAS
Estas
se consideran causa de artrogriposis múltiple congénita en neonatos con
cualquiera de las siguientes características: (1) mayor talla para la edad
gestacional, (2) piel hiperextensible y transparente, (3) esclerótica
azulada, (4) forma del lóbulo auricular anormal y 5) craneosinostosis.
La inteligencia es normal; la hiperelasticidad articular y la restricción
natural de los movimientos, impuesta por la vida intrauterina, originan la
enfermedad, que suele mejorar con el crecimiento. Las entidades más
frecuentes que se presentan con esta combinación son: (1) síndrome de
Beals, (2) síndrome de Antley-Bixer, (3) displasia diastrófica y (4)
artrogriposis distal.
Síndrome de Beals
El
síndrome de Beals es un trastorno del tejido conectivo unido a un sitio
fibrilar en el cromosoma 5q23-31 (FBN2); se transmite en forma autosómica
dominante. Los rasgos que lo distinguen son las orejas arrugadas y
miembros largos y delgados con dedos también largos. Los pacientes con
este síndrome presentan hueso frontal abultado, cuello corto, deformidades
espinales, hipoplasia del músculo de la pantorrilla, metatarso varo, y
talipes equinovaro. La artrogriposis afecta principalmente rodillas, codos
y manos. Los pacientes no presentan anormalidades neurológicas.
Síndrome de
Antley-Bixer
El
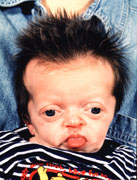
síndrome de Antley-Bixer se observa en neonatos con rasgos faciales
dismórficos (Figura 154.1. A). Muchos de ellos no pueden respirar por la
nariz (atresia coanal). El diagnóstico se confirma mediante radiografías
convencionales que demuestran anormalidades óseas, sobre todo si se
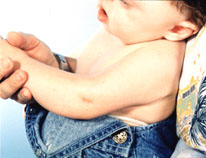
registran sinostosis radiohumeral y encorvadura femoral. La artrogriposis
involucra dedos, muñeca, codos (Figura 154.1. B), caderas, y tobillos. Los
pacientes con este síndrome pueden fallecer durante el período neonatal
por atresia coanal. Los sobrevivientes tienen inteligencia normal. Las
contracturas mejoran con la edad. Este síndrome probablemente se transmite
en forma autosómica recesiva.
|
A |
B |
|
|
Figura 154.1.—Síndrome
de Antley-Bixer. A. Rasgos faciales: braquicefalia, hueso frontal
prominente y abultado; orejas displásticas; hipoplasia mediofacial; puente
nasal deprimido; proptosis; B. Artrogriposis de los codos.
Displasia
diastrófica
Diastrófico
significa encorvado. Los neonatos con displasia diastrófica son más cortos
y con miembros encorvados (Figura 154.2). La displasia diastrófica puede
acompañarse de obstrucción respiratoria por estenosis laríngea. La muerte
ocurre por apnea obstructiva.
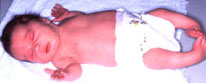
Figura 154.2.—Displasia
diastrófica. Artrogriposis de ambos pies.
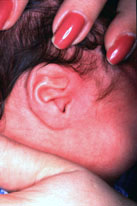
Un rasgo muy característico
de displasia diastrófica es la presencia de masas quísticas blandas en la
aurícula durante el período neonatal (Figura 155.1. A). En estos pacientes
se presenta inestabilidad atlantoaxial. Estas masas a menudo se vuelven
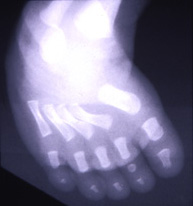
cartílagos hipertróficos en la infancia temprana. Las radiografías de los
miembros distales muestran anormalidades óseas (Figura 155.1. B). La
displasia diastrófica es un trastorno autosómico recesivo. El gen se situa
en el mapa del brazo distal largo del cromosoma 5.
|
A |
B |
|
|
Figura 155.1.—Displasia
diastrófica. A. Masa auricular quística. B. Huesos tubulares cortos y
encorvados.
Síndrome de
artrogriposis distal
Los
neonatos con síndrome de artrogriposis distal nacen a término con un peso
mediano. Su apariencia es sana. La cara no es dismórfica, aunque se han
descrito casos con paladar hendido, labio leporino, microglosia, trismus,
ptosis, y leves pliegues epicantales. La artrogriposis comprende manos y,
en menor grado, pies. Se desconoce la causa de este síndrome. Pero se
sospecha que la causa de la artrogriposis distal es una anormalid del
colágeno, que producen alteraciones de los tendones. La artrogriposis
mejora con la edad. La inteligencia no está afectada. La afección es
autosómica dominante con expresión variable. El gen de la artrogriposis
distal está en la región pericentromérica del cromosoma 9.
