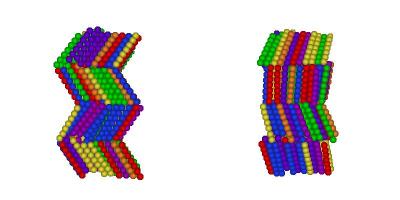
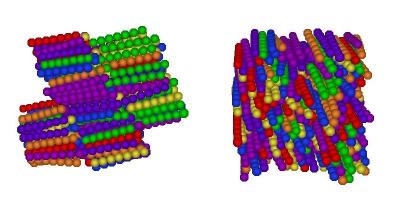
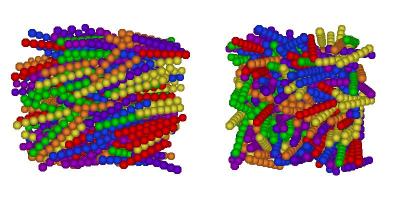
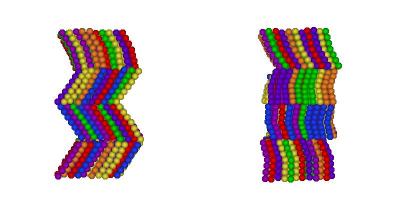
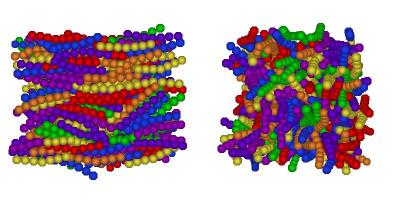
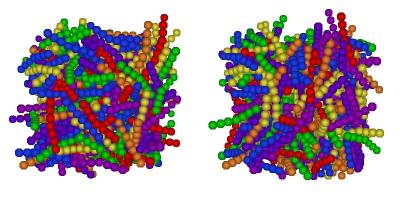
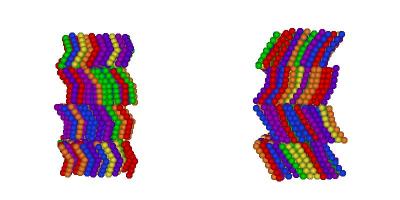
I've finally finished my Phd in theoretical chemistry under the supervision of Dr Philip Camp in the School of Chemistry at Edinburgh University. Below is a brief history and some details of research that I've performed before and during my Phd. My homepage can be found here.