RESTRICCIÓN
FÍSICA DE MOVIMIENTOS
La
artrogriposis congénita múltiple por restricción física ocurre cuando la
capacidad de la cavidad uterina es pequeña y asimétrica o cuando existen
bandas cutáneas que dificultan los movimientos de los miembros fetales. La
capacidad del útero puede disminuir por oligohidramnios, malformaciones
anatómicas, bandas amnióticas, o tumores uterinos. En el síndrome de
Escobar existen bandas cutáneas gruesas que reducen los movimientos
articulares fetales.
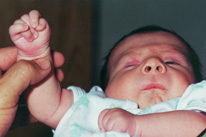
Secuencia del oligohidramnios
El
oligohidramnios es la causa más frecuente de artrogriposis múltiple debida
a contención física. Una reducción de la capacidad uterina expone al feto
a adquirir múltiples anomalías, además de la artrogriposis. La serie de
hallazgos por disminución del líquido amniótico se denomina secuencia de
oligohidramnios. Las características de esta entidad son piel arrugada;
pliegues superficiales que se extienden del ángulo interno del ojo a la
parte superior de la mejilla; nariz chata; orejas grandes de implantación
baja y rotadas hacia atrás; cuello corto y manos grandes, arrugadas. La
artrogriposis suele afectar a las rodillas y a los pies. Las causas más
frecuentes de oligohidramnios son insuficiencia placentaria y ruptura
prematura de membranas. La agenesia renal bilateral es, a menudo,
consecuencia rara, pero severa, de oligohidramnios asociado con
artrogriposis.
Síndrome de
Escobar
El
síndrome de Escobar, o síndrome de pterigios múltiples, se caracteriza por
gruesos pliegues de piel, que producen una apariencia rara y mantienen las
articulaciones en una posición fija. Los pliegues cutáneos suelen situarse
en cuello, axila, áreas antecubitales, poplíteas y digitales. Los pliegues
gruesos limitan la movilidad articular en útero y producen artrogriposis,
que es especialmente marcada en las manos. La inteligencia es normal. A
menudo se desarrolla escoliosis a los cinco años de edad. Este síndrome se
hereda de forma autosómica recesiva. Puede ser necesario el tratamiento
quirúrgico del pterigio.
ANORMALIDADES
NEUROLÓGICAS
Deben
suponerse afecciones neurológicas en cualquier neonato sin signos que
hagan sospechar anormalidad cartilaginosa, secuencia de oligohidramnios o
síndrome de Escobar. La clasificación de las anormalidades neurológicas se
basa en la región del sistema nervioso más afectada.
Síndrome de
la trisomía 13
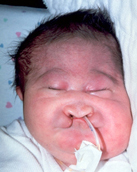
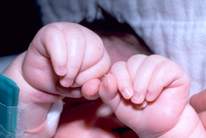
Este
síndrome se caracteriza por microcefalia, lesiones posteriores del cuero
cabelludo, microftalmía, labio leporino (Figura157.1 A), coloboma del
iris, orejas displásticas de baja implantación, sordera, paladar hendido,
polidactilia, manos de típica trisomía 18 (Figura 157.1 B), talones
prominentes, criptorquidia, y escroto anormal.
|
A |
B |
|
|
Figura 157.1.—
Síndrome de trisomía 13. A. Rasgos faciales típicos
de la trisomía 13. B. Posiciones típicas de las manos y artrogriposis
digital.
La
anomalía del sistema nervioso central más frecuente es la
holoprosencefalia (Figura 157.3). La holoprosencefalia puede ser alobar,
semilobar y lobar. La holoprosencefalia alobar se caracteriza por tener un
solo tálamo; ausencia del tercer ventrículo, de la fisura interhemisférica
y del cuerpo calloso; desplazamiento anterior de la masa encefálica; y un
ventrículo en forma de media luna que se une a un quiste dorsal gigante.
La holoprosencefalia semilobar se caracteriza por tener los tálamos
parcialmente separados; un tercer ventrículo pequeño, una fisura
interhemisférica y un cuerpo calloso presentes en la región posterior, una
buena formación de los lóbulos occipitales y temporales, y un ventrículo
telencefálico continuo con un quiste dorsal. La holoprosencefalia lobar se
caracteriza por una hipoplasia falcial anterior y de los lóbulos
frontales, la ausencia del septum pellucidum, la separación de los
tálamos, un tercer ventrículo y una fisura interhemisférica normales y la
presencia del cuerpo calloso en la región posterior.
La
trisomía 13 completa es más frecuente en hijos de madres de mayor edad. La
translocación de material del cromosoma 13 produce un fenotipo similar.
Hay que llevar a cabo estudios cromosómicos en padres de lactantes con
translocación, porque ellos pueden ser portadores asintomáticos de un
translocación balanceada. La probabilidad de recidivas es mayor si
cualquiera de los dos progenitores es portador de una translocación
balanceada.
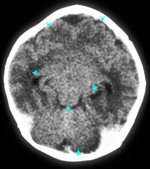
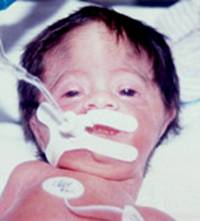
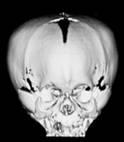
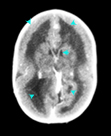
Figura 157.2.—
 TAC
del cerebro muestra holoprosencefalia; lóbulos frontales parcialmente
fusionados; formación parcial de la hendidura interhemisférica y ausencia
de terceros ventrículos. TAC
del cerebro muestra holoprosencefalia; lóbulos frontales parcialmente
fusionados; formación parcial de la hendidura interhemisférica y ausencia
de terceros ventrículos.
Síndrome de
la trisomía 18
El
síndrome se caracteriza por diámetro cefálico bifrontal estrecho, con
occipucio prominente, pliegues epicantales, boca pequeña, labio superior
corto, micrognatia, orejas displásticas de implantación baja, pecho en
escudo, esternón corto, limitación de la abducción de la cadera, y
artrogriposis de manos y pies. La posición de las manos es típica en la
mayoría de los casos. Las anomalías de los pies se caracterizan por
talipes calcáneovalgo, hallux grande y corto en dorsiflexión y talones
prominentes. El sistema nervioso central está afectado en neonatos con
este síndrome; las alteraciones más frecuentes son: mielinización anormal,
microgiria, hipoplasia cerebelosa, agenesia del cuerpo calloso,
hidrocefalia y meningomielocele. Los neonatos mueren a menudo durante el
período neonatal por apnea. Si sobreviven, requieren apoyo respiratorio y
alimentación nasogástrica. La tasa de supervivencia global es muy baja y
los que sobreviven usualmete sufren retardo mental severo. La posibilidad
de limitar las medidas médicas extraordinarias para prolongar la vida debe
discutirse con los padres.
La
trisomía del cromosoma 18 puede ser completa o incompleta (translocación).
La forma completa es más frecuente en hijos de madres viejas y tiene un
riesgo de recidivas de menos de 1%. La forma incompleta (translocación)
tiene un mayor riesgo de recividas. Los padres de un lactante con
translocación pueden ser portadores asintomáticos de una translocación
balanceada. La probabilidad de recidivas es por lo tanto mayor si
cualquiera de los padres es portador de una translocación balanceada.
Síndrome de
Pena-Shokeir I
Se
caracteriza por hipertelorismo, micrognatia, nariz chata, orejas
deformadas de implantación baja, crestas dérmicas hipoplásicas, pulmones
hipoplásicos, criptorquidia, y pie en bastón (Figura 158.1). A menudo son
mortinatos o mueren dentro del primer año de vida.
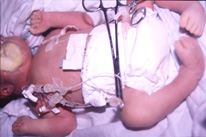
Figura 158.1.—
Síndrome de Pena-Shokeir I. Artrogriposis distal e hipoplasia pulmonar.
La artrogriposis es
prominente de modo especial en las manos; también afecta caderas y
rodillas. Coexisten anormalidades del sistema nervioso central y
periférico. La patología descrita en estos pacientes incluye cambios
atróficos neurogénicos del músculo y alteraciones histológicas de la
médula espinal y cerebelo. La hipoplasia pulmonar se produce por mengua de
la motilidad diafragmática. Los niños nacen prematuros o pequeños para la
edad gestacional. El síndrome ha sido considerado una afección autosómica
recesiva, pero con más probabilidad representa un fenotipo típico que es
consecuencia de la disminución de los movimientos fetales en determinada
edad gestacional.
Síndrome de Pena-Shokeir II
Este
síndrome, también conocido como cerebro-óculo-facio-esquelético, se
caracteriza, como está definido, por anomalías cerebrales, oculares, del
esqueleto y rasgos faciales característicos. Las alteraciones cerebrales
consisten en microcefalia, calcificación de los núcleos lenticulares y de
la sustancia blanca hemisférica, gliosis focal del tercer ventrículo,
microgiria focal, e hipoplasia del tracto óptico y quiasma. Las afecciones
oculares abarcan cataratas, blefarofimosis, y microftalmia y las
esqueléticas comprenden camptodactilia, talones prominentes, pies
achatados y una ranura longitudinal en la planta del pie. La artrogriposis
afecta de modo especial codos y rodillas. Las radiografías de las
extremidades inferiores muestran ángulos acetabulares tenues, coxa valga,
segundo metatarsiano en posición posterior y astrágalo vertical. Los
rasgos faciales característicos son cabello grueso, orejas grandes, raíz
de la nariz prominente, labio superior abultado que se sobrepone al
inferior y micrognatia. Este síndrome es un afección autosómica recesiva.
Síndrome de
trigonocefalia de Opitz
Este
síndrome, también conocido como síndrome C, se caracteriza, por la
presencia de rasgos faciales característicos (trigonocefalia, fisura
palpebral elevada en la región lateral, raiz de la nariz hipoplástica,
hipertrofia de las encias, ohidos con angulación posterior) (Figura
159.1), piel hiperelástica, anomalias cardíacas y artrogriposis distal. El
tamaño de la cabeza es normal al nacer pero el crecimiento postnatal es
pobre. Todos los casos reportados han tenido retraso mental. Este síndrome
es un afección autosómica recesiva. Los pacientes con las características
del síndorme de trigonocefalia de Opitz se deben distinguir de aquellos
con cromosomopatias (especialmente del cromosoma 3), síndrome de Frydman y
síndrome de Say-Meyer.
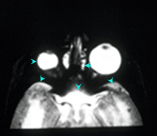
Figura 159.1—
Síndrome de trigonocefalia de Opitz. [A]
trigonocefalia y raiz de la nariz hipoplástica,; [B] Tomografía 3-D
demuestra sinostosis de la sutura metópica y de las suturas coronarias;
[C] Tomografía del cerebro demuestra la configuración anormal del cráneo y
espacio subaracnoideo prominente.
Síndrome de Smith-Lemli-Opitz
Se
caracteriza por microcefalia con frente estrecha, orejas sesgadas o con
implante bajo, ptosis, orificios nasales antevertidos, criptorquidia e
hipospadias. Los rasgos más importantes en los varones son lal
criptorquidia y el hipospadias.
El diagnóstico de este
síndrome es muy difícil en niñas porque falta lo más característico:
criptorquidia e hipospadias. Coexisten malformaciones cerebrales y del
tronco cerebral. La artrogriposis es más evidente en las manos. El
síndrome de Smith-Lemli-Opitz se establece por un notorio defecto de la
enzima 7-deshidrocolesterol reductasa en la biosíntesis del colesterol.
Esto produce niveles altos del precursor del colesterol, el
7-deshidrocolesterol, y concentraciones bajas de colesterol, las cuales, a
su vez, originan alteraciones de la función mitocondrial, síntesis
hormonal, mielinización, y del metabolismo de los ácidos biliares y de la
vitamina D. Dicho síndrome se logra diagnosticar en el período prenatal o
postnatal midiendo el 7-deshidrocolesterol mediante ensayo cromatográfico.
La mayoría de los pacientes fallecen durante el período neonatal; quienes
alcanzan a sobrevivir sufren severo retardo mental. Este síndrome se
transmite en forma autosómica recesiva.
Síndrome de Zellweger
Se
caracteriza por hipotonía severa, braquicefalia, fontanelas amplias y
suturas muy abiertas; hepatomegalia, hipospadias y criptorquidia en
varones (Figure 160.1), e hipertrofia clitoridiana en niñas; frente
prominente, occipucio aplanado, cara redonda, micrognatia, orificios
nasales antevertidos, orejas displásticas de implantación baja,
hipertelorismo, párpados edematosos, pliegues epicantales, glaucoma,
cataratas, córnea opaca y manchas de Brushfield. Puede que la
hepatomegalia no se presente en el momento de nacer, pero suele
desarrollarse durante el primer mes de vida. La artrogriposis afecta sobre
todo las articulaciones distales. La radiografía ósea revela
calcificaciones puntiformes de rótula y acetábulo. La RM muestra: (1)
hipomielinización, (2) malformaciones corticales perisilvianas y
perirolándicas, y (3) quistes germinolíticos.
El síndrome de Zellweger se asocia con altas concentraciones plasmáticas
de ácidos grasos de cadena muy larga.
|
A |
B |
|
|
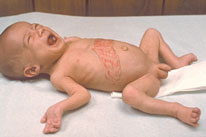
Figura 160.1.—
[A] Mancha rojo cereza en paciente con gangliosidosis GM1. [B]
Hepatoesplenomegalia en un paciente con síndrome de Zellweger.
El diagnóstico del síndrome
de Zellweger se establece por cultivo de fibroblastos y biopsia de hígado,
cuyas células, cuando se tiñen para detectar enzimas peroxisómicas, no
muestran los peroxisomas. En cambio, la electromicroscopía del mismo
tejido sí evidencia membranas peroxisómicas. De esta discrepancia ha
surgido la denominación de "peroxisomas fantasmas”, porque mientras la
membrana aparece en un examen, no lo hace en el otro.
También se relacionan con este síndrome defectos genéticos en 7q11.23 y
1p22-p21. La mayoría de pacientes fallecen durante el primer año de vida y
los que logran sobrevivir sufren retardo mental.
Esta afección se hereda de forma autosómica recesiva.
Síndrome de Walker-Warburg
Esta
afección se caracteriza por hidrocefalia, agiria (cerebral y cerebelosa),
displasia retiniana, y, en ocasiones, encefalocele. Además de la displasia
retiniana, otras anormalidades oculares son microftalmía (Figura 161.1 A),
alteraciones del vítreo, desprendimiento de retina, glaucoma, cataratas, y
opacidades corneales. Los neonatos son hipotónicos y suelen tener
convulsiones. La artrogriposis es distal. La patología cerebral se debe a
desperfectos en la lámina basal externa que, en circunstancias normales,
establece la barrera que detiene el proceso migratorio neuronal. La falta
de desarrollo de esta lámina les da vía libre a las neuronas para cruzar
las cisuras e introducirse en las meninges, originando un cerebro plano y
atascando las meninges. El síndrome es familiar, y es probable que lo sea
por su carácter hereditario autosómico recesivo, aunque cabe la
posibilidad de una infección viral intrauterina persistente, que afecta a
varios miembros de una misma familia. Su diagnóstico se hace mediante la
RM, que muestra aplanadas las superficies cerebrales y cerebelosas,
ventrículos laterales grandes, respecto a la masa cerebral y ausencia del
vermis cerebeloso (Figura 161.1 B, C).
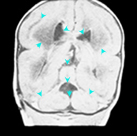
Figura 161.1.—
Síndrome de Walker-Warburg. A: imagen axial en
T2 muestra la microftalmía; B: la imagen axial en T1 evidencia la
lisencefalia con ventrículos laterales grandes; y C: imagen axial en T1
demostrando agenesia del vermis cerebeloso (hemisferios cerebelosos en
aposición sin un vermis intermedio).
Síndrome de
Marden-Walker
Se
caracteriza por expresión facial fija, blefarofimosis, micrognatia y
múltiples contracturas articulares desde el nacimiento. Son frecuentes las
malformaciones cerebrales y de fosa posterior. Los neonatos con el
síndrome de Marden-Walker pueden presentar la anormalidad de Dandy-Walker
y la hipoplasia del tronco cerebral. Se hereda de forma autosómica
recesiva.
MÉDULA ESPINAL
Amioplasia
congénita
Los
neonatos con amioplasia congénita son niños a término y de peso mediano;
su apariencia es saludable; su cara es redonda, se notan los orificios
nasales antevertidos y micrognatia (Figura 162.1 A). Casi siempre se
observan hemangiomas capilares en la parte media de la cara. Tienen
múltiples articulaciones, fijas, simétricas, con posición y distribución
típicas. Los miembros son cilíndricos y los dedos delgados. La posición de
las extremidades superiores es muy característica: hombros en aducción y
rotados medialmente, codos extendidos o flexionados, antebrazos en
pronación, muñecas dobladas con desviación cubital y dedos flexionados. La
postura de la extremidad superior, excepto en los neonatos con codos
encorvados, se parece a la de los pacientes con parálisis del plexo
braquial que afecta el tronco superior. Las extremidades inferiores con
frecuencia están comprometidas y, de ser así, toman una postura
característica: caderas flexionadas; rodillas están dobladas o extendidas;
y pies en posiciones equinovara o calcáneovalga (Figura 162.1 B).
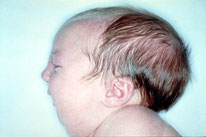
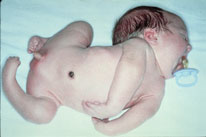
|
A |
B |
|
|
Figura 162.1—
Amioplasia congénita. A: apariencia facial típica. B: posición anómala de
las extremidades.
En los pacientes con
amioplasia congénita, no existen alteraciones cerebrales, cardíacas, o del
tracto genitourinario. Se han informado casos de atresia intestinal y
gastrosquisis. No se conoce la causa de este trastorno. La más probable
explicación es necrosis de la médula espinal fetal debida a hipotensión
sistémica la cual produce isquemia celular del asta anterior. Es
importante reconocer la amioplasia congénita porque, aunque es una
afección esporádica, las probabilidades de recidivas son pocas y el
pronóstico es bueno. Las biopsias del músculo muestran evidencia de
miopatía y neuropatía. El diagnóstico definitivo de amioplasia congénita
comprende cerca de la tercera parte de neonatos con artrogriposis.
Anormalidades de la médula espinal
La
lesión traumática de la médula espinal suele ocurrir durante el parto.
Debe sospecharse cuando no se encuentra causa alguna que explique la
artrogriposis y no hay signos de compromiso del cerebro o del tronco
cerebral. Está indicada la RM de columna. Se han descrito casos de
artrogriposis de extremidades inferiores concomitantes con el meningocele
lumbosacro (Figura 163.1) y con la agenesia del sacro.
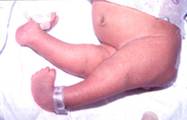
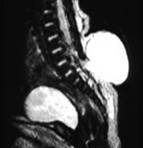
|
A |
B |
|
|
Figura 163.1.—
A:
artrogriposis de extremidades inferiores (después de cirugía). B: la RM de
la columna muestra un meningocele lumbosacro.
MOTONEURONAS
ALFA
Atrofia muscular espinal infantil
La
artrogriposis múltiple congénita se da en 10% a 20% de neonatos con
atrofia muscular espinal infantil.En
neonatos con artrogriposis inexplicable deben realizarse estudios del
ácido desoxirribonucleico para detectar la atrofia muscular espinal
infantil. El pronóstico es pobre. Parece que la afección se hereda de
forma autosómica recesiva y ligada al X.
Degeneración neuronal infantil
La
degeneración neuronal infantil sólo puede diagnosticarse por autopsia,
cuyos resultados revelan atrofia del asta anterior de la motoneurona y
cambios degenerativos en la columna de Clarke, en los tractos
corticoespinales, espinocerebelosos y espinotalámicos, en el estrato
celular de Purkinje, y núcleos talámicos ventral y dentado. Se presenta
con hallazgos en la EMG de atrofia muscular espinal infantil y velocidad
de conducción motora y sensitiva retardada. El estudio del ácido
desoxirribonucleico para precisar la enfermedad de Werdnig-Hoffmann es
normal.
Atrofia muscular espinal focal
La
artrogriposis que abarca sólo las extremidades superiores e inferiores se
presenta en la atrofia focal espinal cervical o lumbar.
Síndrome de
Moebius
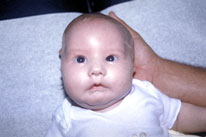
Este
síndrome consiste en debilidad facial bilateral por disfunción de la
unidad motora del VII nervio craneal o nervio facial (Figura 164.1). El
trastorno se establece por hipoplasia o destrucción de este nervio. El
síndrome de Moebius, que también se ha descrito en pacientes con patología
del músculo facial, a menudo se asocia con incapacidad para la abducción
del ojo y con cambios atróficos de la lengua por implicación del par
craneal XII. Con este síndrome se puede presentar la sucesión de Poland.
Hay deficiencia mental en el 15% de los neonatos. En general, el síndrome
de Moebius es esporádico. En algunos casos la transmisión hereditaria es
dominante. Una tercera parte de los neonatos con este síndrome sufren
artrogriposis.
Más sobre el tema...181
|
A |
B |
|
 |
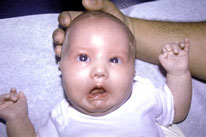 |
Figura 164.1— Síndrome de Möebius. A:
no se nota ninguna asimetría, en reposo. B: durante el llanto, la
asimetría facial es evidente.
NERVIO PERIFÉRICO
Neuropatía hipomielinizante congénita
La
artrogriposis es rara en esta afección. Los neonatos son hipotónicos y
débiles. Los estudios de conducción nerviosa revelan una conducción motora
reducida (5 a 8 metros por segundo). El diagnóstico se establece por la
biopsia del nervio safeno externo, que muestra cantidades pequeñas o falta
de las vainas de mielina y ocasionales formaciones en bulbo de cebolla.
UNIÓN MIONEURAL
Miastenia grave congénita transitoria
Los
neonatos con miastenia grave transitoria pueden tener artrogriposis y
también hipotonía, debilidad, y fatigabilidad. Esta última es el síntoma
cardinal de la miastenia grave. El diagnóstico se establece evaluando a
las madres, quienes tienen antecedentes o signos clínicos de la
enfermedad. El tratamiento con neostigmina mejora la fuerza muscular. La
fisioterapia resulta eficaz en el alivio de las contracturas.
Distrofia miotónica congénita
Los
neonatos con distrofia miotónica congénita manifiestan marcada hipotonía
facial y corporal. Estos neonatos parecen letárgicos dado el alto grado de
hipotonia facial. La artrogriposis tiende a ser más frecuente en las
extremidades inferiores que en las superiores (Figura 165.1). Las
articulaciones distales están más comprometidas que las proximales. El
único rasgo facial distintivo es la atrofia del músculo temporal, pero
ésta rara vez se presenta al nacer. La cabeza es, en general, grande. La
RM del cerebro muestra en muchas ocasiones dilatación ventricular. El
pronóstico es pobre. El retraso mental se va haciendo notorio a medida que
el niño crece, en la mayoría de los casos. La distrofia miotónica
congénita se diagnostica, demostrando la presencia de miotonía en la madre
y se confirma con la prueba del ADN. La causa del trastorno reside en la
expansión del número de repeticiones del trinucleótido en la región del
cromosoma 19q13.3. Se hereda de forma autosómica dominante, pero la forma
neonatal sólo se manifiesta cuando la madre es la portadora del gen
anormal.
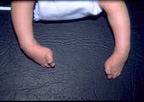
Figura 165.1.—
Artrogriposis distal en neonato con distrofia miotónica congénita.
Distrofia
muscular congénita
Los
neonatos con esta enfermedad se presentan hipotónicos, débiles y con
artrogriposis distal. La concentración sérica de la creatincinasa está
elevada o normal. Los datos electromiográficos son consistentes con el
proceso miopático (potenciales de la unidad motora breves, pequeños y
abundantes).
La biopsia del músculo muestra variaciones de tamaño de sus fibras, núcleo
central y sustitución del músculo por fibrosis y proliferación de tejido
adiposo. En algunos casos se nota deficiencia de merosina. A cada paciente
con distrofia muscular congénita se le debe hacer un estudio con RM del
cerebro, que, en la distrofia muscular congénita del tipo Fukuyama,
determina los errores migratorios (polimicrogiria, lisencefalia, y
heterotopia) e hipomielinización del centro semioval.
Miopatía miotubular
Los
neonatos con esta alteración presentan debilidad facial, ptosis,
oftalmoplejía, debilidad generalizada e hipotonía y, a veces,
artrogriposis. La forma severa cursa con serio compromiso respiratorio que
puede llevar a la asfixia. Esta forma maligna se manifiesta en los varones
(herencia ligada al cromosoma X). La miopatía miotubular se diagnostica en
base a la biopsia muscular que, con la tinción del la ATP-asa, muestra
fibras musculares (con uno o más núcleos centrales), rodeadas por un halo
claro (área desprovista de miofibrillas). El diagnóstico prenatal de la
forma recesiva ligada al X se cumple con biopsia de vellosidades
coriónicas y estudio del marcador de ADN de la región Xq28.
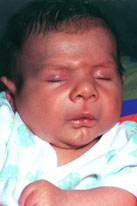
Displasia craneocarpotarsal
Esta
afección también conocida como síndrome de Freeman-Sheldon, y síndrome de
la cara silbante, se caracteriza por rasgos faciales muy peculiares: cara
aplanada con expresión de llanto, philtrum largo y labios fruncidos (como
listos para silbar) surco en la barbilla en forma de H o de V (Figura
166.1 A y B). La artrogriposis es más marcada en las extremidades
superiores que en las inferiores. Se presentan con frecuencia problemas de
la alimentación. La inteligencia se conserva normal. La enfermedad se
transmite en forma autosómica dominante, pero la modalidad recesiva se
manifiesta en algunas familias. Se desconoce la naturaleza de esta
enfermedad. La biopsia del músculo buccinador revela tejido conectivo
fibroso que reemplaza los haces musculares.
|
A |
B |
|
|
Figura 166.1.—
Síndrome de Freeman-Sheldon. A: características típicas de la cara; B:
artrogriposis distal.
Síndrome de Schwartz-Jampel
Es
una afección heterogénea heredada de forma recesiva, determinada por
miotonía, baja estatura y displasia ósea. La artrogriposis se presenta en
un número significativo de pacientes con síndrome de Schawart-Jampel. No
se dispone de pruebas genéticas para diagnosticarlo. El diagnóstico de
este síndrome requiere la presencia de miotonía. La miotonía se
caracteriza por descargas continuas, espontáneas, de bajo voltaje y alta
frecuencia, o descargas en reposo, de bajo voltaje con altibajos (como de
un bombardero en picada). Estas descargas ocurren de forma espontánea o
son desencadenadas por movimientos de la aguja. Para el diagnóstico del
síndrome de Schwartz-Jampel también se requiere la presencia de displasia
ósea. La displasia osea y la remodelación ósea que ocurre como
conscecuencia de la hipotonía muscular (disminución de tracción muscular
normal) producen: cara aplanada, micrognatia, tórax en embudo o en quilla,
fémures encorvados o en rotación externa, coxa vara o valga, displasia de
caderas, rodillas en recurvatum, talipes valgo o plano, artrogriposis y
osteoporosis. El diagnóstico diferencial incluye otras entidades que se
presentan con miotonía neonatal y artrogriposis, tales como la miotonía
del canal del sodio que es una afección heredada de modo dominante y no se
acompaña de displasia ósea y la distrofia miotónica.
DIAGNÓSTICO
DIFERENCIAL DE LA ARTROGRIPOSIS MÚLTIPLE CONGÉNITA
La
artrogriposis múltiple congénita, que abarca ambas extremidades
superiores, se parece a la parálisis bilateral del plexo braquial. La
artrogriposis múltiple congénita se distingue de la parálisis bilateral
del plexo braquial porque en la artrogriposis múltiple congénita la
posición anormal del brazo está presente en todo momento mientras que en
la parálisis bilateral del plexo braquial la posición anormal se establece
sólo cuando el neonato intenta mover el brazo y por la presencia de
hoyuelos superficiales en las muñecas en los pacientes con artrogriposis
múltiple congénita (Figura 167.2).
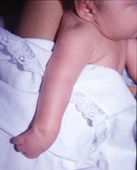
Figura 167.2.—
Posición típica del brazo y hoyuelo en la muñeca propios de la
artrogriposis múltiple congénita.
DIAGNÓSTICO
DIFERENCIAL DEL NEONATO CON ARTROGRIPOSIS MÚLTIPLE CONGÉNITA
Es
probable que los neonatos con artrogriposis proximal tengan amioplasia
congénita. Aquellos con artrogriposis distal entrañan un problema
diagnóstico complejo. En la mayoría, sólo se puede formular un diagnóstico
buscando una constelación de signos que lleven a la definición de un
síndrome; los datos de laboratorio son específicos en algunos de ellos. El
síndrome de Zellweger cursa con elevación de los ácidos grasos de cadena
muy larga en plasma. En el de Smith-Lemli-Opitz se observan niveles altos
del precursor del colesterol 7-deshidrocolesterol y concentraciones bajas
de colesterol. Las trisomías se relacionan con anormalidades cromosómicas.
El síndrome de Walker-Warburg manifiesta anormalidades características en
el estudio de RM. La atrofia muscular espinal se diagnóstica con la prueba
del ADN. La RM de columna detecta las anormalidades de la médula espinal.
La neuropatía hipomielinizante congénita se diagnóstica por estudios de
conducción nerviosa. El síndrome de Schwartz-Jampel se manifiesta con
miotonía electromiográfica y displasia ósea. Pacientes con miopatías por
anomalidades de los canales de sodio y con distrofia miotónica también
pueden tener miotonía.
TRATAMIENTO DE LA ARTROGRIPOSIS MÚLTIPLE
El
tratamiento de la artrogriposis en el período neonatal consiste en
modificar las posturas anormales mediante la colocación de yesos.
REFERENCIAS
Barkovich JA, Peck
WW. MR of Zellweger syndrome. Am J Neuroradiol. 1997;18:1163-1170.
Boylan KB, Ferriero DM, Greco CM, et al. Congenital hypomyelination
neuropathy with arthrogryposis multiplex congenita. Ann Neurol. 1992;31:337-340.
Fenichel GM. Neonatal Neurology. New York, NY: Churchill
Livingstone; 1990.
Giedion A, Boltshauser E, Briner J et al. Heterogeneity in
Schwartz-Jampel chondrodystrophic myotonia. Eur J Pediatr. 1997;214-223.
Gorlin RJ, Cohen MM, Levin LS. Syndromes of the head and neck. New
York, Oxford unniversity press; 1990:889-891.
Hageman G, Jenneckens FGI, Vette JK, et al. The heterogeneity of
distal arthrogryposis. Brain Dev. 1984;6:273-283.
Hageman G, Willemse J. Arthrogryposis multiplex congenita.
Neuropediatrics. 1983;14:6-11.
Hall JC, Reed SD, Driscoll EP. Amyoplasia: a common sporadic
condition with congenital contractures. Am J Med Genet. 1983;15:571-590.
Hall JG. In utero movement and use of limbs are necessary for
normal growth: a study of individuals with arthrogryposis. Prog in Clin
and Biol Res. 1985;200:155-162.
Heymans HS, Wanders RJA, Schutgens RBH. Peroxisomal disorders. In:
Fernandez J, Saudubray JM, Tada K, eds. Inborn Metabolic Diseases.
Diagnosis and Treatment. Berlin: Springer-Verlag; 1990:421-433.
Holmes LB, Driscoll SG, Bradley WG. Contracture in a newborn infant
of a mother with myasthenia gravis. J Pediatr. 1980;96:1067-1069.
Jones HR Jr, Bolton CF, Harpen CM Jr. Pediatric Clinical
Electromyography. Philadelphia, Penn: Lippincott-Raven; 1996.
Jones KL. Smith’s Recognizable Patterns of Human Malformations.
Philadelphia, Penn: WB Saunders; 1997.
Livingstone IR, Sack GH Jr. Arthrogryposis multiplex congenita
occurring with maternal multiple sclerosis. Arch Neurol. 1984;41:1216-1217.
Lyon G. Congenital malformation of the brain. In: Levene MI,
Lilford RJ, eds. Fetal and Neonatal Neurology and Neurosurgery. Edinburg,
Britain: Churchill Livingstone; 1994:193-214.
Papazian O. Transient neonatal myasthenia gravis. J Child Neurol.
1992;7:135-141.
Sarnat HB, Case ME, Graviss R. Sacral agenesis. Neurologic and
neuropathologic features. Neurology. 1976;26:1124-1129.
Schneider V, Cabrera-Meza G, eds. Rudolph's Brief Atlas of the
Newborn. Hamilton, Ontario: Decker Publishing Inc; 1998.
Volpe JJ. Neurology of the Newborn. Philadelphia, Penn: WB
Saunders; 1995.
|